

The immunopathogenesis of hepatitis B virus (HBV) involves a complex interaction of innate and adaptive immune response associated with several cytokines. However, due to experimental limitations, very limited and even no activation of the innate immune response can be demonstrated in acute HBV infection. The role of innate immunity and the key innate effectors for HBV clearance is still uncertain. It is also not clear whether clinical use of TNF inhibitors in treating rheumatoid arthritis and other autoimmune disease patients with chronic HBV infection would affect viral reactivation. However, in a recent paper in Cell Mol Immunol, Chyuan et al. reported that under TNF-α blockade, HBV viral clearance was impaired with persistent elevated HBV viral load in a dose and temporal-dependent manner.1 It demonstrates that a deficiency of TNF-α reduces viral clearance and promotes HBV persistence in a mouse model, and the impairment of HBV clearance under anti-TNF-α occurs at an early time point of HBV infection, suggesting that HBV may reactivate during therapy with TNF inhibitors. The results together with previous reports support that TNF is a key innate cytokine required to clear HBV.2,3 It also provides evidence that therapy with TNF inhibitors may impair immune response to HBV clearance.
Weak or even absent activation of innate immunity seems to be the hallmark of acute HBV infection. Moreover, pro-inflammatory cytokines are often undetectable during the early phases of HBV infection. For over two decades, numerous efforts have been reported in search for critical innate effectors in HBV clearance. It is suggested that an efficient control of HBV infection requires the coordinated actions of innate immune cells and viral specific T cells; however, the innate immunity and key cytokine related to HBV clearance are still not well defined. Acute HBV infection models of both woodchucks and chimpanzees also fails to verify the role of innate immune response in HBV infection. It is an overall scenario of weak or even transcription silence in activation of innate immunity to HBV, in particular, the type I interferon (IFN-I) response. Apparent lack of the IFN-I response is observed in HBV infection and thereby considered as one of the underlying mechanisms for HBV to escape innate recognition.4,5 Different lines of evidence indicate that it is questionable if HBV can actually be sensed by the innate immunity.6 In fact, the mechanisms how innate immune system senses HBV are still not elucidated and it is also unclear which HBV molecules (DNA, RNA or viral proteins) are actually recognized by the pattern recognition receptors (PRR) and thereby trigger the antiviral response.
In order to explore the role of innate sensor in response to HBV, we previously investigated a panel of gene knockout mice with deficiency in innate immune sensors or effectors for their capability in HBV clearance.2 Instead of IFN receptor deficiency, interestingly, TNF-α deficiency in knockout mice resulted in impaired clearance and prolonged persistence of HBV, suggesting that TNF-α and associated innate pathways are crucial in HBV clearance.2 The results are consistent with previous studies supporting that TNF is a key innate effector required to clear HBV.3
TNF-α is a well-known pro-inflammatory cytokine and displays multiple functions as an antitumor, immune modulation and integrating host defense system against infections. As a key cytokine, the direct anti-viral effect of TNF-α also contributes to HBV eradication.7,8,9 A recent study demonstrated that cellular inhibitor of apoptosis proteins (cIAPs) attenuate TNF-α signaling during HBV infection and restrict the death of infected hepatocytes, thus allowing viral persistence.10 In addition, TNF-α delivers non-cytopathic antiviral signals to hepatocytes to degrade the transcript and the nucleocapsid particles of HBV, and supports HBV-specific T cells to abolish HBV replication in hepatocytes.11 TNF-α destabilizes nucleocapsids, reduces nuclear viral DNA,12 and inhibits the transcriptional activity of the HBV core promoter, leading to interfering cccDNA integrity. Furthermore, cleaved c-FLIP mediates the antiviral effect of TNF-α against HBV by dysregulating hepatocyte NF-κB pathway.13 In adoptively transferred HBV-specific CD8+ T cells in HBV-transgenic mice model, TNF-α abolished HBV gene expression and replication in the liver without killing the hepatocytes.8 Moreover, the TNF-α and IFN-γproduced by T cells, reduced HBV cccDNA levels in HBV-infected hepatocytes.14 In addition, TNF-α drives local intrahepatic proliferation of T cells in a way called iMATEs (intrahepatic myeloid-cell aggregates for T-cell population expansion), facilitating the expansion of the cytotoxic T lymphocytes against viral infection.15 Higher intrahepatic level of TNF-α is associated with increased HLA class I expression and enhanced CD8+ T cell response against HBV. Therefore, the TNF-α-induced HBV clearance may be due to its direct anti-HBV effect, while the induced innate immunity by HBV and TNF-α-triggered cytopathic and noncytopathic mechanisms are also involved. The role of TNF in HBV clearance is evidenced not only by impaired T cell responses against HBV and prolonged viral persistence in TNF receptor-1 (TNFR1) knockout mice 3 and TNF-α blockaded model mice,1 but also by the genetic studies, in which TNF-α gene polymorphisms, in particular the promoter region, affect susceptibility to chronicity and outcome of HBV infection.16 All these data support TNF-α as a key cytokine in the immune response to HBV infection.
