

Significance
Aggregation of amyloidogenic proteins is implicated in a number of debilitating human diseases. A mechanistic understanding of their aggregation behavior requires a quantitative description of the multistep equilibria involved in the self-assembly processes. Here we report the application of an integrative aggregation assay combining 19F-NMR and turbidity measurements to reveal the kinetics and energetics of the human transthyretin aggregation pathway. A highly sensitive trifluoroacetyl probe enables direct observation and quantification of a weakly populated aggregation intermediate and reveals the importance of hydrophobic interactions for self-association. Extension of these studies to common pathogenic variants that are associated with early onset familial disease provides insights into the effect of the mutations on the kinetics and energetics of transthyretin aggregation.
Keywords: amyloidogenic protein, real-time NMR, aggregation kinetics, low-population intermediate, hydrophobic interaction
Abstract
Aggregation of transthyretin (TTR) is the causative agent for TTR cardiomyopathy and polyneuropathy amyloidoses. Aggregation is initiated by dissociation of the TTR tetramer into a monomeric intermediate, which self-assembles into amyloid. The coupled multiple-step equilibria and low-concentration, aggregation-prone intermediates are challenging to probe using conventional assays. We report a 19F-NMR assay that leverages a highly sensitive trifluoroacetyl probe at a strategic site that gives distinct 19F chemical shifts for the TTR tetramer and monomeric intermediate and enables direct quantification of their populations during the aggregation process. Integration of real-time 19F-NMR and turbidity measurements as a function of temperature allows kinetic and mechanistic dissection of the aggregation pathway of both wild-type and mutant TTR. At physiological temperature, the monomeric intermediate formed by wild-type TTR under mildly acidic conditions rapidly aggregates into species that are invisible to NMR, leading to loss of the NMR signal at the same rate as the turbidity increase. Lower temperature accelerates tetramer dissociation and decelerates monomer tetramerization and oligomerization via reduced hydrophobic interactions associated with packing of a phenylalanine (F87) into a neighboring protomer. As a result, the intermediate accumulates to a higher level, and formation of higher-order aggregates is delayed. Application of this assay to pathogenic (V30M, L55P, and V122I) and protective (T119M) mutants revealed significant differences in behavior. A monomeric intermediate was observed only for V122I: aggregation of V30M and L55P proceeds without an observable monomeric intermediate, whereas the protective mutant T119M remains resistant to tetramer dissociation and aggregation.
Aberrant protein aggregation is implicated in several devastating human diseases, including Alzheimer’s disease, type 2 diabetes, and transthyretin amyloidosis (ATTR) (1). Driven by spontaneous self-assembly, amyloidogenic proteins form a variety of aggregated species, some of which are cytotoxic (2, 3). In ATTR, amyloid formation is initiated by dissociation of the TTR tetramer to form an aggregation-prone monomer that self-associates to form small oligomers, amorphous aggregates, and fibrils (4–6). The aggregation of wild-type (WT) TTR results in senile systemic amyloidosis, which affects as much as 25% of the population over age 80 (7). Pathogenic TTR variants with weakened tetramer stabilities lead to an earlier age of onset in familial amyloid cardiomyopathy and polyneuropathy (8). The most common pathogenic mutation associated with ATTR neuropathy is V30M (9), with an estimated disease onset age between 30 and 60 y (10); L55P is a highly destabilized tetramer variant conferring a very early onset in the second decade of life (11); and V122I is involved in ATTR cardiomyopathy with an onset age of ∼60 y (12). Protective mutations also exist. T119M, for instance, is a dissociation-resistant mutation that protects compound heterozygotes, even where the other allele is V30M (13) (see Fig. 1 for the locations of these mutation sites). Extensive insight into the relative tetramer stabilities in TTR variants at neutral pH has been obtained from urea unfolding experiments (14, 15). What remains unclear are the quantitative details of the TTR aggregation pathway at mildly acidic pH, where tetramer dissociation is enhanced and the tetramer–monomer equilibrium is kinetically coupled with the downstream aggregation equilibria. Many questions regarding the fundamentals of these multistep equilibria are unknown: How many intermediates can be kinetically resolved? What are the rates and equilibrium constants and how do they vary with temperature? How do pathogenic mutations affect these coupled processes and their temperature dependences? Answering these questions requires simultaneous kinetic monitoring of tetramers, intermediates and aggregates in real time.
Fig. 1.

The TTRF tetramer showing the BTFA labels at S85C and the mutation sites. (A) Modeled TTRF tetramer is based on PDB 1BMZ (51), where BTFA is conjugated at S85C with Cys side chain atoms shown in gray spheres and the three fluorine atoms highlighted in magenta. The four protomers are colored individually. Cα locations of pathogenic (V30M, L55P, and V122I) and protective (T119M) mutations are shown in cyan spheres for the yellow protomer. The Cα of F87 from the pink protomer is drawn as a red sphere. (B) Expanded view of the dashed region in A. The side chain heavy atoms of V30, L55, T119, and V122 from the yellow protomer are shown as cyan spheres, and the side chain heavy atoms of F87 from the pink protomer are shown as red spheres. The chemical structure of BTFA conjugated to the thiol in the Cys side chain is shown at the bottom right.
A wide range of biophysical techniques have been applied to study protein aggregation kinetics in vitro (2). The self-assembly of aggregates can be followed by aggregation-sensitive optical signals, morphologic changes, or alterations in secondary structure. Thioflavin T fluorescence and turbidity are perhaps the most commonly used techniques for characterizing the kinetics of aggregated products. However, neither is able to directly detect amyloidogenic intermediates that have optical readout that is indistinguishable from that of mature fibrils. As a result, most theoretical modeling has focused on fibrillar formation kinetics (16, 17). It remains a major challenge to directly visualize and accurately quantitate the aggregation intermediates, whose concentrations are typically low [hundreds of nM estimated for TTR (18), for example] and whose populations are highly heterogeneous and constantly exchanging with protein reactants and aggregates.
Real-time 19F-NMR has the potential to detect aggregation intermediates at low concentrations and distinguish them from higher-order aggregates (19, 20). Here we have used 19F-NMR to follow time-dependent amyloid formation by human TTR, labeled by reaction of 3-bromo-1,1,1-trifluoroacetone, BTFA (Fig. 1) with a reactive cysteine side chain, to form a highly sensitive trifluoroacetyl probe at a site adjacent to the TTR subunit interface. The covalently attached probe enables simultaneous quantification of the time-dependent changes in population of the NMR-visible TTR tetramer, monomeric intermediate, and small oligomers during the aggregation process. Integration of the NMR with turbidity measurements allowed the monitoring of NMR-invisible aggregates. A two-step reversible kinetic scheme constructed on the basis of these data models TTR aggregation and provides quantitative temperature-dependent energetic insight into the multiple-step TTR aggregation pathway.
Results
Design of a BTFA-Tagged TTR Variant for Aggregation Studies.
