

Summary
The mucosal surface of the respiratory tract encounters microbes, such as fungal particles, with every inhaled breath. When pathogenic fungi breach the physical barrier and innate immune system within the lung to establish an infection, adaptive immunity is engaged, often in the form of helper CD4 T‐cell responses. Type 1 responses, characterized by interferon‐γ production from CD4 cells, promote clearance of Histoplasma capsulatum and Cryptococcus neoformans infection. Likewise, interleukin‐17A (IL‐17A) production from Th17 cells promotes immunity to Blastomyces dermatitidis and Coccidioides species infection by recruiting neutrophils. In contrast the development of T helper type 2 responses, characterized by IL‐5 production from T cells and eosinophil influx into the lungs, drives allergic bronchopulmonary aspergillosis and poor outcomes during C. neoformans infection. Experimental vaccines against several endemic mycoses, including Histoplasma capsulatum, Coccidioides, Cryptococcus and Blastomyces dermatitidis, induce protective T‐cell responses and foreshadow the development of vaccines against pulmonary fungal infections for use in humans. Additionally, recent work using antifungal T cells as immunotherapy to protect immune‐compromised patients from opportunist fungal infections also shows great promise. This review covers the role of T‐cell responses in driving protection and pathology in response to pulmonary fungal infections, and highlights promising therapeutic applications of antifungal T cells.
Keywords: adaptive immunity, fungal infection, lung, mucosal immunity, T cell
Fungal recognition and T‐cell priming the lung
The lungs represent a massive environment‐exposed surface in the body, which is challenged with microbes and microbial products with every breath.1 Fungi represent a medically important class of pathogenic microbes, and 4–11% of the fine‐particle mass inhaled into the lungs contains fungal spores.2 To preserve epithelial integrity and prevent colonization by infectious organisms including fungi, the host has developed several immunological mechanisms in the lungs.1 Epithelial cells provide barrier immunity, preventing inhaled particles access to deeper tissues and vasculature, while mucus and antimicrobial peptides further deter colonization.1 Resident alveolar macrophages remove debris from the lungs,3 maintaining clear airways and preventing the establishment of infection.
The mammalian host employs a suite of pattern recognition receptors (PRRs) that are capable of recognizing fungal ligands and initiating innate inflammatory responses.4, 5 One of the best‐characterized PRR–fungal ligand relationships is the recognition of fungal β‐glucan by the prototypical C‐type lectin receptor (CLR) dectin‐1. Likewise, heterodimers of Toll‐like receptors (TLR) TLR2 and TLR1 recognize triacylated lipoprotein, whereas TLR2/TLR6 heterodimers recognize diacylated lipoprotein.5 Mannose receptor recognizes mannose and other sugar moieties on the surface of microbial cells.6 The CLR dectin‐2 has previously been shown to respond to mannan stimulation,5 and recent work has also identified the glycoprotein Bl‐Eng2 of Blasotmyces dermatitidis as a bona‐fide ligand for this receptor.7 A newly defined CLR, MelLec enables the host to sense melanin on pathogens including Aspergillus spores.8 These PRR–fungal ligand interactions represent a major means by which the immune system recognizes and initiates immune responses against inhaled pulmonary fungal pathogens.
When these early mechanisms fail and fungal infection is established in the airway, the host responds with a coordinated immune response. Monocyte‐derived macrophages and dendritic cells are often recruited into the airway.9, 10 Likewise, other myeloid cell populations, including neutrophils,11, 12 monocytes13 and eosinophils,14, 15 infiltrate the airways in an effort to combat the infection. In almost all cases of pulmonary fungal infection, the development of adaptive immunity and the engagement of CD4 T‐cell help is a key determinant of the outcome on infection.