As one of the breakthroughs in clinic, blockage of TNF-α pathway with associated inhibitors has been widely used to treat many autoimmune inflammatory diseases, including rheumatoid arthritis, ankylosing spondylitis, psoriasis and psoriatic arthritis, as well as inflammatory bowel disease. The use of TNF-α inhibitors paradoxically illustrated the crucial role of this pathway in host defense against infections since TNF-α blockade could induce reactivation of quiescent or latent infections such as tuberculosis. Typically, accumulated lines of evidence support that TNF-α inhibitors increase risk of HBV reactivation in patient with chronic HBV;17,18 however, strictly controlled studies are needed to ensure the TNF-α blockage-triggered HBV reactivation. In HBV model mice, TNF-α knockout or TNF-α inhibitors reduce viral clearance, which is associated with increased number of intrahepatic PD-1highCD127low exhausted CD8+ T cells,1,2 footnoting one of the potential mechanisms for the induced HBV reactivation. In addition, the HBV viral load was increased by anti-TNF-α Ab in a dose-dependent manner. Sufficient blockage of TNF-α at an early phase significantly impaired subsequent HBV clearance and resulted in its persistence with an elevated viral load, indicating that TNF-α deficiency impairs immune response to HBV at or from an early phase. Furthermore, TNF-α blockade also abolishes the effects of TLR9 ligand-induced facilitation of HBV clearance.1 Taken together, these results indicate that anti-TNF-α therapy impairs T cell functions and delays HBV clearance.
So far, with the lack of type I IFN response, the TNF pathway is the only innate response demonstrated to play a key role in HBV clearance. Nevertheless, it is necessary to clarify how innate immune cells are triggered and regulated to produce TNF-α during HBV infection. As one possible mechanism that we have previously reported, HBV core protein is sensed by the innate immune system to induce TNF-α production and thereby viral clearance; which can also be abolished by TNF-α blockade.2 Although the cellular source of TNF-α remains to be elucidated, several studies have reported that TNF-α is released by activated monocytes, T cells, NK cells, mast cells, B cells and Kupffer cells in the liver.19,20 Moreover, the intra-hepatic leukocytes (IHLs), but not the hepatocytes, are the cell source responsible for TNF-α production induced by HBV core. These results suggest innate immunity is able to sense HBV and induce TNF-α via HBV core (Figure 1). The innate sensors binding with HBV and the recognized molecular pattern of HBV by the innate immune system remain to be identified. Failure of sufficient TNF-α production by the immune system and early TNF-α blockade during HBV infection, results in reduced viral clearance and elevated viral load. It suggests that HBV may reactivate during anti-TNF therapy. Taken together, anti-TNF therapy can reactivate chronic HBV infection by impairing the immune response to HBV and suitable countermeasures may be necessary.
Figure 1.
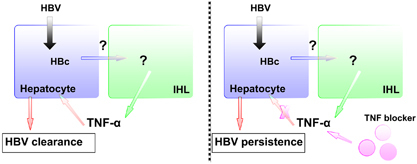
Association between host innate immune system and HBV to induce TNF-α. Innate immune system senses HBV core through unidentified sensors and then produces TNF-α. Insufficient TNF-α production and blockade of TNF-α pathway impair HBV clearance and result in its persistence.