An ideal site for placement of a BTFA-reactive Cys would display distinct 19F chemical shifts for tetramer and monomeric intermediate TTR. Serine 85 was chosen because it is not a known pathogenic site (9), is not conserved in transthyretin-related proteins (21), and is surface exposed to facilitate reaction with BTFA. S85 is close to but not directly involved in the subunit interface (Fig. 1A) and is therefore expected to give rise to oligomerization-dependent 19F chemical shifts, as verified by 19F-NMR (Fig. 2A). To avoid undesired labeling of the native C10 residue, the S85C mutation was introduced into a C10S background. The 15N-filtered diffusion-ordered NMR spectroscopy (DOSY) shows that these mutations do not alter the translational diffusion coefficient compared with that of WT TTR (Materials and Methods) and hence do not alter the oligomerization state. The 1H–15N heteronuclear single quantum coherence (HSQC) spectra show that the chemical shift perturbations in C10S-S85C relative to WT TTR are small and localized to residues near residue 10 (SI Appendix, Fig. S1 A, C, and D). Reaction of C10S-S85C with BTFA is stoichiometric, as confirmed by electrospray ionization mass spectrometry, and the presence of the BTFA label causes only very small backbone amide chemical shift changes (<0.1 ppm) in the immediate proximity of S85 (SI Appendix, Fig. S1B). No 19F resonance could be observed for a C10S negative control (SI Appendix, Fig. S2A), showing that the BTFA labeling is site-specific to S85C. The kinetic traces of acid-mediated aggregation of WT TTR and C10S-S85C-BTFA-TTR are nearly indistinguishable (SI Appendix, Fig. S2 B–D). Collectively, these control experiments firmly establish that C10S-S85C-BTFA TTR (abbreviated as TTRF hereafter) is suitable for 19F-NMR aggregation studies.
Fig. 2.

Monitoring aggregation kinetics of TTRF at pH 4.4 by integrating 19F-NMR and turbidity measurements. (A) Contour plot of time-dependent 19F-NMR spectra of TTRF at 298 K with a time resolution of 1 h. Relative intensity from high to low is colored from red to blue. A representative one-dimensional 19F-NMR spectrum recorded from the 9th to 10th hour is shown below with tetramer (T) and intermediate (I) peaks labeled. The 19F-NMR peak areas (left black axis) and optical density at 330 nm (OD330, right orange axis) plotted as a function of aggregation time at (B) 298 K, (C) 310 K, and (D) 277 K. The peak area data points are colored blue for tetramer (T) and red for intermediate (I). Green points represent the missing signal amplitude for the ensemble of aggregates (A), derived by subtracting the sum of (T + I) from 1. The maximal OD330 (see SI Appendix, Fig. S8B, for a full range of time points) is normalized to the maximum of the A signal for convenience of comparison. The black lines show the fits of the NMR kinetic data based on Scheme 1. The rate of formation of higher order aggregates at 310 K was approximated by fitting the normalized OD330 data to a single exponential function (orange line). The orange connecting lines in OD330 at 298 and 277 K are for eye guidance. Error bars in OD data represent 1 SD from three independent measurements in C but are not shown in B and D because they are smaller than the size of data markers.
Kinetics of TTR Aggregation Monitored by 19F-NMR.
The aggregation of TTRF at a monomer concentration of 10 µM, within the physiological concentration range of 7–21 µM in plasma (22), was followed by real-time 19F-NMR. Aggregation was initiated by lowering the pH from 7.0 to 4.4, the pH at which WT TTR fibril formation rate is maximal (5). At 298 K, the tetramer resonance at −84.19 ppm decays over time, and a new 19F resonance appears 0.28 ppm upfield at −84.47 ppm (Fig. 2A). The translational diffusion coefficient of the species giving rise to this new resonance, measured in a 19F-DOSY experiment, is 1.55 times larger than that of tetrameric TTR (SI Appendix, Fig. S2E). This is in good agreement with the ratio (1.53) predicted for monomeric and tetrameric TTR (Eqs. 1 and 2), indicating that the upfield resonance arises from a monomeric TTR species.
The normalized peak area for this new resonance rapidly builds to a maximum of 15% over 6 h and then slowly decays, showing that it is a kinetic intermediate in the aggregation process (red points in Fig. 2B). The decay of the tetramer peak is biphasic with an ∼75% slow component (blue points in Fig. 2B), hinting at a kinetic process involving at least two steps and three species (23). At pH 7.0, the area of the 19F peak of the tetramer does not change over 66 h (SI Appendix, Fig. S2F), showing that aggregation does not occur under these conditions. At pH 4.4, however, the peak areas for both tetramer and monomer decrease over this time period, and there is an overall loss of the 19F-NMR signal (missing amplitude, green points in Fig. 2B), which is attributed to formation of an ensemble of aggregated species that tumble too slowly to be detected by NMR.
Taken together, these lines of evidence led us to model TTRF aggregation by the following two-step reversible kinetic scheme as a maximum parsimony model:
where T stands for tetramer, I stands for intermediate, and A stands for an ensemble of aggregated species, which accounts for the missing NMR signal so that the total TTR mass is conserved. This model is based on four kinetic rates: the tetramer dissociation rate k1, monomer tetramerization rate k−1, forward oligomerization rate k2, and reverse oligomerization rate k−2 (see SI Appendix, SI Text, for full modeling details). The TTRF aggregation data at 298 K are fitted well by the model (solid black lines in Fig. 2B; see Table 1 for statistics).
Table 1.