CD4 T‐cell responses can only occur after a carefully orchestrated process involving precise interactions between stromal, myeloid and lymphoid cells. The exact mechanisms underpinning the priming of T‐cell responses have been extensively reviewed elsewhere,16, 17, 18, 19 and are only briefly summarized here. Upon uptake of foreign antigen by professional antigen‐presenting cells at mucosal sites, the antigen‐presenting cells traffic to draining lymph nodes in order to locate and prime naive T cells.18 CD4 T‐cell priming occurs not only by activation of the T‐cell receptor by binding its cognate antigen in the context of MHCII, but also through co‐stimulatory signals19 and the cytokine milieu20 present during T‐cell priming. Once T‐cell priming is complete, the effector T cells depart the lymph node to enter into the mucosal tissue and carry out their effector function.17
There are three major CD4 helper T‐cell subsets that we will discuss in the context of fungal immunity.21, 22, 23, 24 Type 1 helper T cells (Th1 cells) are characterized by interferon‐γ (IFN‐γ) production, and are broadly effective at clearing intracellular pathogens.25, 26 In contrast, Th17 cells produce interleukin‐17 (IL‐17) and protect against extracellular pathogens in part through the recruitment of neutrophils.25, 27 Production of IL‐5 and IL‐13 is characteristic of Th2 cells, which are generally believed to promote clearance of helminth worms and other large parasitic organisms, but may also help to clear select fungi or alternatively mediate allergic inflammation in response to inhaled mould and related products (summarized in Fig. 1).25, 26, 28, 29, 30, 31 This review article will be largely organized along these lines: discussing pulmonary fungal infections grouped by the predominant/protective response of either Th1, Th17 or Th2.
Figure 1.
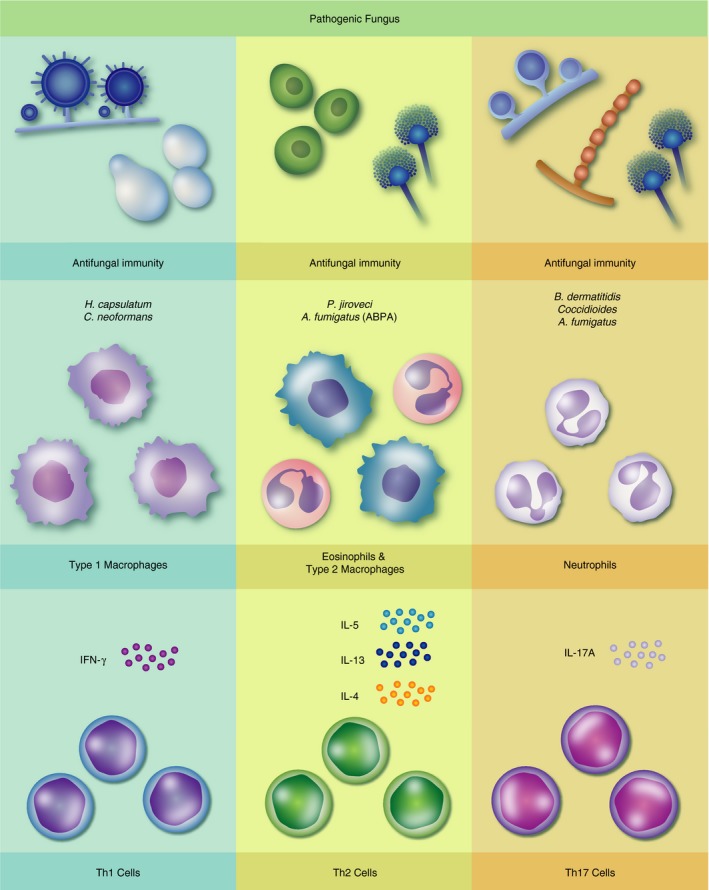
T‐cell responses to pulmonary fungal infections. Left column: Type 1 responses, characterized by interferon‐γ (IFN‐γ) production from CD4 cells and Type 1/classically activated macrophages, mediate antifungal immunity to Histoplasma capsulatum and Cryptococcus neoformans infection. Centreal column: Eosinophil and alternatively activated macrophages supported by interleukin‐4 (IL‐4), IL‐5 and IL‐13 production from T cells protect against Pneumocystis jiroveci infection and drive pathology during allergic bronchopulmonary aspergillosis (ABPA). Right column: Host immunity to Blastomyces dermatitidis, Aspergillus fumigatus and Coccidioides species is mediated in part by T helper type 17 cells by IL‐17A and neutrophil‐dependent mechanisms.
