

Abstract
NagR, belonging to the GntR/HutC family, is a negative regulator that directly represses the nagP and nagAB genes, which are involved in GlcNAc transport and utilization in Bacillus subtilis. Our previous work confirmed that the chitinase B gene (chiB) of Bacillus thuringiensis strain Bti75 is also negatively controlled by YvoABt, the ortholog of NagR from B. subtilis. In this work, we investigated its regulatory network in Bti75 and found that YvoABt is an N-acetylglucosamine utilization regulator primarily involved in GlcNAc catabolism; therefore YvoABt is renamed as NagRBt. The RNA-seq data revealed that 27 genes were upregulated and 14 genes were downregulated in the ΔnagR mutant compared with the wild-type strain. The regulon (exponential phase) was characterized by RNA-seq, bioinformatics software, electrophoretic mobility shift assays, and quantitative real-time reverse transcription PCR. In the Bti75 genome, 19 genes that were directly regulated and 30 genes that were indirectly regulated by NagRBt were identified. We compiled in silico, in vitro, and in vivo evidence that NagRBt behaves as a repressor and activator to directly or indirectly influence major biological processes involved in amino sugar metabolism, nucleotide metabolism, fatty acid metabolism, phosphotransferase system, and the Embden–Meyerhof–Parnas pathway.
Keywords: N-acetylglucosamine, NagRBt, pleiotropic, repressor, activator, Bacillus thuringiensis
Introduction
N-Acetylglucosamine (GlcNAc) is a nitrogen-containing monosaccharide that is a preferred nutrient source for the growth and development of many microorganisms because it is highly abundant and provides both carbon and nitrogen (Mobley et al., 1982). After being absorbed, GlcNAc is funneled into the glycolysis shunt pathway for catabolism or directed to peptidoglycan synthesis for anabolism (Bertram et al., 2011). GlcNAc is exploited for both catabolic and anabolic purposes; therefore, its correct utilization requires rigorous control.
DasR (deficient in aerial hyphae and spore formation), a member of the gluconate operon repressor (GntR) family transcriptional repressor, was initially reported to be related to the complex morphological differentiation in Streptomyces griseus (Seo et al., 2002). Subsequently, Rigali et al. found the role of DasR in GlcNAc metabolism using Streptomyces coelicolor as a model organism (Rigali et al., 2004). Later studies revealed that DasR is a global regulator that plays a pivotal role in the regulation of antibiotic synthesis, morphological differentiation, and GlcNAc transport and metabolism (Rigali et al., 2006, 2008; Swiatek et al., 2012; Tenconi et al., 2015). In addition, it had also been shown to act as a repressor and an activator in S. coelicolor (Nazari et al., 2013) (Swiatek-Polatynska et al., 2015). The DNA-binding site of DasR has been identified as a conserved 16-bp consensus sequence in S. coelicolor, and was named the DasR-responsive element (dre) (Colson et al., 2007). DasR can specifically bind the dre sites in the upstream regions of some genes to control their expressions.
Bertram et al. found that YvoA, an ortholog of DasR, is a less prominent regulator than DasR in Bacillus subtilis and only directly represses the nagP and nagAB genes, which are involved in GlcNAc transport and utilization (Bertram et al., 2011). Moreover, as the distribution of predicted YvoA-binding sites (dreBacillus) was limited to nagP and the nagAB-yvoA locus within the chromosome of B. subtilis, they suggested renaming YvoA as NagR, for the GlcNAc utilization regulator.
In our previous work, we found that, in addition to nagP and nagAB, chitinase A and chitinase B genes are also negatively controlled by YvoABt (Jiang et al., 2015). Since the main genes controlled by YvoABt are involved in GlcNAc catabolism, we renamed YvoABt as NagRBt. Moreover, we predicted some other genes that might have dre-like sites on their promoter region or in nearby areas using the PREDetector software program. Consequently, we speculated that NagRBt might act as a pleiotropic transcriptional regulator to modulate more genes in various metabolic pathways.
In the present study, we demonstrate that NagRBt acts as a pleiotropic transcriptional regulator that controls at least 19 genes directly and 30 genes indirectly, and these genes are involved in amino sugar metabolism, nucleotide metabolism, fatty acid metabolism, phosphotransferase system, and the Embden–Meyerhof–Parnas (EMP) pathway. Moreover, our study indicates that NagRBt is also a dual transcription regulator that acts as both a repressor and an activator in Bti75. Finally, a regulatory model network of NagRBt is presented and discussed.
Materials and methods
Bacterial strains and culture conditions
The wild-type Bacillus thuringiensis strain Bti75 and its mutants were used in this study. These strains were cultivated at 30°C in an orbital shaker (200 rpm). Escherichia coli DH5α, and E. coli BL21 (DE3) were generally grown at 37°C with shaking at 200 rpm. All the strains were cultured in lysogeny broth (LB) medium. The culture medium contained the following appropriate concentrations of antibiotics: 50 μg mL−1 of erythromycin (Erm), 100 μg mL−1 of ampicillin (Amp), 10 μg mL−1 of chloramphenicol (Cm), and 100 μg mL−1 of kanamycin (Kan). The bacterial strains and plasmids used in this study are listed in Table 1.
Table 1.
Bacterial strains and plasmids used in this study.
| Plasmid or strain | Relevant characteristic(s)a | Source or reference |
|---|---|---|
| STRAINS | ||
| Bti75 | Wide-type strain | Laboratory collection |
| Bti75ΔnagR | Bti75ΔnagR | Laboratory collection |
| Bti75ΔnagR-pPerm-nagR | Bti75ΔnagR-pPerm-nagR | This study |
| E. coli DH5α | Laboratory collection | |
| E. coli BL21-pET- nagR | Kanr; nagR gene cloned into pET-28a(+), His tag binding C terminus | Laboratory collection |
| PLASMIDS | ||
| pKSV7 | Ampr Emr Cmr; Bacillus-E. coli shuttle vector, temp sensitive | Laboratory collection |
| pKSV7-Perm-nagR (pPerm-nagR) | Ampr Emr Cmr; 732bp fragment of nagR overlapped with the promoter of erm cloned into the pKSV7 EcoRI/XbaI site | This study |
| pCB | Ampr Emr; the purpose promoter-probe vector containing the β-galactosidase gene bgaB; Bacillus-E. coli shuttle vector, temp sensitive | Laboratory collection |
| pCB-Ppgi | Ampr Emr; the promoter of pgi without its dre site cloned into the pCB BamHI/SalI site | This study |
| pCB-dre+Ppgi | Ampr Emr; the promoter of pgi with its dre site cloned into the pCB BamHI/SalI site | This study |
| pCB-Ppgi+dre | Ampr Emr; the position interchange between the promoter of pgi and its dre site cloned into the pCB BamHI/SalI site | This study |
Ampr, ampicillin resistance; Emr, erythromycin resistance; Cmr, chloramphenicol resistance; Kanr, kanamycin resistance.
Construction of nagR deletion mutants and complemented strains
The nagR (formerly termed yvoA) deletion mutant, ΔnagR, was constructed in our laboratory and reported previously (Jiang et al., 2015). To construct the complemented mutant strain of nagR, the fragment of nagR was amplified from Bti75. The fragment was overlapped with the erm promoter, digested with EcoRI and XbaI, and inserted into the corresponding sites of pKSV7, to create the pKSV7-Perm-nagR (pPerm-nagR) complementing plasmid. The resulting plasmid was transferred into Bti75 by electroporation (Lecadet et al., 1992). The desired complementary strains were screened by Cm-resistance screening, PCR detection, and sequencing verification. All primers used in this study are shown in Table S1.
Electrophoretic mobility shift assay
The expression and purification of NagRBt (formerly termed YvoABt) were performed according to previously described methods (Jiang et al., 2015). DNA probes (16 bp) obtained using software prediction were extended by 12 bp on both sides. The ~40 bp DNA probes were generated by annealing primers. The DNA probes were heated at 98°C for 5 min and then incubated at room temperature for 20 min. The upstream regions of each differentially expressed gene (~300 bp upstream of the ATG start site) from RNA-seq were generated using PCR reactions. The 5′ ends of some primers were labeled with biotin for non-specific and specific competition assays. The concentrations of proteins and DNA probes were determined using a NanoDrop 2000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and are indicated in the corresponding figures.