Aggregation kinetics for 10 µM TTR mutants by 19F-NMR at three temperatures and pH 4.4
| Construct | Temperature | k1 (h−1)* | k−1 (h−1)* | k2 (h−1)* | k−2 (h−1)* | γ1† (h−1)* | γ2† (h−1)* | RMSE‡ |
| TTRF | 310 K | 0.10 ± 0.01 | 0.75 ± 0.12 | 0.73 ± 0.03 | 0.03 ± 0.01 | 1.54 ± 0.11 (4%) | 0.06 ± 0.01 (96%) | 0.011 |
| TTRF | 298 K | 0.13 ± 0.02 | 0.63 ± 0.10 | 0.06 ± 0.01 | 0.01 ± 0.01 | 0.81 ± 0.12 (25%) | 0.02 ± 0.01 (75%) | 0.013 |
| TTRF | 277 K§ | 0.35 ± 0.01 | 0.15 ± 0.01 | 0.03 ± 0.01 | 0.02 ± 0.01 | 0.51 ± 0.01 (78%) | 0.04 ± 0.01 (22%) | 0.014 |
| V30MF | 310 K¶ | kslow = 0.06 ± 0.01 (100%) | 0.015 | |||||
| V30MF | 298 K¶ | kslow = 0.07 ± 0.01 (100%) | 0.014 | |||||
| V30MF | 277 K¶ | kfast = 0.69 ± 0.08 (77%) and kslow = 0.09 ± 0.01 (23%) | 0.009 | |||||
| F87AF | 310 K | 1.60 ± 0.16 | 0.18 ± 0.04 | 0.56 ± 0.01 | 0.09 ± 0.01 | 1.87 ± 0.20 (81%) | 0.57 ± 0.02 (19%) | 0.038 |
| F87AF | 298 K | 0.46 ± 0.02 | 0.04 ± 0.01 | 0.06 ± 0.01 | 0.03 ± 0.01 | 0.51 ± 0.03 (92%) | 0.08 ± 0.01 (8%) | 0.022 |
| F87AF | 277 K | 0.50 ± 0.02 | 0.02 ± 0.01 | 0.03 ± 0.01 | 0.09 ± 0.01 | 0.53 ± 0.02 (98%) | 0.11 ± 0.01 (2%) | 0.020 |
| L55PF | 310 K¶ | kfast = 0.31 ± 0.05 (41%) and kslow = 0.04 ± 0.01 (59%) | 0.010 | |||||
| L55PF | 298 K¶ | kfast = 0.39 ± 0.02 (64%) and kslow = 0.07 ± 0.01 (36%) | 0.008 | |||||
| L55PF | 277 K# | 3.86 ± 0.60 | 1.06 ± 0.17 | 0.05 ± 0.01 | 0.03 ± 0.01 | 4.94 ± 0.77 (86%) | 0.07 ± 0.01 (14%) | 0.020 |
| V122IF | 310 K | 0.86 ± 0.14 | 1.86 ± 0.40 | 0.95 ± 0.04 | 0.07 ± 0.01 | 3.45 ± 0.52 (22%) | 0.30 ± 0.01 (78%) | 0.020 |
| V122IF | 298 K | 0.90 ± 0.05 | 0.43 ± 0.03 | 0.15 ± 0.01 | 0.04 ± 0.01 | 1.38 ± 0.07 (68%) | 0.13 ± 0.01 (32%) | 0.018 |
| V122IF | 277 K§ | 0.97 ± 0.11 | 0.09 ± 0.01 | 0.02 ± 0.01 | 0.07 ± 0.01 | 1.06 ± 0.12 (98%) | 0.09 ± 0.01 (2%) | 0.015 |
For mutants with observable intermediates, four fitted rates (k1, k−1, k2, and k−2) and two relaxation rates (γ1 and γ2) in Scheme 1 () are reported; otherwise, single or double exponential fits for the tetramer signals are tabulated.
Uncertainties were calculated as 1 SD from 50 bootstrap datasets.
See SI Appendix, Eq. S3, for the definitions of γ1 (fast) and γ2 (slow) relaxation rates. The relative amplitudes are shown in parentheses.
Root-mean-square error.
Peak areas of monomeric intermediate and low molecular weight intermediate(s) were combined as the I signal.
Exponential fits (kfast and kslow) for tetramer signal decays with the corresponding amplitudes are shown in parentheses. Due to the absence of observable aggregation intermediates, only the exponential fits (kfast and kslow) are reported, which approximate the relaxation rates of γ1 and γ2, respectively.
Peak areas of broad peaks of low molecular weight intermediate(s) were combined as the I signal.
Temperature Dependence of Aggregation Kinetics of TTRF.
TTR aggregation is favored by elevated temperature (5). To probe the temperature dependence of kinetic rates in these coupled dissociation–aggregation equilibria at pH 4.4, we monitored TTRF aggregation at additional temperatures of 310 and 277 K (SI Appendix, Figs. S3A and S4A). The tetramer dissociation rate (k1) is faster at low temperature (Table 1) and is correlated (SI Appendix, Fig. S5A) with the subunit exchange rates measured at pH 7.4 or 7.6 under nonaggregating conditions (24, 25). However, at each temperature, k1 at pH 4.4 is consistently threefold to fourfold faster than the subunit exchange rate at pH 7.4 or 7.6, consistent with the role of acid in facilitating tetramer dissociation (4, 8). Temperature also alters the relative amplitudes of the two processes that contribute to the biphasic tetramer 19F resonance decay (Fig. 2 C and D). The slow component dominates at 310 K (96%), whereas its relative amplitude diminishes to 22% at 277 K (Table 1 and SI Appendix, Fig. S6A). The decreased amplitude of the slowly decaying component at 277 K is largely due to the 24- and 5-fold reduction in the forward oligomerization rate k2 and monomer tetramerization rate k−1, respectively, compared with 310 K (Table 1).
The steady-state population of the aggregation intermediate (Isteady) at each temperature was calculated using SI Appendix, Eq. S6. Compared with its population at 298 K (8%), Isteady is decreased at 310 K (3%), whereas it is greatly enhanced (33%) at 277 K (Fig. 2 C and D). These phenomenological outcomes are again rooted in temperature-dependent changes in the kinetics: the rates of both processes that consume the intermediate (k2 and k−1) decline markedly from 310 to 277 K, whereas k1 increases, and k−2 is little changed (Table 1). The observed Isteady at 310 K (3% or 300 nM) is comparable to the estimated critical concentration range (360–870 nM) for aggregation of a designed monomeric TTR (M-TTR) at pH 4.4 (18) and is consistent with the population of monomeric intermediate (<5%) determined by analytical ultracentrifugation (AUC) for WT TTR (6). Additionally, the time at which the I signal reaches a maximum (tmax) is delayed from 2.5 h at 310 K to 6.7 h at 277 K (SI Appendix, Fig. S6B) because the increase in k1 (by 0.25 h−1 from 310 K to 277 K), which could shorten tmax, is outweighed by the decrease in k2 and k−1 (1.3 h−1) that lengthens tmax.
Integrating 19F-NMR and Turbidity Assays Offers Kinetic Insight into Aggregation.
The missing amplitude of the overall 19F-NMR signal increases with time at all three temperatures studied (A signal; green points in Fig. 2 B–D). To determine whether such signal loss is indeed linked to the formation of fibrils or amorphous aggregates that are sufficiently large to scatter light at 330 nm (OD330), we compared the kinetics of A signal increase and the increase in OD330 (orange points in Fig. 2 B–D) at each temperature. At 310 K, the normalized A signal and OD330 kinetic traces are nearly superimposable. Lowering the temperature to 298 K leads to a lag period of at least 1 d, during which the overall 19F signal amplitude decreases without a concomitant increase in OD330. At 277 K, aggregation is further slowed; no significant increase in OD330 was seen during the first 3 d, but the protein eventually aggregated within a period of 2 wk, with a lag time of approximately 1 wk (SI Appendix, Fig. S2D). These results indicate that the missing amplitude of the 19F-NMR signal is correlated with formation of at least two types of particles: high-molecular weight soluble oligomers that do not effectively scatter light at 330 nm and insoluble fibrils or amorphous aggregates that do scatter light. At 310 K, the former rapidly convert into the latter, leading to a time-dependent increase in OD330 that tracks with the missing NMR signal. This observation is consistent with the highly efficient downhill self-assembly pathway of M-TTR at 310 K (18) and with AUC results, which show that the concentration of large soluble oligomers of WT TTR is very low at 310 K (6). Cooling to 298 or 277 K, however, substantially slows down the conversion of soluble oligomers to insoluble forms, so that a lag period appears in OD330. The relationship between the increased lag time and a reduction in the forward rate to convert oligomers into fibrils or other forms of large aggregates is supported by numerical simulations (SI Appendix, Fig. S7).