Type 1 responses to pulmonary mycoses
Pneumocystis jiroveci (previously carinii) is an opportunistic pathogen and causes Pneumocystis pneumonia almost exclusively in immunocompromised hosts. Upon entry into the lung, Pneumocystis binds to lung epithelial cells (LECs).32, 33 The LECs play a significant role in the host response to Pneumocystis.30, 34, 35, 36 In vitro studies have demonstrated that LECs can directly respond to Pneumocystis by activating the nuclear factor‐κB (NF‐κB) pathway,36 and that chemokine production by LECs in response to the fungus is myeloid differentiation primary response 88 (MyD88) ‐dependent and IL‐1R‐dependent.34 Furthermore, the chemokine CCL2 is expressed by LECs in response to infection in vivo,35 and the ablation of NF‐κB selectively within LECs significantly impairs clearance of Pneumocystis from the lungs,30 emphasizing the essential role of LECs in mediating a protective response to this fungus.
Though there is robust evidence from both mouse37 and human38 studies that strongly suggest that CD4 T‐cell responses are required for clearance of Pneumocystis infection, the exact CD4 T‐cell phenotype(s) required for protection remains a subject of open debate. Numerous groups have suggested a role for IL‐17A and Th17 cells in host protection against Pneumocystis.30, 39, 40 Interleukin‐17A‐producing CD4 T cells are recruited to the lungs of infected animals,39 and neutralization of IL‐17A or the Th17‐promoting cytokine IL‐23 both significantly increase lung fungal burden at later time‐points.40 Furthermore, impaired fungal clearance was associated with significantly reduced IL‐17+ CD4 T‐cell numbers in the lungs of IKK∆LEC mice30 (which lack NF‐κB signalling specifically within the lung epithelium), suggesting a potential mechanism by which the lung epithelium marshals protective immunity to Pneumocystis.
Other studies have implicated Type 1 and Type 2 immunity in protecting against Pneumocystis infection. Some studies have correlated robust Type 2 responses and M2‐polarized macrophage responses with protection against Pneumocystis and fungal killing.23, 41 Conversely, artificial induction of IFN‐γ responses during Pneumocystis infection in the absence of CD4 T‐cell help restores control of infection,42 suggesting that Type 1 responses at least have the potential to mediate protection against Pneumocystis. Though the precise mechanisms of anti‐Pneumocystis immunity remain to be fully elucidated, collectively these studies emphasize the critical role of CD4 T‐cell‐mediated responses in immunity to this fungus.
Histoplasma capsulatum is a dimorphic, primary fungal pathogen capable of causing pulmonary histoplasmosis in immunocompetent hosts.11, 43 As is common among the dimorphic fungi, the spore is the infectious particle, and upon entry into the lung undergoes a phase‐transition to the yeast phase to mediate disease.11, 21 Elegant studies using green fluorescent protein‐expressing H. capsulatum strains reported that dendritic cells were the most likely phagocyte population to be associated with H. capsulatum at 1 day post infection,44 suggesting that dendritic cell–H. capsulatum interactions are a key early event in the host response to infection. An important subset includes CD103+ conventional dendritic cells, which are critical for TLR7/9‐dependent host defence against H. capsulatum.45 Innate cytokine production is also important in early control of infection, as the neutralization of granulocyte–macrophage colony‐stimulating factor (GM‐CSF) results in an order of magnitude higher fungal burden in the lungs by 1 week post infection.46 Ablation of GM‐CSF also impairs the early production of tumour necrosis factor‐α (TNF‐α) and IFN‐γ,46 two cytokines that are likewise critical for the control of H. capsulatum infection.47, 48 CCR2‐dependent inflammatory cell recruitment is also crucial for early control of infection, as CCR2−/− mice show a significant increase in lung fungal burden by 7 days post infection,49 which involves tilting T helper cell immunity away from Th1 responses.
Numerous studies have demonstrated that Th1 responses are protective against H. capsulatum infection.50, 51 Mice deficient in IFN‐γ signalling are exquisitely susceptible to experimental H. capsulatum infection, dying in little more than a week after experimental infection.47 In immunocompetent mice, the kinetics of IFN‐γ production from CD4 T cells correlates well with the clearance of the fungus from the lungs.52 Furthermore, recall of these anti‐H. capsulatum T cells is dependent upon TNF‐α, as the neutralization of TNF‐α early after re‐challenge of immune mice ablates protective immunity and leads to significant mortality.53 Hence, Th1 cells and their products are paramount in mediating effective immunity against H. capsulatum infection.