Electrophoretic mobility shift assays (EMSAs) were carried out at 30°C for 30 min in a binding reaction buffer containing 10 mM Tris HCl (pH 8.0), 1 mM MgCl2, 50 mM NaCl, 0.5 mM DTT, 0.5 mM EDTA, and 5% glycerol. After binding, the samples were separated using a non-denaturing polyacrylamide gel in an ice bath of 1 × Tris-borate-EDTA (TBE) buffer at 50 V for 4 h. Then the gel was exposed to ultraviolet light after EB staining for preliminarily screening. For DNA probes, which gave an obviously shifted band in the direct assay, unlabeled specific fragments (400-fold) and non-specific competitor DNA (0.5 mg mL−1 sheared salmon sperm DNA) were used. In this case, biotin-labeled probes were incubated with NagRBt. After binding, the samples were loaded and separated on non-denaturating PAGE gels, and then transferred onto a nylon membrane. The biotin-labeled probes were detected using a biotin chromogenic detection kit (Thermo Fisher Scientific Inc.).
RNA preparation and transcriptome assay
Starting with an independent colony from the LB agar plate, Bti75 and Bti75 ΔnagR were grown overnight in LB medium at 30°C. They were diluted 1:100 (vol/vol) into LB and shaken at 200 rpm at 30°C for ~9 h, at which point they reached the exponential phase. The bacteria were harvested, immediately frozen in liquid nitrogen, and stored at −80°C until use. The two samples (Bti75 and Bti75 ΔnagR) were shipped to GENEWIZ (www.genewiz.com) for mRNA-seq library construction and transcriptome sequencing. The mRNA-seq library construction comprised the following steps: RNA extraction and quality control (Agilent Eukaryote Total RNA Nano Kit, Agilent 2100), library construction (NEBNext Ultra RNA Library Prep Kit), library purification (Beckman AMPure XP beads), library detection (Agilent High Sensitivity DNA Kit, Agilent 2100), quantitative analysis of the library (ABI 7500 RealTime PCR System, KAPA SYBR Green FAST Universal 2 × qPCR Master Mix), and cBOT automatic clusters (TruSeq PE Cluster Kit v3). The process of each step was strictly controlled and then Solexa sequencing was performed using an Illumina HiSeq™ 2500 instrument for each sample that passed the quality test. The process of RNA-Seq data analysis was as follows. The original data were identified using CASAVA (v1.8.2) (Hosseini et al., 2010) and the excess adaptors and low-quality sequences were removed to obtain clean reads using Trimmomatic (v0.30) (Bolger et al., 2014). High-quality sequences were further analyzed by downstream processing and mapped according to the reference sequence (ftp://ftp.ncbi.nih.gov/genomes/Bacteria/Bacillus_thuringiensis_HD_789_uid173860/) using bowtie2 (2.1.0) (Langdon, 2015). The assembled sequences were analyzed based on a series of databases to classify and annotate the obtained reads. The normalization method of fragment per kilobase per million mapped reads (FPKM) (Mortazavi et al., 2008) was used to calculate gene expression.
cDNA synthesis and quantitative real-time reverse transcription PCR analysis
cDNA was synthesized from the RNA of each sample using a PrimeScript™ RT Reagent Kit with gDNA Eraser (Takara, Dalian, China), according to the manufacturer's instructions. Quantitative real-time reverse transcription PCR (qRT-PCR) was performed using TB Green Premix Ex Taq II (Takara) and the PCR products were detected using a StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) based on the manufacturer's instructions. The relative expression level of each gene was normalized by that of 16S rRNA, as an endogenous control, and calculated according to the 2−ΔΔCT method (Livak and Schmittgen, 2001). Experiments using independent biological and technical replicates of each biological sample for each gene were repeated three times to ensure reliability and reproducibility. The primers for qRT-PCR are presented in Table S1.
Promoter/dre position swapping experiments
Promoter/dre position swapping experiments were carried out using a promoter-probe vector pCB containing the reporter gene β-galactosidase (bgaB) (Xie et al., 2012). This vector was used to determine the effect of the changes in the relative position between the promoter and the dre site on the expression of the reporter gene. The pgi gene was activated by NagRBt and we chose its upstream region to construct the corresponding plasmids. Different fragments carrying the pgi promoter with and without the dre operator were amplified: 1, promoter without a dre “Ppgi” (oligos Ppgi-F and Ppgi-R); 2, promoter with its own dre “dre+Ppgi” (oligos Pdre+Ppgi-F and Ppgi-R); 3, promoter with dre downstream “Ppgi+dre” (oligos Ppgi-F and Ppgi+dre-R) (Figure 4A). F oligos carry BamHI sites and R oligos carry SalI sites. Ppgi+dre-R also has an extension with a copy of the dre site. Fragments were digested with SalI and BamHI enzymes and inserted into pCB digested with the same enzymes creating pCB-Ppgi, pCB-drepgi+Ppgi, and pCB-Ppgi+drepgi, respectively. The resulting plasmids were transferred into Bti75 using electroporation, and then the expression of bgaB was determined using qRT-PCR.
Figure 4.

Influence of the relative position of the promoter and the dre site in pgi gene. (A) Binding site of NagRBt and promoter in the upstream region of pgi gene. The putative−35 and−10 positions (predicted by Neural Network Promoter Prediction, http://www.fruitfly.org/seq_tools/promoter.html, score 0.94) are shaded. The corresponding predicted +1 is indicated by a bent arrow and the dre site is boxed. The pgi promoter region (underlined) was amplified with primers (Ppgi-F and Ppgi-R). The fragment with the dre site before the promoter (i.e., native configuration) was amplified with primers (Pdre+Ppgi-F and Ppgi-R) and the fragment with dre site after the promoter with primers (Ppgi-F and Ppgi+dre-R). (B) Schematic diagram of the change in the relative position with promoter and dre site. (C) qRT-PCR was used to examine the expression of bgaB in three plasmids (pdrepgi+Ppgi, pPpgi+drepgi, and pPpgi) in wild-type strain Bti75. (D) β-galactosidase activity assay of three plasmids (pdrepgi+Ppgi, pPpgi+drepgi, and pPpgi) in wild-type strain Bti75. Significance was calculated by two-sample t-test (*p < 0.05, **p < 0.01, and ***p < 0.001).
β-galactosidase activity assay
β-galactosidase activity of Bt strains were performed according to the protocols as previously described (Hirata et al., 1984) and expressed in Miller units. All the reactions were run at least in three independent assays.
Computational prediction of dre sequences in Bti75
NagRBt-binding upstream sequences of five dre targets (drenagA: GCACGAGTAGTTGTCT; drenagP: ACACATCTATACAACT; drechiB: AGACTTCGTGATGTCT; drechiA: ATACATCTAGACAACT; drechitin: AGTTGGCTAGTCATCT) in Bti75 were used to predict the NagRBt-binding sites in B. thuringiensis subsp. israelensis strain HD-789 using PREDetector (Hiard et al., 2007). The position weight matrix fitted out by these five binding sequences was used to predict the potential dre sites, which are listed in Table S2; the cutoff score was greater than 7.0.
Results
Transcriptome sequencing
To study the regulatory range of NagRBt, we used Solexa/Illumina sequencing to perform a transcriptome sequencing analysis for two samples (Bti75 and Bti75-ΔnagR) in the exponential phase (~9 h). In the gene expression analysis, a mathematic algorithm was used to identify differentially expressed genes [false discovery rate (FDR) < 0.01 and absolute log2 values ratio ≥ 2]. This analysis identified 27 upregulated genes and 15 downregulated genes in Bti75-ΔnagR. The RNA-seq data were deposited at the NCBI Sequence Read Archive (submission no. SUB2391258).