Aggregation Kinetics of TTR Mutants.
Having established the basis of this integrated aggregation assay using TTRF, we then applied the approach to study the temperature-dependent aggregation of three pathogenic (V30M, L55P, and V122I) and one protective (T119M) TTR mutant at 310 K (SI Appendix, Fig. S3), 298 K (Fig. 3 A–D), and 277 K (SI Appendix, Fig. S4). All mutations were made within the TTRF framework and are termed V30MF, L55PF, V122IF, and T119MF.
Fig. 3.

Temperature dependence of aggregation kinetics of pathogenic and protective mutants at pH 4.4. The 19F-NMR contour plots of (A) V30MF, (B) L55PF, (C) V122IF, and (D) T119MF are shown at 298 K. A representative 19F NMR spectrum recorded from the 9th to 10th hour is shown below each contour plot (see SI Appendix, Figs. S3 and S4, for contour plots at 310 and 277 K, respectively). The time-dependent changes in the tetramer (T) resonance areas of (E) V30MF, (F) L55PF, (G) V122IF, and (H) T119MF are plotted at three temperatures. For each plot, the first data point is normalized to 1. Exponential fits are in black solid lines (see SI Appendix, Fig. S9, for fitting details).
As expected, T119MF is resistant to tetramer dissociation (Fig. 3 D and H and SI Appendix, Figs. S3F and S4F), and no aggregate is detectable within a period of 60 h, although some aggregation occurs over 400 h at 298 and 310 K (SI Appendix, Fig. S8F). The decays of the tetramer resonances of the three pathogenic mutants are similar to or more rapid than TTRF (Fig. 3 A–C and SI Appendix, Figs. S3 C–E and S4 C–E) and are accelerated at low temperature (Fig. 3 E–G). The tetramer dissociation rates of the mutants, relative to TTRF, measured by 19F-NMR at pH 4.4/277 K, are strongly correlated with the rates measured by tryptophan fluorescence at pH 7.0/298 K (14) (SI Appendix, Fig. S5B). These results indicate that the relative kinetic stabilities of these TTR variants are comparable under these two conditions.
Among the three pathogenic mutants, an aggregation-prone, monomeric intermediate was detected for V122IF (Fig. 3C and SI Appendix, Figs. S3E and S4E) at all three temperatures but not for V30MF (Fig. 3A and SI Appendix, Figs. S3C and S4C) or L55PF (Fig. 3B and SI Appendix, Figs. S3D and S4D). At 277 K, broad 19F resonances that are upfield-shifted by ∼0.1–0.2 ppm from the tetramer resonances were observed for V30MF and L55PF (SI Appendix, Fig. S4 C and D), similar to TTRF (SI Appendix, Fig. S4A). These broad resonances likely arise from soluble low-molecular weight oligomers that accumulate at low temperature, where aggregation is slower (SI Appendix, Fig. S8H).
For TTRF and each of the mutants, the rate of 19F-NMR signal loss is very similar to the rate of aggregation obtained from turbidity measurements at 310 K (SI Appendix, Fig. S5C). The similarity of these rates indicates that any downstream steps following the formation of NMR-invisible particles are not rate limiting at 310 K. In contrast, lowering of the temperature profoundly decelerates the downstream processes and increases the lag between the loss of 19F-NMR signal and OD330 (SI Appendix, Fig. S6C).
Role of F87 Hydrophobic Interactions in Temperature-Dependent Aggregation Rates.
The anti-Arrhenius behavior of the cold-enhanced tetramer dissociation of TTRF (Table 1) and the previously observed destabilization of WT TTR at low temperature (24) suggest that hydrophobic interactions are important for stabilizing the tetramer (26). A critical role in stabilizing the tetramer is played by the side chain of F87, which packs into a hydrophobic cavity in the neighboring protomer across the strong dimer interface (Fig. 1B) (27). We therefore created an F87A mutant within the TTRF framework (F87AF) to investigate the role of the F87 side chain in temperature-dependent tetramer dissociation and aggregation.
The tetramer dissociation rate (k1) of F87AF at pH 4.4 is enhanced compared with TTRF. Although k1 for F87AF is larger than that for TTRF at all temperatures, the temperature dependence is reversed such that k1 for the mutant TTR becomes smaller at lower temperatures (Fig. 4A). The reversal of the temperature dependence suggests that the intersubunit hydrophobic interactions mediated by the bulky F87 side chain make a substantial contribution to the anti-Arrhenius behavior of WT TTR dissociation. As expected, the monomer tetramerization rate k−1 is smaller for F87AF than TTRF (Fig. 4B), again reflecting the loss of the F87-mediated hydrophobic interactions that favor tetramer formation. The forward and backward rates (k2 and k−2) of the oligomerization step exhibit similar temperature-dependent trends for TTRF and F87AF. Lowering the temperature from 310 to 298 K reduces k2 by around 10-fold in both constructs (Fig. 4C). Similar to k1, k−2 is consistently greater for F87AF than TTRF (Fig. 4D), suggesting another role of F87 in stabilizing the ensemble of aggregated states. A minimum in k−2 is observed at 298 K for TTRF and F87AF (also for V122IF; Discussion) indicating a curved temperature-dependent free energy function such as is associated with transfer of nonpolar solutes to aqueous solvent (28).
Fig. 4.

Comparison of kinetic parameters for TTRF and F87AF. (A) Tetramer dissociation rate k1. (B) Monomer tetramerization rate k−1. (C) Forward oligomerization rate k2. (D) Reverse oligomerization rate k−2. Uncertainty was estimated as 1 SD from 50 bootstrap datasets.
V122IF and F87AF Favor Formation of Monomeric Intermediate.
The monomeric intermediate with a 19F signal upfield-shifted by ∼0.3 ppm is present in F87AF samples at all three temperatures studied (Fig. 5A and SI Appendix, Figs. S3B and S4B). Kinetics analysis shows that Isteady is higher for F87AF than for TTRF (Fig. 5B). V122IF also forms a higher steady-state population of intermediate than TTRF at all three temperatures (Fig. 5B). Accordingly, the tetramer dissociation rate k1 is enhanced in V122IF relative to TTRF (Table 1), consistent with the destabilized V122I tetramer revealed by urea unfolding at pH 7.0 (29, 30). The apparent equilibrium constant for the tetramer–intermediate equilibrium (Kapp,1 = k1/k−1) reveals a clear ranking with F87AF > V122IF > TTRF at all three temperatures (Fig. 5C). The ratio between k2 and k−2 represents the apparent equilibrium constant for the oligomerization step (Kapp,2 = k2/k−2). In contrast to Kapp,1, Kapp,2 follows the reverse trend, TTRF > V122IF > F87AF, except for nearly identical values of Kapp,2 for V122IF and F87AF at 277 K (Fig. 5D). Because Kapp,2 is inversely proportional to the steady-state concentration of [I], both Kapp,1 and Kapp,2 favor the intermediate state in V122IF and F87AF compared with TTRF. The sign of the slopes in Fig. 5 C and D indicates that tetramer dissociation is exothermic, whereas the aggregation process is endothermic.