Although Type 1 immunity is critical for control and clearance of H. capsulatum infection, Type 2 responses are uniformly detrimental in this context. Overexpression of IL‐4 in transgenic mice was associated with increased lung fungal burden at day 7 post infection, but minimal alteration in the induction of IFN‐γ of TNF‐α responses,54 suggesting that enhanced Type 2 responses directly benefit pathogen growth in the absence of impaired Type 1 immunity. Loss of CCR2‐dependent signalling is a major determinant driving Type 2 immunity in response to H. capsulatum.49 Though CCR2−/− mice exhibit no defects in lung IFN‐γ production, IL‐4 levels are markedly increased and associated with significant mortality in a normally non‐lethal model of infection.49 Collectively, these studies indicate that Type 2 immunity is not only inefficient at clearing H. capsulatum, but that it actively promotes progression of the infection.
Cryptococcus neoformans is an encapsulated, ubiquitous fungus capable of causing lung disease and meningitis in immunocompromised patients.55 Initial interactions with resident phagocytes in the lung are crucial for control of C. neoformans, as depletion of CD11c+ myeloid cells results in significant and rapid mortality in otherwise non‐fatal C. neoformans lung infection.56 Likewise, ablation of TNF‐α signalling early in infection leads to increased skewing towards a Th2 response with associated impaired clearance of the fungus, probably due to defective maturation of dendritic cells.57 Collectively, these studies highlight how interactions early in the innate immune response can be critical for host survival and influence the later development and polarization of adaptive immune responses.
While C. neoformans infection rarely occurs in the immunocompetent host, infection is common among HIV/AIDS patients,55 underscoring a role for CD4 T cells in mediating immunity against this fungus. Numerous studies have shown a protective benefit of IFN‐γ production and Type 1 immunity during C. neoformans infection.58, 59 Chen and colleagues demonstrated that mice deficient in the IFN‐γ receptor showed impaired clearance of lung C. neoformans, increased dissemination, and increased mortality compared with wild‐type animals.58 This phenotype was largely attributed to decreased fungicidal activity in macrophages in the absence of IFN‐γ‐mediated activation.58 Additionally, monocyte recruitment is essential for the development of Th1 immunity, as CCR2−/− mice mount significantly weaker IFN‐γ responses.60 Furthermore, pulmonary infection with a transgenic strain of C. neoformans that produces mammalian IFN‐γ results in enhanced fungal clearance and survival of an otherwise fatal infection,59 highlighting the benefit of Type‐1 immunity in this infection.
In contrast to Type 1 immunity, Type 2 immune responses are associated with poor outcome during C. neoformans infection.22, 28, 61 A comparison of several mouse strains infected with the same strain of C. neoformans showed that while strong IFN‐γ responses were associated with fungal clearance, enhanced IL‐4 production and lung eosinophil recruitment was associated with compromised fungal clearance and increased dissemination to the spleen and brain.61 Additional studies have added further mechanistic insight into the role of Th2 responses in this infection. Mice deficient in the receptor for IL‐33, an important signal for Th2 cell function and differentiation, mount a weaker Type 2 response to C. neoformans and are consequently better able to control lung fungal colonization and survive infection.22 Ablation of IL‐4 signalling is also associated with reduced eosinophil recruitment and decreased lung fungal burden at later time‐points during C. neoformans pulmonary infection,28 further demonstrating the largely detrimental role of Type 2 responses in this context.
Type 17 responses to pulmonary mycoses
Blastomyces dermatitidis is a dimorphic fungus and the causative agent of blastomycosis, a potentially fatal pulmonary infection seen in immunocompetent individuals.62, 63, 64, 65 Innate immune cells play a critical role in regulating the pathogenesis of this infection. Neutrophil recruitment helps to limit the initial growth of the pathogen, as depletion of neutrophils yields an increase in the lung fungal burden by 2 days post infection.66 Recent studies by Hernandez‐Santos et al.67 have also demonstrated a key role for lung epithelial cells in orchestrating early responses to B. dermatitidis infection. Specifically, NF‐κB signalling within lung epithelial cells restrains fungal growth in part through the recruitment of IL‐17A‐ and GM‐CSF‐producing innate lymphoid cells, such as “natural” Th17 (nTh17) cells and γδ T cells, in the first 2 days of infection. This innate IL‐17A and GM‐CSF production in turn is required to activate recruited neutrophils and other myeloid cells and enhance their ability to kill fungal cells, highlighting the complex and multifaceted interactions of stromal, myeloid and lymphoid cells in antifungal immunity.