As shown in Table 2, data from the RNA-seq analysis revealed that the expression of genes nagAB, nagP, chiA, chiB, and chitin-binding protein were all increased in the ΔnagR mutant relative to the wild-type strain. These genes, which are involved in amino sugar metabolism, were previously shown to have a dre site. In addition, we found other differentially expressed genes (DEGs) in Bti75-ΔnagR. Cytidine deaminase (BTF1_06850), pyrimidine-nucleoside phosphorylase (BTF1_06845), and pyrimidine nucleoside transporter (BTF1_06840) involved in nucleotide metabolism might be on the same operon. The expression of genes, lipoprotein (BTF1_26870), acyl-dehydrogenase (BTF1_25115 and BTF1_08960), and acyl carrier protein (BTF1_17540) involved in fatty acid metabolism were decreased in the Bti75-ΔnagR mutant, suggesting that NagRBt could activate these genes, rather than repress them. Glucose-specific phosphotransferase enzyme IIA (BTF1_24980), phosphoenolpyruvate-protein phosphotransferase (BTF1_18555), phosphocarrier protein HPr (BTF1_18560), and the PTS lactose cellobiose family IIC subunit (BTF1_24425) involved in the phosphotransferase system showed enhanced expression as previously reported in S. coelicolor (Rigali et al., 2004, 2006).
Table 2.
Genes controlled by NagRBt in Bti75.
| Locus_taga | Gene | Description | Positionsb | dre motifc | Scored | Fold changee | qRT-PCRf |
|---|---|---|---|---|---|---|---|
| GENES UPREGULATED IN THE ΔnagR MUTANT COMPARED TO WILD-TYPE STRAIN Bti75 | |||||||
| BTF1_00220 | nagP | PTS system subunit IIBC | −107 | acacatctatacaact | 12.12 | 17.301 | 21.967 ± 0.785 |
| BTF1_01610 | Outer surface protein | −156 | agatgtatagacttgt | 9.8 | 20.046 | 6.046 ± 0.280 | |
| BTF1_01615 | licR | BigG family transcription antiterminator | / | / | / | 7.751 | NT |
| BTF1_06840 | nupC | nucleoside transporter NupC | / | / | / | 4.738 | NT |
| BTF1_06845 | pdp | Pyrimidine-nucleoside phosphorylase | / | / | / | 14.614 | NT |
| BTF1_06850 | cdd | Cytidine deaminase | / | / | / | 19.753 | 45.010 ± 2.977 |
| BTF1_07625 | Hypothetical protein | / | / | / | 12.407 | NT | |
| BTF1_09020 | oxlT | Oxalate formate antiporter | / | / | / | 10.024 | NT |
| BTF1_11065 | Hypothetical protein | / | / | / | 12.690 | NT | |
| BTF1_11665 | lpmo | Chitin-binding protein | −56 | agttggctagtcatct | 9.56 | 25.204 | NT |
| BTF1_16665 | chiA | Extracellular exochitinase Chi36 | −36 | atacatctagacaact | 12.76 | 6.679 | 3.551 ± 0.270 |
| BTF1_18555 | ptsI | Phosphoenolpyruvate-protein phosphotransferase | / | / | / | 6.284 | NT |
| BTF1_18560 | ptsH | Phosphocarrier protein HPr | −52 | agttgtatagacgtga | 9.82 | 6.395 | 4.716 ± 0.299 |
| BTF1_18585 | nagB | Glucosamine-6-phosphate deaminase | / | / | / | 11.365 | NT |
| BTF1_18590 | nagA | N-acetylglucosamine-6-phosphate deacetylase | −90 | gcacgagtagttgtct | 8.05 | 21.012 | 36.413 ± 1.955 |
| BTF1_19070 | Hypothetical protein | −78 | acatgtcaagacaact | 9.82 | ND | NT | |
| BTF1_19250 | Hypothetical protein | / | / | / | 7.169 | NT | |
| BTF1_20050 | ispH | 4-hydroxy-3-methylbut-2-enyl diphosphate reductase | / | / | / | 7.307 | NT |
| BTF1_23785 | dps | DNA protection during starvation protein 2 | / | / | / | 7.428 | NT |
| BTF1_24425 | chbC | PTS system cellobiose-specific transporter subunit IIC | / | / | / | 4.807 | NT |
| BTF1_24980 | crr | PTS system glucose-specific transporter subunit IIA | −251 | acttgtctagaggtct | 8.87 | 75.026 | 23.846 ± 1.973 |
| BTF1_25170 | XRE family transcriptional regulator | / | / | / | 33.600 | NT | |
| BTF1_25685 | lrgB | Antiholin-like protein LrgB | / | / | / | 19.299 | NT |
| BTF1_25690 | lrgA | Murein hydrolase regulator LrgA | / | / | / | 10.165 | NT |
| BTF1_26820 | Group-specific protein | / | / | / | 13.357 | NT | |
| BTF1_26825 | glmS | Glucosamine-fructose-6-phosphate aminotransferase | / | / | / | 5.506 | NT |
| BTF1_28050 | chiB | Chitinase B | −113 | agacatcacgaagtct | 9.57 | 39.320 | NT |
| GENES DOWNREGULATED IN THE ΔnagR MUTANT COMPARED TO WILD-TYPE STRAIN Bti75 | |||||||
| BTF1_00565 | pphA | Serine/threonine protein phosphatase | / | / | / | 0.0943 | NT |
| BTF1_00720 | nprE | Bacillolysin, neutral protein Npr599 | / | / | / | 0.153 | 0.649 ± 0.058 |
| BTF1_04880 | liaI | Hypothetical protein | / | / | / | 0.0542 | NT |
| BTF1_04885 | PspA/IM30 family protein | / | / | / | 0.0584 | NT | |
| BTF1_06780 | nheA | Non-hemolytic enterotoxin lytic component L2 | / | / | / | 0.207 | 0.391 ± 0.056 |
| BTF1_08960 | Acyl-dehydrogenase | / | / | / | 0.214 | 0.4480 ± 0.046 | |
| BTF1_12180 | Metallo-beta-lactamase, hydrolase | / | / | / | 0.133 | 0.335 ± 0.027 | |
| BTF1_16395 | Mutt/nudix family protein | / | / | / | 0.116 | NT | |
| BTF1_17540 | acpP | Acyl carrier protein | / | / | / | 0.206 | NT |
| BTF1_18580 | nagR | GntR family transcriptional regulator NagR | / | / | / | 0.0312 | NT |
| BTF1_19025 | Hypothetical protein | −107 | aatcatctagacaact | 10.1 | 0.0187 | 0.570 ± 0.030 | |
| BTF1_23020 | pgi | Glucose-6-phosphate isomerase | −168 | agatgtatatacatca | 7.08 | 0.188 | 0.6781 ± 0.038 |
| BTF1_24870 | nuoC | NADH dehydrogenase subunit C | / | / | / | 0.180 | 0.199 ± 0.020 |
| BTF1_25115 | mmgC | Acyl-CoA dehydrogenase | / | / | / | 0.172 | NT |
| BTF1_26870 | Lipoprotein | / | / | / | 0.133 | 0.817 ± 0.038 | |
| GENES WITH RECOGNIZED dre SITES BUT NOT DIFFERENTIALLY EXPRESSED IN ΔnagR MUTANT COMPARED TO WILD-TYPE STRAIN Bti75 | |||||||
| BTF1_00745 | Transcription antiterminator, LytR family protein | −45 | atacatttagaagtgt | 7.03 | NS | NT | |
| BTF1_01335 | Peptidase, M23/M37 family protein | −35 | agttgtatagttgtaa | 7.58 | NS | NT | |
| BTF1_02575 | Major facilitator superfamily protein | −133 | agatatctaaataact | 8.35 | NS | NT | |
| BTF1_03055 | TetR family transcriptional regulator | −194 | agatgtatagtcgact | 10.7 | NS | NT | |
| BTF1_19055 | pepT | Peptidase T | −116 | acatgtctagataacc | 8.88 | NS | NT |
| BTF1_23845 | Wall-associated protein | −276 | cgtcatctatacatct | 8.62 | NS | NT | |
| BTF1_24430 | chbB | PTS system, lichenan-specific IIB component | −249 | agttgtctagttatgc | 9.2 | NS | NT |
| BTF1_26160 | bofA | SigmaK-factor processing regulatory protein BofA | −39 | atacaaatagatgtat | 7.19 | NS | NT |
The BTF1 number of genes are the open reading frames based on the genome annotation of B. thuringiensis HD-789 (Doggett et al., 2013).