Fig. 5.

Comparison of the populations of the intermediates formed by TTRF, V122IF, and F87AF at pH 4.4. (A) Time-dependent 19F-NMR contour plot of F87AF at 298 K. A representative spectrum recorded from the 9th to 10th hour is shown below. Compared with TTRF (Fig. 2A) and V122IF (Fig. 3C), the intermediate population predominates over tetramer during this time period. (B) The relative population of the intermediate at steady state (Isteady) for TTRF (black), V122IF (cyan), and F87AF (red). van’t Hoff plot for (C) the apparent tetramer–intermediate equilibrium constant (Kapp,1 = [I]/[T]) and (D) the apparent intermediate–aggregate equilibrium constant (Kapp,2 = [A]/[I]) for TTRF, V122IF, and F87AF. In C and D, black lines are linear fits to the van’t Hoff equation, and dashed lines denote zero. Uncertainties in B–D were estimated as 1 SD from 50 bootstrap datasets. Some uncertainties in B and D are comparable to or smaller than the size of data marker.
Apparent Free Energy Diagram for TTR Aggregation.
Based on the apparent equilibrium constants and four rates in the TTRF, F87AF, and V122IF aggregation equilibria, we plotted the apparent energy level diagram for each protein (Fig. 6). Both mutations destabilize the TTR tetramer. The V122IF tetramer is destabilized relative to TTRF by 0.8–1.4 kcal/mol between 310 and 277 K, in good agreement with ΔΔG < 1 kcal/mol estimated previously by an SDS separation assay at 298 K (30). The tetramer of F87AF is further destabilized relative to V122IF by 0.4–1.8 kcal/mol between 310 and 277 K.
Fig. 6.

Schematic free energy level diagram at pH 4.4 of TTRF (black), V122IF (cyan), and F87AF (red) at (A) 310 K, (B) 298 K, and (C) 277 K based on Scheme 1. The apparent free energy of the I state is set to 0 as reference for all three temperatures (dashed line). The transition states between T and I and between I and A are designated 1* and 2*, respectively.
Decreasing temperature destabilizes the tetramer relative to the intermediate. Thus, the TTRF tetramer is 1.2 and 0.9 kcal/mol more stable than the monomeric intermediate at 310 and 298 K, respectively, but is 0.5 kcal/mol less stable than the intermediate at 277 K. For V122IF, the tetramer is less stable than the intermediate at temperatures of 298 K and lower, whereas the F87AF tetramer is 1.3–1.7 kcal/mol less stable than the intermediate at all three temperatures.
The ensemble of aggregated states is also destabilized at lower temperatures. The TTRF aggregates are destabilized by 1.8 kcal/mol between 310 and 277 K but remain more stable than the intermediate over this temperature range. In contrast, the aggregates of F87AF and V122IF are less stable than their intermediates by 0.6–0.7 kcal/mol at 277 K and are less stable than TTRF by 0.2–1 kcal/mol at each temperature studied (Fig. 6).
At 310 K, the transition state for the tetramer–intermediate reaction (1*) of V122IF is about 0.6 kcal/mol more stable than that of TTRF when both are referenced to the intermediate. Together with a 0.8 kcal/mol destabilization of the ground state of the V122IF tetramer, this results in a nearly ninefold increase in tetramer dissociation rate, k1 relative to TTRF. In contrast, the transition state of T119M is of higher energy than that of WT TTR, leading to a greatly slowed tetramer dissociation rate at neutral pH (31). At lower temperatures, the V122IF 1* transition state is destabilized by 0.2–0.3 kcal/mol relative to that of TTRF. Nevertheless, V122IF tetramer dissociation remains faster than TTRF because of the 0.8–1.4 kcal/mol destabilization of the V122IF tetramer. Similar results were found for F87AF. Because the relative changes in k2 across mutations are generally small (onefold to twofold) at each temperature, the energetic variations for the transition states in the intermediate–aggregate reaction (2*) are relatively minor (0.3–0.6 kcal/mol) when referenced to the intermediate (Fig. 6).
Discussion
In the current model of the TTR aggregation pathway, the tetramer must dissociate to form monomer before the downstream aggregation events (5, 8, 32). The kinetic details of the coupled dissociation–aggregation equilibria have remained unknown due to the lack of suitable methods to monitor the formation and consumption of the aggregation-prone intermediates. We have addressed this problem by introducing a highly sensitive 19F-NMR probe that simultaneously reports on the populations of both the TTR tetramer, the monomeric intermediate, and from missing signal amplitude, the aggregated states. Based on this assay, we propose a two-step kinetic scheme (Scheme 1) that affords a direct comparison across TTR mutants and at variable temperatures.
Scheme 1.
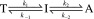
The temperature dependence of the kinetic rates reveals rich details of the TTR aggregation pathway. At pH 4.4, the TTRF tetramer dissociation rate (k1) increases at low temperature (Table 1). This anti-Arrhenius behavior points toward the critical role of intersubunit hydrophobic interactions in stabilization of the TTR tetramer (26), which can prevent certain hydrophobic residues from forming aberrant intersubunit contacts in amyloidogenic self-assembly. Low temperature reduces the strength of hydrophobic interactions (33), destabilizing tetrameric TTR and accelerating dissociation. Remarkably, cold-enhanced dissociation is greatly attenuated in V122IF (Table 1), and the sign of the temperature dependence is reversed in the F87AF mutant (Fig. 4A). Structurally, the hydrophobic side chains of F87 and V122 form a part of the strong dimer interface of the tetramer (Fig. 1B). Our data show that perturbations at this interface attenuate hydrophobic interactions between subunits and thus destabilize the tetramers of V122IF and F87AF with respect to TTRF (Fig. 6). The hydrophobic interactions mediated by the F87 and V122 side chains also appear to play a role in stabilization of aggregates because ΔΔG values show that the aggregates of V122IF and F87AF are destabilized relative to TTRF at each temperature studied. As the temperature is lowered, the rates of the reactions that convert intermediates into tetramers (k−1) and oligomers (k2) decrease for TTRF, V122IF, and F87AF. These observations could be accounted for by the hydrophobic effect or by the reduced thermal energy available to overcome activation barriers at low temperature. However, the reverse oligomerization rates (k−2) of these proteins exhibit a curved temperature dependence, with a minimum at 298 K (Fig. 4D and Table 1), which implies a curved free energy surface associated with heat capacity changes that arise from transfer of nonpolar residues to aqueous solution (34, 35). At the middle temperature, exposure of hydrophobic groups buried in the aggregates to solvent is least favored, and the corresponding rate of formation of the monomeric intermediate is slowest.