Acknowledgements
This work was financially supported by a grant from the Ministry of Science and Technology (MOST), Taiwan
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Chyuan IT, Tsai HF, Tzeng HT, Sung CC, Wu CS, Chen PJ, et al. Tumor necrosis factor-alpha blockage therapy impairs hepatitis B viral clearance and enhances T-cell exhaustion in a mouse model. Cell Mol Immunol. 2015;12:317–325. doi: 10.1038/cmi.2015.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tzeng HT, Tsai HF, Chyuan IT, Liao HJ, Chen CJ, Chen PJ, et al. Tumor necrosis factor-alpha induced by hepatitis B virus core mediating the immune response for hepatitis B viral clearance in mice model. PLoS One. 2014;9:e103008. doi: 10.1371/journal.pone.0103008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang PL, Althage A, Chung J, Maier H, Wieland S, Isogawa M, et al. Immune effectors required for hepatitis B virus clearance. Proc Natl Acad Sci U S A. 2010;107:798–802. doi: 10.1073/pnas.0913498107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wieland SF, Chisari FV. Stealth and cunning: hepatitis B and hepatitis C viruses. J Virol. 2005;79:9369–9380. doi: 10.1128/JVI.79.15.9369-9380.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertoletti A, Ferrari C. Innate and adaptive immune responses in chronic hepatitis B virus infections: towards restoration of immune control of viral infection. Gut. 2012;61:1754–1764. doi: 10.1136/gutjnl-2011-301073. [DOI] [PubMed] [Google Scholar]
- 6.Durantel D, Zoulim F. Innate response to hepatitis B virus infection: observations challenging the concept of a stealth virus. Hepatology. 2009;50:1692–1695. doi: 10.1002/hep.23361. [DOI] [PubMed] [Google Scholar]
- 7.Guidotti LG, Chisari FV. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu Rev Immunol. 2001;19:65–91. doi: 10.1146/annurev.immunol.19.1.65. [DOI] [PubMed] [Google Scholar]
- 8.Guidotti LG, Ishikawa T, Hobbs MV, Matzke B, Schreiber R, Chisari FV. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity. 1996;4:25–36. doi: 10.1016/S1074-7613(00)80295-2. [DOI] [PubMed] [Google Scholar]
- 9.Biermer M, Puro R, Schneider RJ. Tumor necrosis factor alpha inhibition of hepatitis B virus replication involves disruption of capsid Integrity through activation of NF-kappaB. J Virol. 2003;77:4033–4042. doi: 10.1128/JVI.77.7.4033-4042.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ebert G, Preston S, Allison C, Cooney J, Toe JG, Stutz MD, et al. Cellular inhibitor of apoptosis proteins prevent clearance of hepatitis B virus. Proc Natl Acad Sci U S A. 2015;112:5797–5802. doi: 10.1073/pnas.1502390112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chisari FV. Cytotoxic T cells and viral hepatitis. The Journal of clinical investigation. 1997;99:1472–1477. doi: 10.1172/JCI119308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puro R, Schneider RJ. Tumor necrosis factor activates a conserved innate antiviral response to hepatitis B virus that destabilizes nucleocapsids and reduces nuclear viral DNA. J Virol. 2007;81:7351–7362. doi: 10.1128/JVI.00554-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park YK, Park ES, Kim DH, Ahn SH, Park SH, Lee AR, et al. Cleaved c-FLIP mediates the antiviral effect of TNF-alpha against hepatitis B virus by dysregulating hepatocyte nuclear factors. J Hepatol. 2016;64:268–277. doi: 10.1016/j.jhep.2015.09.012. [DOI] [PubMed] [Google Scholar]
- 14.Xia Y, Stadler D, Lucifora J, Reisinger F, Webb D, Hosel M, et al. Interferon-gamma and Tumor Necrosis Factor-alpha Produced by T Cells Reduce the HBV Persistence Form, cccDNA, Without Cytolysis. Gastroenterology. 2016;150:194–205. doi: 10.1053/j.gastro.2015.09.026. [DOI] [PubMed] [Google Scholar]
- 15.Huang LR, Wohlleber D, Reisinger F, Jenne CN, Cheng RL, Abdullah Z, et al. Intrahepatic myeloid-cell aggregates enable local proliferation of CD8(+) T cells and successful immunotherapy against chronic viral liver infection. Nature immunology. 2013;14:574–583. doi: 10.1038/ni.2573. [DOI] [PubMed] [Google Scholar]
- 16.Xiao Q, Fu B, Chen P, Liu ZZ, Wang W, Ye Q. Three polymorphisms of tumor necrosis factor-alpha and hepatitis B virus related hepatocellular carcinoma: A meta-analysis. Medicine (Baltimore) 2016;95:e5609. doi: 10.1097/MD.0000000000005609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ye H, Zhang XW, Mu R, Fang LK, Gu JR, Lin J, et al. Anti-TNF therapy in patients with HBV infection—analysis of 87 patients with inflammatory arthritis. Clinical rheumatology. 2014;33:119–123. doi: 10.1007/s10067-013-2385-1. [DOI] [PubMed] [Google Scholar]
- 18.Perrillo RP, Gish R, Falck-Ytter YT. American Gastroenterological Association Institute technical review on prevention and treatment of hepatitis B virus reactivation during immunosuppressive drug therapy. Gastroenterology. 2015;148:221–244 e223. doi: 10.1053/j.gastro.2014.10.038. [DOI] [PubMed] [Google Scholar]
- 19.Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/S0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 20.Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nature reviews Rheumatology. 2016;12:49–62. doi: 10.1038/nrrheum.2015.169. [DOI] [PMC free article] [PubMed] [Google Scholar]