The position of the outermost end of a dre site relative to the respective translation start point of the gene.
The dre-motifs were detected after using the PREDetector software program.
The scores were determined after using the PREDetector software program.
Fold change gene expression is given for a gene in the ΔnagR mutant strain compared to the wild-type in RNA-seq.
Fold change gene expressions are shown as the mean ± SD from three independent biological and technical replicates of each biological sample for each gene in the ΔnagR mutant strain compared to the wild-type by qRT-PCR.
ND, no expression was detected in wild-type strain. NS, No significant difference between wild-type and ΔnagR mutant. NT, No test. /, No result was offered by the PREDetector software program.
To validate the reliability of the Solexa/Illumina sequencing technology, 10 DEGs were selected randomly from different metabolic pathways for qRT-PCR analysis. As shown in Figure 1, the results of the qRT-PCR analysis were consistent with those of the Solexa/Illumina sequencing analysis, although there were quantitative differences.
Figure 1.

qRT-PCR validation of RNA-seq data. (A) Five DEGs that showed increased expression were selected randomly, namely BTF1_01610 (outer surface protein), BTF1_06850 (cdd), BTF1_16665 (chiA), BTF1_18590 (nagA), and BTF1_24980 (crr). (B) Five DEGs that showed decreased expression were selected randomly, namely BTF1_00720 (nprE), BTF1_06780 (nheA), BTF1_19025 (hypothetical protein), BTF1_24870 (nuoC), and BTF1_26870 (Lipoprotein).
Complemented mutant strain
The complementing vector pKSV7-Perm-nagR was constructed and its structure confirmed by PCR and restriction enzyme digestion analysis. Subsequently, the recombinant plasmid was transferred into Bti75ΔnagR by electroporation. The complemented clones with resistance to chloramphenicol were confirmed by PCR and DNA sequencing. The expression of gene nagR was detected by using qRT-PCR in Bti75ΔnagR, Bti75ΔnagR-pPerm-nagR, and the wild-type strain. The result showed that no expression of nagR was detected in Bti75ΔnagR and the fold-change of gene nagR was similar in the wild-type strain and Bti75ΔnagR-pPerm-nagR (Figure S1). Moreover, randomly selected genes were assessed using qRT-PCR to determine the influence of nagR. The results revealed that the fold-change of each gene in Bti75ΔnagR-pPerm-nagR was between the value for the wild-type and Bti75ΔnagR. These results suggested that changes in genes expression were indeed caused by the deletion of nagR (Figure 2).
Figure 2.

qRT-PCR validation of Bti75ΔNagR, Bti75ΔnagR-pPerm-nagR, and the wild-type (WT). Expression profiles of selected random DEGs (BTF1_00220, BTF1_01610, BTF1_16665, BTF1_18560, BTF1_08960, BTF1_19025, BTF1_23020, BTF1_26870) as determined by qRT-PCR.
Distribution of NagRBt-binding sites in the genome
To identify the regulons controlled by NagRBt in Bti75, we predicted its binding sites in the Bt HD-789 genome using PREDetector (Hiard et al., 2007). The genome of HD-789 served as the reference genome because of its high sequence similarity with Bti75. Five known dre-binding sites from Bti75 were used as the input to the program to construct the binding site recognition profile. Table S2 shows the output for the predicted NagRBt-binding sites (67 candidates) with scores >7.0. Furthermore, compared with transcriptome data, 11 DEGs were found to harbor the predicted binding sites, implying that these genes might be regulated by NagRBt directly (Table 2).
Identification of genes directly controlled by NagRBt
All the DEGs were tested using EMSAs. If the DEG possessed a predicted dre site, the predicted 16 bp sequence was extended by 12 bp on both sides and the 5′ end labeled with biotin (for competition experiments) to use for EMSAs with NagRBt. If not, ~300 bp of the upstream sequence from the DEG was amplified by PCR with unlabeled or labeled biotin primers.
In Figure 3A (DNA was detected by ethidium bromide staining), we show all the 18 positive targets whose migration was retarded by NagRBt respectively. Since the dre site of chiB has been reported by Jiang et al. (2015), it is not shown in Figure 3. To further validate the specific binding between NagRBt and dre, 0.5 mg mL−1 of salmon sperm DNA (non-specific competition) and a 400-fold excess of unlabeled dre sequence (specific competition) were added to the reaction mixture of NagRBt with biotin-labeled dre (Figure 3B, Biotin-labeled probes were detected after transfer to nylon membranes). The positive results indicated that the salmon sperm DNA failed to block the binding of NagRBt to biotin labelled dre, while the 400-fold excess of unlabelled dre sequence could eliminate or reduce the specific shift band. The results from these experiments revealed that 11 site sequences with significantly different expression could bind specifically with NagRBt and these dre sites were also found by computational prediction. However, the addition of NagRBt to the DNA fragments covering the 300 bp upstream of the DEG without recognizable dre sequences did not cause any band shift (data not shown). Furthermore, other NagRBt-binding sites (from genes that did not show differential expression) from the computational prediction were detected by EMSAs and we found that seven sites could be bound by NagRBt specifically. All the different positive dre sequences are listed in Table 2.
Figure 3.
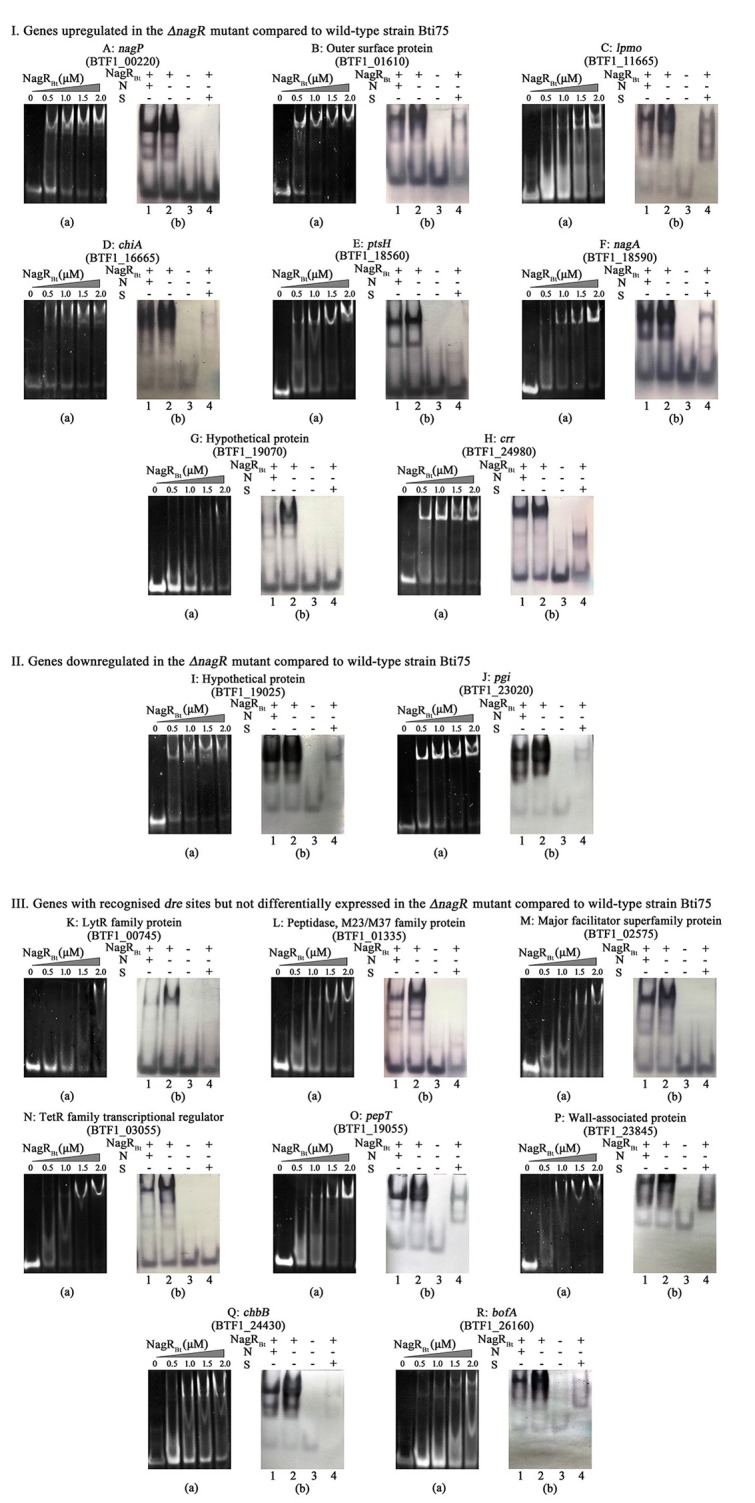