A high population of the monomeric amyloidogenic intermediate is not necessarily correlated with fast TTR aggregation, because aggregation is a complex multistep process involving tetramers, intermediates, oligomers, and higher-order aggregates. Likewise, the forward and reverse rates for the oligomerization step (k2 and k−2) alone do not correlate with the overall aggregation kinetics; the upstream (tetramer dissociation and monomer tetramerization, k1 and k−1) and downstream (formation and dissolution of higher order aggregates) steps are also important in determining the overall aggregation kinetics. The increased lag period between the 19F-NMR signal loss and OD330 at 277 K (SI Appendix, Fig. S6C) indicates that the rates of formation and dissociation of NMR-invisible, higher-order aggregates are more pivotal in determining the slow aggregation kinetics than the rates of preceding steps in the equilibrium (k1/−1 or k2/−2). At 310 K, however, k1/−1 and k2/−2 are the major determinant of the overall aggregation because the rate at which OD330 increases closely matches the rate of 19F-NMR signal loss (SI Appendix, Fig. S5C).
At physiological temperature and concentration, the absence of a detectable monomeric intermediate for V30MF or L55PF suggests that the oligomerization and tetramerization rates that deplete the intermediate outweigh the corresponding reverse rates that accumulate it at pH 4.4. The side chains of V30 and L55 are in close contact in the TTR structure (Fig. 1B) and induce subtle conformational perturbations that destabilize the V30M and L55P tetramer and monomer relative to WT TTR (15). Solid-state NMR has shown that local structural changes occur at the short d-strand (residues G53-L55) in the insoluble aggregates of V30M and L55P (36), whereas the native-like β-sheet structures remain intact in WT TTR amyloids (37). Molecular dynamics simulations have also suggested displacement of the d-strand in the unfolding pathway of V30M and L55P monomers (38). Given the millisecond timescale conformational fluctuations observed in M-TTR (39), it is likely that conformational dynamics in the monomeric intermediates of WT or pathogenic TTR mutants could contribute to misfolding and amyloidogenic self-assembly (40).
As aggregation slows at 277 K (SI Appendix, Fig. S8H), some soluble TTR oligomers become NMR-visible in TTRF, V30MF, and L55PF (SI Appendix, Fig. S4 A, C, and D), whereas they are absent at 298 K (Figs. 2A and 3 A and B) or 310 K (SI Appendix, Fig. S3 A, C, and D). Analogous results have been reported using AUC (6). It is likely that the formation of TTR oligomers is strongly dependent upon experimental conditions. TTR oligomers have been observed when aggregation is initialized by unfolding at pH 2 followed by addition of NaCl (41, 42). Moreover, cell culture medium has been reported to facilitate formation of oligomeric intermediates by M-TTR at pH 7.4 but not by the V30M tetramer (43). We anticipate that our aggregation assay will be readily applicable to the detection and quantitation of small TTR oligomers under many different solution conditions. In particular, the absence of an endogenous 19F signal suggests that our approach may have the potential to observe small oligomers and measure real-time aggregation of 19F-tagged TTR variants in the presence of human plasma ex vivo.
At 298 K and above, the monomeric TTR intermediates are converted rapidly to higher-order aggregates without accumulation of soluble oligomers that are small enough for detection by 19F-NMR. TTR is similar in this regard to the islet amyloidogenic polypeptide IAPP (44) but differs from the Aβ (1–40) peptide and the prion protein PrP, which aggregate to form NMR-detectable, low-molecular weight oligomers on the pathway to fibrils (19, 20)
Direct detection of the amyloidogenic monomeric intermediate provides the missing kinetic evidence for the TTR aggregation mechanism at acidic pH (5, 8, 32). Our two-step kinetic model is consistent with published data for aggregation under the same conditions (14) and offers additional quantitative kinetic insight into the multistep equilibria involved in the TTR aggregation process. Like other amyloidogenic proteins, the kinetics and mechanism of TTR aggregation are undoubtedly influenced by pathogenic mutations and dependent upon solution conditions. For example, different aggregation kinetics have been reported for TTR at pH 2–3 (41, 45), where the major species is the largely unfolded TTR monomer before aggregation is initiated. The partially denaturing conditions used in our current work, where aggregation is initialized by the TTR tetramer at a physiological concentration and the solution pH 4.4 is close to that of a lysosome (pH 4.7), are likely more physiologically relevant. The high efficiency of our real-time 19F-NMR assay enables determination of TTR aggregation kinetics, equilibrium constants, and free energies in one simple experiment, opening the way to detailed mapping of the aggregation landscape of WT, variant, and mixed tetrameric TTRs and its dependence on solution conditions. Such studies will provide new insights into the factors that promote TTR aggregation and lead to debilitating disease.
Materials and Methods
Protein Expression, Purification, and Labeling.
TTR mutants were expressed as described previously (46). See SI Appendix for details of protein purification and labeling.
NMR and Turbidity Experiments.
HSQC spectra for ∼100 µM [1H, 15N]-C10S-S85C in GF buffer were recorded on a Bruker Avance 900-MHz spectrometer at 298 K before and after coupling to BTFA. Chemical shift differences between C10S-S85C and WT TTR and between C10S-S85C and TTRF are shown in SI Appendix, Fig. S1. The translational diffusion coefficient of C10S-S85C was measured on the 900-MHz spectrometer using 15N-filtered DOSY at pH 7.0 and 298 K. The measured translational diffusion coefficient is (5.1 ± 0.1) 10−7 cm2/s for C10S-S85C, which agrees with the reported data of WT TTR as (5.1 ± 0.2) 10−7 cm2/s (46). BTFA labeling is site-specific to S85C because the observed tetramer 19F peak at −84.19 ppm in TTRF at 298 K is absent in a C10S control that was treated with BTFA the same way as C10S-S85C (SI Appendix, Fig. S2A).
Unless otherwise noted, aggregation assays were conducted for 10 µM TTR mutants (monomer concentration) in 50 mM sodium acetate and 100 mM KCl at pH 4.4 (aggregation buffer) at 310, 298, or 277 K. Optical density (OD) measurements were performed as previously described (5). Real-time 19F-NMR aggregation experiments were performed on a Bruker Avance 601 spectrometer with a QCI 1H/19F–13C/15N quadruple resonance cryoprobe and a shielded z-gradient coil. Corrections were made for the measurement dead time (∼10 min, required for mixing, matching, tuning, and shimming), so that time 0 in the kinetics data analyses corresponded to the time when the pH was lowered from 7.0 to 4.4. Typically, 100 µM TTR in GF buffer was mixed with 8× (vol/vol) aggregation buffer and 1× (vol/vol) D2O. Each spectrum was recorded with 4k complex points using a pulse length of 10–12 µs and a recycle delay of 1 s. A line broadening factor of 1 Hz was applied to raw free induction decays, which then were zero-filled to 16k before Fourier transformation in NMRPipe (47).