NagRBt-binding sites are detected by EMSAs. (I) Genes upregulated in the ΔnagR mutant compared to wild-type strain Bti75; (II) Genes downregulated in the ΔnagR mutant compared to wild-type strain Bti75; (III) Genes with recognized dre sites but not differentially expressed in the ΔnagR mutant compared to wild-type strain Bti75. (a): Each 0.2 μM 40 bp-Pdre was electrophoresed after incubation with various concentrations of NagRBt. (b): EMSAs to determine the specific binding of NagRBt. Lane 1, Pdre (Bio), NagRBt, and 0.5 μg/μL salmon sperm DNA for non-specific competitor (N); Lane 2, Pdre (Bio), NagRBt; Lane 3, Pdre (Bio); Lane 4, Pdre (Bio), NagRBt, and a 400-fold excess of unlabeled Pdre for specific competitor (S). All the non-specific and specific competition assays used 3.0 μM NagRBt and 0.2 μM Pdre (Bio). Different letters represent different genes. A: nagP, BTF1_00220; B: outer surface protein, BTF1_01610; C: lpmo, BTF1_11665; D: chiA, BTF1_16665; E: ptsH, BTF1_18560; F: nagA, BTF1_18590; G: hypothetical protein, BTF1_19070; H: crr, BTF1_24980; I: hypothetical protein, BTF1_19025; J: pgi, BTF1_23020; K: lytR family protein, BTF1_00745; L: peptidase, BTF1_01335; M: major facilitator superfamily protein, BTF1_02575; N: TetR family transcriptional regulator, BTF1_03055; O: pepT, BTF1_19055; P: wall-associated protein, BTF1_23845; Q: chbB, BTF1_24430; R: bofA, BTF1_26160.
NagRBt activates the expression of some genes directly
The gene encoding a hypothetical protein (BTF1_19025) and the pgi (glucose-6-phosphate isomerase, BTF1_23020) gene both possess a predicted dre site, and NagRBt can specifically bind to them (Figures 3Ia,b,Ja,b). Most members of the GntR/HutC family are repressors, including NagRBt. However, comparative transcriptome analysis showed that the expression of these two genes are downregulated rather than upregulated in the NagRBt deletion mutant (Table 2). qRT-PCR data were qualitatively consistent with NagRBt acting as a positive regulator at these two genes. BTF1_19025 and BTF1_23020 expression in Bti75ΔnagR decreased to ~0.57 and 0.68 of the transcription observed in the wild-type respectively (Figure 2). These results suggested that NagRBt could activate the expression of these two genes.
To further verify this, we chose the upstream region of the pgi gene (Figure 4A) to construct the corresponding plasmids. The promoter-probe vector was used to detect the influence of changing the relative position of the promoter and the dre site on the expression of a downstream reporter gene (Figure 4B). The upstream sequence of pgi was selected to construct corresponding plasmids, and then transferred into Bti75. The qRT-PCR analysis showed (Figure 4C) that the expression of bgaB was ~1.3-fold increase in pdrepgi+Ppgi and 0.28-fold decrease in pPpgi+drepgi compared with that in pPpgi respectively. In addition, the corresponding regulatory sequences were analyzed by evaluating β-galactosidase activity (Figure 4D). The result showed that the trend of β-galactosidase activity was consistent with qRT-PCR results.
Consensus sequence of NagRBt-responsive elements (dre) in Bti75
The consensus sequence of dre was generated using WebLogo (http://weblogo.berkeley.edu/) (Crooks et al., 2004) based on the NagRBt-binding sequences in Bti75 (Figure 5). The overall height of the stack indicates the sequence conservation at that position, while the height of symbols within the stack indicates the relative frequency of each nucleic acid at that position. Compilation of known and newly identified NagRBt-binding sites led us to construct a NagRBt consensus sequence. It is not a perfect palindrome compared with previous graphical representations of Bacillus dre sites.
Figure 5.
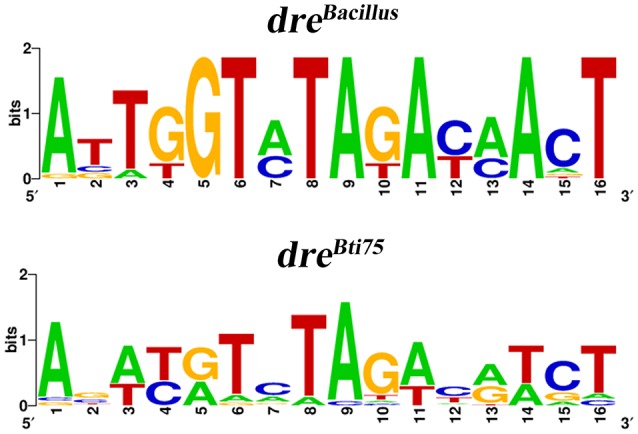
WebLogo representation of Bacillus and Bti75 consensus dre sites. WebLogo representation of dreBti75 was generated with all confirmed NagRBt-binding sequences and the WebLogo representation of dreBacillus was generated with the dre sites from the upstream regions of the nagAB and nagP genes in Bacillus species (Bertram et al., 2011).
Discussion
In B. subtilis, NagR only directly represses the nagP and nagAB genes (Bertram et al., 2011). According to the phylogenetic analysis (Figure S2), NagRBt is an ortholog to NagR in Bacillus species. However, previous studies had identified five NagRBt-binding sites in Bti75 (Jiang et al., 2015), suggesting that the regulation of NagRBt is broader than NagRBs. To identify new NagRBt-binding sites, we exploited the PREDetector software program to build a matrix model to scan for potential binding sites in the complete genome of Bt HD-789, which is available from the GenBank database (Benson et al., 2007). To gain a better understanding of regulatory factor NagRBt, the transcriptomes of Bti75-ΔnagR and wild-type Bti75 in the logarithmic phase were compared to identify potential target genes of nagR. To confirm the interaction of NagRBt with the newly identified dre sequences, we tested the dre-like sequences using EMSAs. The differentially expressed genes and confirmed dre sequences are listed together in Table 2. The main NagRBt-related pathways are summarized in Figure 6. Three different groups of NagRBt-influenced genes are distinguished according to their expression patterns.
Figure 6.

Summary of pathways and genes influenced by NagRBt. Proteins whose genes have a NagRBt-binding site in their upstream region are indicated by yellow shading. Arrows in green or red show genes whose expression is upregulated or downregulated, respectively, in the ΔnagR mutant compared with their expression in the wild-type strain Bti75. Key metabolites are shown in blue boxes and different metabolic pathways are indicated in magenta ovals. Gene annotations are based on the KEGG database: BTF1_00220, nagP; BTF1_01610, outer surface protein; BTF1_11665, lpmo; BTF1_16665, chiA; BTF1_18555, ptsI; BTF1_18560, ptsH; BTF1_18585, nagB; BTF1_18590, nagA; BTF1_19070, hypothetical protein; BTF1_24980, crr; BTF1_28050, chiB; BTF1_19025, hypothetical protein; BTF1_23020, pgi; BTF1_00745, lytR family protein; BTF1_01335, peptidase; BTF1_02575, major facilitator superfamily protein; BTF1_03055, TetR family transcriptional regulator; BTF1_19055, pepT; BTF1_23845, wall-associated protein; BTF1_24430, chbB; BTF1_26160: bofA; PRPP, 5-phosphoribosyl diphosphate.
Group 1 contains the upregulated genes that are divided into two categories: those that contain a dre sequence (verified by EMSAs) and those without a dre sequence. nagAB (BTF1_18590 and BTF1_18585) and nagP (BTF1_00220) in amino sugar metabolism have dre sequences and were upregulated in Bti75ΔnagR according to the in silico, in vitro, and in vivo results. These results were consistent with the reports that nagAB and nagE2 are repressed by DasR in streptomycetes (Rigali et al., 2006; Li et al., 2008; Nothaft et al., 2010) and by NagR in B. subtilis (Bertram et al., 2011). From the transcriptome data, nagR deletion resulted in changes to the expression levels of other genes. Subsequently, dre-like sites were identified in the corresponding upstream regions using PREDetector and verified using EMSAs, and suggested that NagRBt could directly influence their expression.
Among these genes, chiA (BTF1_16665), chiB (BTF1_28050), and chitin-binding protein gene (BTF1_11665) in chitin metabolism are negatively regulated by NagRBt, as confirmed using RNA-seq. This was consistent with the results obtained by Jiang et al. (2015), who reported that chiB is regulated not only by CcpABt but also by NagRBt. Similarly, the promoter region of chitinase gene contains a W-box binding element that is specifically bound by WRKY, a class of transcription factors in plants (Gao et al., 2014). This suggested that the expression of chiB is strictly regulated by multiple transcription factors.
A close inspection of the promoter regions revealed dre-binding sites upstream of some phosphotransferase (PTS) system-related genes, such as crr (BTF1_24980), chbBC (BTF1_24430, BTF1_24425), and ptsH (BTF1_18560). This result was consistent with the conclusion of Rigali et al. (2004), demonstrating that the sugar phosphotransferase system is regulated by DasR regulator in S. coelicolor (in vitro and in vivo experiments).
The other subcategory included upregulated genes that did not have dre sequences, such as cytidine deaminase (BTF1_06850), pyrimidine-nucleoside phosphorylase (BTF1_06845), nucleoside transporter nupC (BTF1_06840) involved in pyrimidine metabolism, glucosamine-fructose-6-phosphate aminotransferase, glmS (BTF1_26825) involved in amino sugar metabolism, and so on. These genes are believed to be indirectly regulated by NagRBt. For example, the expression of glmS (BTF1_26825) and its riboswitch gene (BTF1_26820) showed increased expression in this study. The enzyme glutamine-fructose-6-phosphate amidotransferase, encoded by the glmS gene, catalyzes the reaction between fructose-6-phosphate (Fru-6-P) and glutamine to generate glucosamine-6-phosphate (GlcN-6-P) (Plumbridge et al., 1993). This reaction usually occurs in the process of cell wall biosynthesis, which involves the production of UDP-GlcNAc (Komatsuzawa et al., 2004). We speculated that the inactivation of the nagR gene relieved its inhibitory effect on nagAB, resulting in their increased expression. GlcN-6-P deaminase, the nagB gene product, converts GlcN-6-P to Fru-6-P and can decrease the intracellular GlcN-6-P level in the absence of an exogenous amino sugar supply. It has been reported that GlcN-6-P-induced riboswitch self-cleavage is coupled to the intracellular stability of the glmS transcript (Winkler et al., 2004; Collins et al., 2007). When the content of GlcN-6-P is below a certain threshold, the riboswitch is not activated. Consequently, glmS transcripts increase to maintain a balanced level of intracellular GlcN-6-P.
The genes of group 2 are not differentially expressed between the ΔnagR mutant strain and the wild-type but possess specific dre sequences that are identified by EMSAs. Some researchers have reported that transcription factors bind thousands of genomic locations in the vicinity of both active and inactive regions in eukaryotes (Li et al., 2008; Biggin, 2011). A number of weakly bound sites detected this way failed to drive transgenic reporter expression (Fisher et al., 2012). These findings lead to the notion of “non-functional” or “spurious” binding. However, some of the apparently “non-functional” or “spurious” binding sites might turn out to play pivotal roles in maintaining adequate transcriptional regulation under the condition of genetic or environmental abnormalities (Spivakov, 2014). In addition, an increasing number of examples of transcription factor binding sites are not associated with specific gene regulation in prokaryotes (Grainger et al., 2005; Qian et al., 2016). Eight genes have recognized dre sites but are not differentially expressed; we speculated that some of them might be involved in the type of specific non-functional interactions, such as gene bofA (BTF1_26160). Its gene product, BofA, exerts negative regulation by controlling the level of the SpoIVEA protein in pro-σK processing during sporulation (Resnekov, 1999; Zhou and Kroos, 2004). We speculated that bofA itself barely expresses in the exponential growth stage and is only activated in the process of sporulation. This suggested that NagRBt might have spatiotemporal influence, for instance in the mother cell during the late stages of sporulation, to regulate the expression of other genes. This aspect of NagRBt regulation requires further research.
Another gene, tetR (BTF1_03055), is one of the TetR family of regulators, members of which regulate genes involved in carbon metabolism, amino acid metabolism, signal transduction systems, antibiotic production, and other physiological metabolism (Ramos et al., 2005; Cuthbertson and Nodwell, 2013). Bioinformatics analysis showed that only one member of the TetR family of regulators has a dre-binding site in its upstream promoter region. However, the expression level of tetR was unchanged after deletion of nagR, as determined by qRT-PCR. A plausible explanation is that there might be another more powerful transcription factor than NagRBt that also regulates tetR. Some suspected transcription factors, such as SigL and Xre, which were found by using DBTBS prediction (Sierro et al., 2008), might play a role in regulating tetR. Verification of the connections among these regulators requires further experimental support.
The genes in group 3 are downregulated in the ΔnagR strain and are thus believed to be indirectly regulated by NagRBt because most GntR/HutC family regulators are repressors. Most of the 12 downregulated genes lacking binding sites are involved in fatty acid metabolism. Surprisingly, the EMSA experiments revealed that two of these genes (BTF1_19025 and BTF1_23020) were downregulated and had dre sites. Moreover, qRT-PCR showed that the expression levels of both genes were downregulated in the ΔnagR strain compared with that in the wild-type. Together, our findings highlight that NagRBt can activate these two genes. The gene of BTF1_19025 encodes a hypothetical protein belonging to a conserved protein domain family (DUF3932) and proteins in this family are functionally uncharacterized by searching NCBI's Conserved Domain Database (CDD) (Marchler-Bauer et al., 2017). The pgi gene (BTF1_23020) encodes the enzyme glucose-6-phosphate isomerase that participates in many pathways in the interconversion of glucose 6-phosphate and fructose-6-phosphate. The dre sites in these two genes were located upstream of the predicted promoter region just as in the classic activation mechanism in bacteria (Lee et al., 2012). It has been reported that the different functions (promotion or inhibition) of CcpA are caused by the different position of the cre sites in each gene (Deutscher et al., 2006). When the cre site of major genes is located downstream of the promoter, gene expression is often repressed by the CcpA complex. However, if the cre site is located upstream of the promoter, the CcpA complex stimulates the transcription to activate gene expression, such as in ackA (Moir-Blais et al., 2001), pta (Presecan-Siedel et al., 1999), ilv-leu (Shivers and Sonenshein, 2005; Tojo et al., 2005), and pepQ (Luesink et al., 1998). Other examples of dual function transcription activators and repressors are known in bacteria e.g., Cra and Crp (Pérez-Martín and de Lorenzo, 1997; Cozzone, 1998).