The relative translational diffusion coefficients of tetramer (T) and monomeric intermediate (I) were determined using a 19F longitudinal encode–decode diffusion-ordered NMR experiment (19F-DOSY) (48). To enhance the signal/noise ratio, the concentration of TTRF in the aggregation buffer was increased to 80 µM, and the temperature was set at 277 K to slow down aggregation so that more I species could accumulate. A diffusion delay of 100 ms and 10 evenly spaced relative z-gradient strengths from 5 to 50% were used. All 10 diffusion-ordered experiments were collected in an interleaved manner to average out time-dependent signal changes during acquisition (∼14 h). The fitted slope for the logarithms of peak areas plotted against the squares of relative z-gradient strengths was compared (SI Appendix, Fig. S2E).
A standard inversion recovery pulse sequence was used to measure the 19F longitudinal relaxation time constant (T1) of the T and I forms of TTRF in aggregation buffer at 298 K. Delays of 0.02, 0.05, 0.07, 0.1, 0.15, 0.2, 0.3, 0.5, 0.8, 1, and 2 s were used. The recycle delay was set to 4 s. All spectra were collected in an interleaved manner. The peak areas were fitted using a single exponential function, and the uncertainty was estimated as 1 SD from 50 bootstrap datasets (49).
Estimates of Molecular Size from Stokes–Einstein Equation.
The Stokes–Einstein equation for the translational diffusion coefficient (D) is
| [1] |
where kB is the Boltzmann constant, T is temperature, r is hydrodynamic radius, and η is viscosity.
Because the 19F-DOSY measurements were performed for tetramer and monomer TTR in the same NMR tube, it is only necessary to consider the difference in hydrodynamic radius (r) of these two states, which can be estimated using the equation
| [2] |
where is the specific volume (0.73 cm3/g), M is the molecular weight of either TTR tetramer (55.0 kDa) or monomer (13.8 kDa), NA is the Avogadro’s constant, and rw is the thickness of the hydration shell (assumed to be 1.6 Å) (50). By combining Eqs. 1 and 2, the ratio between the translational diffusion coefficients of monomer and tetramer was determined to be 1.53, close to the experimentally determined ratio of 1.55.
Kinetics Analysis.
A set of ordinary differential equations (ODEs) based on Scheme 1 was used to describe the time-dependent peak area changes. The summed squared residuals between the trial ODEs solved by ode23 and the experimental data were minimized by lsqcurvefit in MATLAB. See SI Appendix for details.
Supplementary Material
Acknowledgments
We thank Gerard Kroon for assistance in NMR experiments, Euvel Manlapaz for technical support, and Ke Yang for electrospray ionization mass spectrometry measurements. This work was supported by National Institutes of Health Grant DK34909 (to P.E.W.), the Skaggs Institute for Chemical Biology, and American Heart Association Grant 17POST32810003 (to X.S.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1807024115/-/DCSupplemental.
References
- 1.Knowles TP, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol. 2014;15:384–396. doi: 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- 2.Bemporad F, Chiti F. Protein misfolded oligomers: Experimental approaches, mechanism of formation, and structure-toxicity relationships. Chem Biol. 2012;19:315–327. doi: 10.1016/j.chembiol.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 3.Walsh DM, Selkoe DJ. Aβ oligomers–A decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 4.Colon W, Kelly JW. Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry. 1992;31:8654–8660. doi: 10.1021/bi00151a036. [DOI] [PubMed] [Google Scholar]
- 5.Lai Z, Colón W, Kelly JW. The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate that can self-assemble into amyloid. Biochemistry. 1996;35:6470–6482. doi: 10.1021/bi952501g. [DOI] [PubMed] [Google Scholar]
- 6.Lashuel HA, Lai Z, Kelly JW. Characterization of the transthyretin acid denaturation pathways by analytical ultracentrifugation: Implications for wild-type, V30M, and L55P amyloid fibril formation. Biochemistry. 1998;37:17851–17864. doi: 10.1021/bi981876+. [DOI] [PubMed] [Google Scholar]
- 7.Pitkänen P, Westermark P, Cornwell GG., 3rd Senile systemic amyloidosis. Am J Pathol. 1984;117:391–399. [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson SM, Connelly S, Fearns C, Powers ET, Kelly JW. The transthyretin amyloidoses: From delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J Mol Biol. 2012;421:185–203. doi: 10.1016/j.jmb.2011.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Connors LH, Lim A, Prokaeva T, Roskens VA, Costello CE. Tabulation of human transthyretin (TTR) variants, 2003. Amyloid. 2003;10:160–184. doi: 10.3109/13506120308998998. [DOI] [PubMed] [Google Scholar]
- 10.Miller SR, Sekijima Y, Kelly JW. Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab Invest. 2004;84:545–552. doi: 10.1038/labinvest.3700059. [DOI] [PubMed] [Google Scholar]
- 11.Jacobson DR, McFarlin DE, Kane I, Buxbaum JN. Transthyretin Pro55, a variant associated with early-onset, aggressive, diffuse amyloidosis with cardiac and neurologic involvement. Hum Genet. 1992;89:353–356. doi: 10.1007/BF00220559. [DOI] [PubMed] [Google Scholar]
- 12.Jacobson DR, et al. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med. 1997;336:466–473. doi: 10.1056/NEJM199702133360703. [DOI] [PubMed] [Google Scholar]
- 13.Hammarström P, Schneider F, Kelly JW. Trans-suppression of misfolding in an amyloid disease. Science. 2001;293:2459–2462. doi: 10.1126/science.1062245. [DOI] [PubMed] [Google Scholar]
- 14.Hammarström P, Jiang X, Hurshman AR, Powers ET, Kelly JW. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc Natl Acad Sci USA. 2002;99:16427–16432. doi: 10.1073/pnas.202495199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sekijima Y, et al. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121:73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 16.Morris AM, Watzky MA, Finke RG. Protein aggregation kinetics, mechanism, and curve-fitting: A review of the literature. Biochim Biophys Acta. 2009;1794:375–397. doi: 10.1016/j.bbapap.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 17.Knowles TP, et al. An analytical solution to the kinetics of breakable filament assembly. Science. 2009;326:1533–1537. doi: 10.1126/science.1178250. [DOI] [PubMed] [Google Scholar]
- 18.Hurshman AR, White JT, Powers ET, Kelly JW. Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry. 2004;43:7365–7381. doi: 10.1021/bi049621l. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki Y, et al. Resolution of oligomeric species during the aggregation of Aβ1-40 using 19F NMR. Biochemistry. 2013;52:1903–1912. doi: 10.1021/bi400027y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larda ST, Simonetti K, Al-Abdul-Wahid MS, Sharpe S, Prosser RS. Dynamic equilibria between monomeric and oligomeric misfolded states of the mammalian prion protein measured by 19F NMR. J Am Chem Soc. 2013;135:10533–10541. doi: 10.1021/ja404584s. [DOI] [PubMed] [Google Scholar]