To further confirm the activation role of NagRBt, we constructed a shuttle promoter-probe vector incorporating the thermostable gene bgaB with the promoter of pgi. qRT-PCR revealed that the expression of bgaB was ~1.3-fold increased by dre+Ppgi compared with Ppgi and ~0.28-fold decreased by Ppgi+dre compared with Ppgi. Furthermore, the trend of β-galactosidase activity was consistent with qRT-PCR results. It demonstrates effectively that a downstream dre site acts as an effective repressor, whereas its inactivation produces a relatively small decrease in pgi expression. So far, there has been no report of this gene being directly regulated by NagRBt; it has been reported this gene was regulated by CcpA in Streptococcus suis (Willenborg et al., 2014).
In addition, the NagRBt-binding site consensus sequence was generated by compiling known and newly identified binding sites using the in silico, in vitro, and in vivo data (Figure 5). The consensus sequence of the NagRBt-binding site is not a perfect palindrome and is different from the Bacillus species dre sites (Bertram et al., 2011). It has obvious conserved recognition sites at positions 5, 6, 8, 9, 11, 14, and 16 in dreBacillus, but the bases in the same position are not strictly conserved in dreBti75. To avoid the omission of NagRBt-binding sites, our future research will include the use of chromatin immunoprecipitation sequencing.
Compared with the wide-ranging regulation by DasR in Streptomycetes (Swiatek-Polatynska et al., 2015), where hundreds of dre-binding sites have been identified, NagRBt-binding sites have a narrower distribution and are found in genes encoding proteins involved in amino sugar metabolism, the phosphotransferase system, chitin metabolism, and other pathways. By contrast, NagRBs-binding sites are limited to the nagP and nagAB-nagR locus. This suggests that although NagRBt is a less prominent regulator than DasR, its regulation is more widespread than that of NagRBs.
Overall, our results demonstrated that NagRBt functions as an important regulator of metabolism, not only by directly regulating the amino sugar metabolism and phosphotransferase system, but also by directly or indirectly influencing other biological processes, such as nucleotide metabolism, fatty acid metabolism, and the EMP pathway. In addition, NagRBt is not only a pleiotropic transcriptional regulator but also a dual transcription regulator that acts as both a repressor and an activator. This adds to our understanding of the role of the GntR/HutC family transcription factors.
Author contributions
ZC and JC designed the research. ZC performed the experimental work and drafted the manuscript. TT performed the RNA-seq analysis. YZ and LH constructed nagR-complemented strain and carried out promoter/dre position swapping experiments. XH purified NagRBt protein. HM performed the β-galactosidase assay. All authors read and approved the final manuscript.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by grants from the National Key R&D Program of China (No. 2017YFD0200400), and the National Natural Science Foundation of China (No. 31670081 and 31371979).
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01899/full#supplementary-material
References
- Benson D. A., Karsch-Mizrachi I., Lipman D. J., Ostell J., Wheeler D. L. (2007). GenBank. Nucleic Acids Res. 35, D21–D25. 10.1093/nar/gkl986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram R., Rigali S., Wood N., Lulko A. T., Kuipers O. P., Titgemeyer F. (2011). Regulon of the N-acetylglucosamine utilization regulator NagR in Bacillus subtilis. J. Bacteriol. 193, 3525–3536. 10.1128/JB.00264-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggin M. D. (2011). Animal transcription networks as highly connected, quantitative continua. Dev. Cell 21, 611–626. 10.1016/j.devcel.2011.09.008 [DOI] [PubMed] [Google Scholar]
- Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins J. A., Irnov I., Baker S., Winkler W. C. (2007). Mechanism of mRNA destabilization by the glmS ribozyme. Genes Dev. 21, 3356–3368. 10.1101/gad.1605307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colson S., Stephan J., Hertrich T., Saito A., van Wezel G. P., Titgemeyer F., et al. (2007). Conserved cis-acting elements upstream of genes composing the chitinolytic system of Streptomycetes are DasR-responsive elements. J. Mol. Microbiol. Biotechnol. 12, 60–66. 10.1159/000096460 [DOI] [PubMed] [Google Scholar]
- Cozzone A. J. (1998). Regulation of acetate metabolism by protein phosphorylation in enteric bacteria. Annu. Rev. Microbiol. 52, 127–164. 10.1146/annurev.micro.52.1.127 [DOI] [PubMed] [Google Scholar]
- Crooks G. E., Hon G., Chandonia J.-M., Brenner S. E. (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbertson L., Nodwell J. R. (2013). The TetR family of regulators. Microbiol. Mol. Biol. Rev. 77, 440–475. 10.1128/MMBR.00018-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutscher J., Francke C., Postma P. W. (2006). How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol. Mol. Biol. Rev. 70, 939–1031. 10.1128/MMBR.00024-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doggett N. A., Stubben C. J., Chertkov O., Bruce D. C., Detter J. C., Johnson S. L., et al. (2013). Complete genome sequence of Bacillus thuringiensis serovar Israelensis strain HD-789. Genome Announc. 1:e01023-13. 10.1128/genomeA.01023-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher W. W., Li J. J., Hammonds A. S., Brown J. B., Pfeiffer B. D., Weiszmann R., et al. (2012). DNA regions bound at low occupancy by transcription factors do not drive patterned reporter gene expression in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 109, 21330–21335. 10.1073/pnas.1209589110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y., Zan X.-L., Wu X.-F., Yao L., Chen Y.-L., Jia S.-W., et al. (2014). Identification of fungus-responsive cis-acting element in the promoter of Brassica juncea chitinase gene, BjCHI1. Plant Sci. 215–216, 190–198. 10.1016/j.plantsci.2013.11.008 [DOI] [PubMed] [Google Scholar]
- Grainger D. C., Hurd D., Harrison M., Holdstock J., Busby S. J. W. (2005). Studies of the distribution of Escherichia coli cAMP-receptor protein and RNA polymerase along the E. coli chromosome. Proc. Natl. Acad. Sci. U.S.A. 102, 17693–17698. 10.1073/pnas.0506687102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiard S., Maree R., Colson S., Hoskisson P. A., Titgemeyer F., van Wezel G. P., et al. (2007). PREDetector: a new tool to identify regulatory elements in bacterial genomes. Biochem. Biophys. Res. Commun. 357, 861–864. 10.1016/j.bbrc.2007.03.180 [DOI] [PubMed] [Google Scholar]
- Hirata H., Negoro S., Okada H. (1984). Molecular basis of isozyme formation of beta-galactosidases in Bacillus stearothermophilus: isolation of two beta-galactosidase genes, bgaA and bgaB. J. Bacteriol. 160, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini P., Tremblay A., Matthews B. F., Alkharouf N. W. (2010). An efficient annotation and gene-expression derivation tool for Illumina Solexa datasets. BMC Res. Notes 3:183. 10.1186/1756-0500-3-183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang K., Li L., Pan J., Wang T., Chen Y., Cai J. (2015). YvoA and CcpA Repress the Expression of chiB in Bacillus thuringiensis. Appl. Environ. Microbiol. 81, 6548–6557. 10.1128/AEM.01549-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsuzawa H., Fujiwara T., Nishi H., Yamada S., Ohara M., McCallum N., et al. (2004). The gate controlling cell wall synthesis in Staphylococcus aureus. Mol. Microbiol. 53, 1221–1231. 10.1111/j.1365-2958.2004.04200.x [DOI] [PubMed] [Google Scholar]
- Langdon W. B. (2015). Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. Biodata Min. 8:1. 10.1186/s13040-014-0034-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecadet M. M., Chaufaux J., Ribier J., Lereclus D. (1992). Construction of novel Bacillus thuringiensis strains with different insecticidal activities by transduction and transformation. Appl. Environ. Microbiol. 58, 840–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D. J., Minchin S. D., Busby S. J. W. (2012). Activating transcription in bacteria. Annu. Rev. Microbiol. 66, 125–152. 10.1146/annurev-micro-092611-150012 [DOI] [PubMed] [Google Scholar]
- Li X., MacArthur S., Bourgon R., Nix D., Pollard D. A., Iyer V. N., et al. (2008). Transcription factors bind thousands of active and inactive regions in the Drosophila blastoderm. PLoS Biol. 6:e27. 10.1371/journal.pbio.0060027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K. J., Schmittgen T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Luesink E. J., van Herpen R. E., Grossiord B. P., Kuipers O. P., de Vos W. M. (1998). Transcriptional activation of the glycolytic las operon and catabolite repression of the gal operon in Lactococcus lactis are mediated by the catabolite control protein CcpA. Mol. Microbiol. 30, 789–798. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A., Bo Y., Han L., He J., Lanczycki C. J., Lu S., et al. (2017). CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 45, D200–D203. 10.1093/nar/gkw1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobley H. L., Doyle R. J., Streips U. N., Langemeier S. O. (1982). Transport and incorporation of N-acetyl-D-glucosamine in Bacillus subtilis. J. Bacteriol. 150, 8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir-Blais T. R., Grundy F. J., Henkin T. M. (2001). Transcriptional activation of the Bacillus subtilis ackA promoter requires sequences upstream of the CcpA binding site. J. Bacteriol. 183, 2389–2393. 10.1128/JB.183.7.2389-2393.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A., Williams B. A., McCue K., Schaeffer L., Wold B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. 10.1038/nmeth.1226 [DOI] [PubMed] [Google Scholar]
- Nazari B., Kobayashi M., Saito A., Hassaninasab A., Miyashita K., Fujii T. (2013). Chitin-induced gene expression in secondary metabolic pathways of Streptomyces coelicolor A3(2) grown in soil. Appl. Environ. Microbiol. 79, 707–713. 10.1128/AEM.02217-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nothaft H., Rigali S., Boomsma B., Swiatek M., McDowall K. J., van Wezel G. P., et al. (2010). The permease gene nagE2 is the key to N-acetylglucosamine sensing and utilization in Streptomyces coelicolor and is subject to multi-level control. Mol. Microbiol. 75, 1133–1144. 10.1111/j.1365-2958.2009.07020.x [DOI] [PubMed] [Google Scholar]
- Pérez-Martín J., de Lorenzo V. (1997). Clues and consequences of DNA bending in transcription. Annu. Rev. Microbiol. 51, 593–628. 10.1146/annurev.micro.51.1.593 [DOI] [PubMed] [Google Scholar]
- Plumbridge J. A., Cochet O., Souza J. M., Altamirano M. M., Calcagno M. L., Badet B. (1993). Coordinated regulation of amino sugar-synthesizing and -degrading enzymes in Escherichia coli K-12. J. Bacteriol. 175, 4951–4956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presecan-Siedel E., Galinier A., Longin R., Deutscher J., Danchin A., Glaser P., et al. (1999). Catabolite regulation of the pta gene as part of carbon flow pathways in Bacillus subtilis. J. Bacteriol. 181, 6889–6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z., Trostel A., Lewis D. E. A., Lee S. J., He X., Stringer A. M., et al. (2016). Genome-wide transcriptional regulation and chromosome structural arrangement by GalR in E. coli. Front. Mol. Biosci. 3:74. 10.3389/fmolb.2016.00074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos J. L., Martinez-Bueno M., Molina-Henares A. J., Teran W., Watanabe K., Zhang X., et al. (2005). The TetR family of transcriptional repressors. Microbiol. Mol. Biol. Rev. 69, 326–356. 10.1128/MMBR.69.2.326-356.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnekov O. (1999). Role of the sporulation protein BofA in regulating activation of the Bacillus subtilis developmental transcription factor sigmaK. J. Bacteriol. 181, 5384–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigali S., Nothaft H., Noens E. E. E., Schlicht M., Colson S., Muller M., et al. (2006). The sugar phosphotransferase system of Streptomyces coelicolor is regulated by the GntR-family regulator DasR and links N-acetylglucosamine metabolism to the control of development. Mol. Microbiol. 61, 1237–1251. 10.1111/j.1365-2958.2006.05319.x [DOI] [PubMed] [Google Scholar]
- Rigali S., Schlicht M., Hoskisson P., Nothaft H., Merzbacher M., Joris B., et al. (2004). Extending the classification of bacterial transcription factors beyond the helix-turn-helix motif as an alternative approach to discover new cis/trans relationships. Nucleic Acids Res. 32, 3418–3426. 10.1093/nar/gkh673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigali S., Titgemeyer F., Barends S., Mulder S., Thomae A. W., Hopwood D. A., et al. (2008). Feast or famine: the global regulator DasR links nutrient stress to antibiotic production by Streptomyces. EMBO Rep. 9, 670–675. 10.1038/embor.2008.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J.-W., Ohnishi Y., Hirata A., Horinouchi S. (2002). ATP-binding cassette transport system involved in regulation of morphological differentiation in response to glucose in Streptomyces griseus. J. Bacteriol. 184, 91–103. 10.1128/JB.184.1.91-103.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivers R. P., Sonenshein A. L. (2005). Bacillus subtilis ilvB operon: an intersection of global regulons. Mol. Microbiol. 56, 1549–1559. 10.1111/j.1365-2958.2005.04634.x [DOI] [PubMed] [Google Scholar]
- Sierro N., Makita Y., de Hoon M., Nakai K. (2008). DBTBS: a database of transcriptional regulation in Bacillus subtilis containing upstream intergenic conservation information. Nucleic Acids Res. 36, D93–D96. 10.1093/nar/gkm910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spivakov M. (2014). Spurious transcription factor binding: non-functional or genetically redundant? Bioessays 36, 798–806. 10.1002/bies.201400036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiatek M. A., Tenconi E., Rigali S., van Wezel G. P. (2012). Functional analysis of the N-acetylglucosamine metabolic genes of Streptomyces coelicolor and role in control of development and antibiotic production. J. Bacteriol. 194, 1136–1144. 10.1128/JB.06370-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiatek-Polatynska M. A., Bucca G., Laing E., Gubbens J., Titgemeyer F., Smith C. P., et al. (2015). Genome-wide analysis of in vivo binding of the master regulator DasR in Streptomyces coelicolor identifies novel non-canonical targets. PLoS ONE 10:e0122479. 10.1371/journal.pone.0122479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenconi E., Urem M., Swiatek-Polatynska M. A., Titgemeyer F., Muller Y. A., van Wezel G. P., et al. (2015). Multiple allosteric effectors control the affinity of DasR for its target sites. Biochem. Biophys. Res. Commun. 464, 324–329. 10.1016/j.bbrc.2015.06.152 [DOI] [PubMed] [Google Scholar]
- Tojo S., Satomura T., Morisaki K., Deutscher J., Hirooka K., Fujita Y. (2005). Elaborate transcription regulation of the Bacillus subtilis ilv-leu operon involved in the biosynthesis of branched-chain amino acids through global regulators of CcpA, CodY and TnrA. Mol. Microbiol. 56, 1560–1573. 10.1111/j.1365-2958.2005.04635.x [DOI] [PubMed] [Google Scholar]
- Willenborg J., de Greeff A., Jarek M., Valentin-Weigand P., Goethe R. (2014). The CcpA regulon of Streptococcus suis reveals novel insights into the regulation of the streptococcal central carbon metabolism by binding of CcpA to two distinct binding motifs. Mol. Microbiol. 92, 61–83. 10.1111/mmi.12537 [DOI] [PubMed] [Google Scholar]
- Winkler W. C., Nahvi A., Roth A., Collins J. A., Breaker R. R. (2004). Control of gene expression by a natural metabolite-responsive ribozyme. Nature 428, 281–286. 10.1038/nature02362 [DOI] [PubMed] [Google Scholar]
- Xie C.-C., Luo Y., Chen Y.-H., Cai J. (2012). Construction of a promoter-probe vector for Bacillus thuringiensis: the identification of cis-acting elements of the chiA locus. Curr. Microbiol. 64, 492–500. 10.1007/s00284-012-0100-0 [DOI] [PubMed] [Google Scholar]
- Zhou R., Kroos L. (2004). BofA protein inhibits intramembrane proteolysis of pro-sigmaK in an intercompartmental signaling pathway during Bacillus subtilis sporulation. Proc. Natl. Acad. Sci. U.S.A. 101, 6385–6390. 10.1073/pnas.0307709101 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.