- 21.Eneqvist T, Lundberg E, Nilsson L, Abagyan R, Sauer-Eriksson AE. The transthyretin-related protein family. Eur J Biochem. 2003;270:518–532. doi: 10.1046/j.1432-1033.2003.03408.x. [DOI] [PubMed] [Google Scholar]
- 22.Johnson SM, et al. Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: A focus on the transthyretin amyloidoses. Acc Chem Res. 2005;38:911–921. doi: 10.1021/ar020073i. [DOI] [PubMed] [Google Scholar]
- 23.Cantor CR, Schimmel PR. Biophysical Chemistry: Part III: The Behavior of Biological Macromolecules. W. H. Freeman; San Francisco: 1980. [Google Scholar]
- 24.Schneider F, Hammarström P, Kelly JW. Transthyretin slowly exchanges subunits under physiological conditions: A convenient chromatographic method to study subunit exchange in oligomeric proteins. Protein Sci. 2001;10:1606–1613. doi: 10.1110/ps.8901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rappley I, et al. Quantification of transthyretin kinetic stability in human plasma using subunit exchange. Biochemistry. 2014;53:1993–2006. doi: 10.1021/bi500171j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hammarström P, Jiang X, Deechongkit S, Kelly JW. Anion shielding of electrostatic repulsions in transthyretin modulates stability and amyloidosis: Insight into the chaotrope unfolding dichotomy. Biochemistry. 2001;40:11453–11459. doi: 10.1021/bi010673+. [DOI] [PubMed] [Google Scholar]
- 27.Jiang X, et al. An engineered transthyretin monomer that is nonamyloidogenic, unless it is partially denatured. Biochemistry. 2001;40:11442–11452. doi: 10.1021/bi011194d. [DOI] [PubMed] [Google Scholar]
- 28.Privalov PL, Gill SJ. Stability of protein structure and hydrophobic interaction. Adv Protein Chem. 1988;39:191–234. doi: 10.1016/s0065-3233(08)60377-0. [DOI] [PubMed] [Google Scholar]
- 29.Hurshman Babbes AR, Powers ET, Kelly JW. Quantification of the thermodynamically linked quaternary and tertiary structural stabilities of transthyretin and its disease-associated variants: The relationship between stability and amyloidosis. Biochemistry. 2008;47:6969–6984. doi: 10.1021/bi800636q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang X, Buxbaum JN, Kelly JW. The V122I cardiomyopathy variant of transthyretin increases the velocity of rate-limiting tetramer dissociation, resulting in accelerated amyloidosis. Proc Natl Acad Sci USA. 2001;98:14943–14948. doi: 10.1073/pnas.261419998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hammarström P, Wiseman RL, Powers ET, Kelly JW. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science. 2003;299:713–716. doi: 10.1126/science.1079589. [DOI] [PubMed] [Google Scholar]
- 32.Kelly JW. Amyloid fibril formation and protein misassembly: A structural quest for insights into amyloid and prion diseases. Structure. 1997;5:595–600. doi: 10.1016/s0969-2126(97)00215-3. [DOI] [PubMed] [Google Scholar]
- 33.Dill KA. Dominant forces in protein folding. Biochemistry. 1990;29:7133–7155. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 34.Baldwin RL. Temperature dependence of the hydrophobic interaction in protein folding. Proc Natl Acad Sci USA. 1986;83:8069–8072. doi: 10.1073/pnas.83.21.8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schindler T, Schmid FX. Thermodynamic properties of an extremely rapid protein folding reaction. Biochemistry. 1996;35:16833–16842. doi: 10.1021/bi962090j. [DOI] [PubMed] [Google Scholar]
- 36.Lim KH, et al. Pathogenic mutations induce partial structural changes in the native beta-sheet structure of transthyretin and accelerate aggregation. Biochemistry. 2017;56:4808–4818. doi: 10.1021/acs.biochem.7b00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lim KH, et al. Solid-state NMR studies reveal native-like β-sheet structures in transthyretin amyloid. Biochemistry. 2016;55:5272–5278. doi: 10.1021/acs.biochem.6b00649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang M, Yordanov B, Levy Y, Brüschweiler R, Huo S. The sequence-dependent unfolding pathway plays a critical role in the amyloidogenicity of transthyretin. Biochemistry. 2006;45:11992–12002. doi: 10.1021/bi0609927. [DOI] [PubMed] [Google Scholar]
- 39.Lim KH, Dyson HJ, Kelly JW, Wright PE. Localized structural fluctuations promote amyloidogenic conformations in transthyretin. J Mol Biol. 2013;425:977–988. doi: 10.1016/j.jmb.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lim KH, et al. Structural changes associated with transthyretin misfolding and amyloid formation revealed by solution and solid-state NMR. Biochemistry. 2016;55:1941–1944. doi: 10.1021/acs.biochem.6b00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Faria TQ, et al. A look into amyloid formation by transthyretin: Aggregation pathway and a novel kinetic model. Phys Chem Chem Phys. 2015;17:7255–7263. doi: 10.1039/c4cp04549a. [DOI] [PubMed] [Google Scholar]
- 42.Lindgren M, Sörgjerd K, Hammarström P. Detection and characterization of aggregates, prefibrillar amyloidogenic oligomers, and protofibrils using fluorescence spectroscopy. Biophys J. 2005;88:4200–4212. doi: 10.1529/biophysj.104.049700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reixach N, Deechongkit S, Jiang X, Kelly JW, Buxbaum JN. Tissue damage in the amyloidoses: Transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc Natl Acad Sci USA. 2004;101:2817–2822. doi: 10.1073/pnas.0400062101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suzuki Y, Brender JR, Hartman K, Ramamoorthy A, Marsh EN. Alternative pathways of human islet amyloid polypeptide aggregation distinguished by 19F nuclear magnetic resonance-detected kinetics of monomer consumption. Biochemistry. 2012;51:8154–8162. doi: 10.1021/bi3012548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Groenning M, Campos RI, Hirschberg D, Hammarström P, Vestergaard B. Considerably unfolded transthyretin monomers preceed and exchange with dynamically structured amyloid protofibrils. Sci Rep. 2015;5:11443. doi: 10.1038/srep11443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun X, Dyson HJ, Wright PE. Fluorotryptophan incorporation modulates the structure and stability of transthyretin in a site-specific manner. Biochemistry. 2017;56:5570–5581. doi: 10.1021/acs.biochem.7b00815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Delaglio F, et al. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 48.Altieri AS, Hinton DP, Byrd RA. Association of biomolecular systems via pulsed field gradient NMR self-diffusion measurements. J Am Chem Soc. 1995;117:7566–7567. [Google Scholar]
- 49.Efron B. 1977 Rietz lecture—Bootstrap methods—Another look at the jackknife. Ann Stat. 1979;7:1–26. [Google Scholar]
- 50.Cavanagh J, Fairbrother WJ, Palmer AG, Rance M, Skelton NJ. Protein NMR Spectroscopy: Principles and Practice. Elsevier Science; San Diego: 2007. [Google Scholar]
- 51.Peterson SA, et al. Inhibiting transthyretin conformational changes that lead to amyloid fibril formation. Proc Natl Acad Sci USA. 1998;95:12956–12960. doi: 10.1073/pnas.95.22.12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.