Several studies have interrogated host immunity in response to fungal spores,66, 68, 69 but most animal studies of immunity to dimorphic fungal infection to date have been performed with the yeast‐like form of the fungi. This is in large part due to technical difficulties in generating pure populations of spores and biosafety concerns of handling infectious spores when performing animal infections. Fungal spores probably represent the infectious particle in most naturally occurring infections. The different biochemical composition and metabolic activity of the spore compared to the yeast probably influences initial interactions with the immune system, including but not limited to ligation of PRRs and interactions with resident phagocytes. Although this discrepancy does not diminish the findings of studies using yeast, future studies delineating the impact of fungal particles on early pathogenesis and immunity will provide further illumination.
While experimental pulmonary infection with B. dermatitidis fails to elicit protective adaptive immune responses,70, 71 vaccine models using inoculation with live recombinant, attenuated B. dermatitidis have yielded key insights into the protective contribution of CD4 T cells.70, 72, 73, 74 Vaccine‐elicited CD4 T cells can produce both IFN‐γ and IL‐17A. However, the protective effects of these T cells are mediated more so by IL‐17A production.73, 74 Indeed, the ability of vaccination to protect against lethal challenge with wild‐type B. dermatitidis is significantly impaired in the absence of IL‐17A receptor signalling or the ablation of IL‐17A signal directly.73 Vaccine immunity is dependent upon robust antifungal response from the myeloid compartment, as both phox‐deficient72 and neutrophil‐depleted73 mice show impaired ability to clear B. dermatitidis from the lungs of vaccinated animals following an infectious challenge.
Interestingly, CD4 T cells are not the only T‐cell subset capable of mediating vaccine immunity against B. dermatitidis. In the absence of CD4 T‐cell help, CD8 T cells compensate and are sufficient to protect CD4‐deficient hosts from otherwise lethal infection.75 Strikingly, these CD8 T cells produce IL‐17A, and mediate immunity against lethal fungal infection by this IL‐17A production.76, 77, 78 Impairment of either IL‐17A signalling or neutrophil recruitment significantly ablates vaccine immunity in CD4‐deficient animals,77 underscoring the protective role of IL‐17A production by CD8 T cells. Furthermore, these IL‐17A‐producing CD8 T cells (Tc17 cells) display many of the phenotypic characteristics of Th17 cells, including increased expression of Retinoid‐related orphan receptor γ T (RORγT) and increased surface CCR6 expression.77 Hence, both CD4 and CD8 T cells are capable of promoting vaccine‐induced clearance of B. dermatitidis by the production of IL‐17A and the recruitment and activation of neutrophils.
Aspergillus fumigatus is a saprophytic mould found throughout the environment and the causative agent of pulmonary aspergillosis.79 Humans most commonly encounter A. fumigatus by inhaling conidia from the conidiating mould. Immunocompetent hosts rapidly clear conidia through the combined action of the mucociliary escalator, alveolar macrophages and neutrophil recruitment.79, 80 Neutrophil recruitment in particular is essential for the control of A. fumigatus, as the germination of A. fumigatus conidia is increased in lungs of mice deficient in neutrophils or neutrophil recruitment; for example, in MyD88‐deficient or caspase recruitment domain‐containing protein 9 (CARD‐9) ‐deficient animals81 or in CXCR2 deficiency, each of which contribute to neutrophil recruitment.80 The recruitment of neutrophils also is an inflammasome‐dependent process, with IL‐1α in particular playing a dominant role in driving initial neutrophil responses.82
Although immunocompetent individuals exposed to A. fumigatus conidia are usually able to clear these fungal particles without the engagement of adaptive immunity,79 patients with severe asthma or cystic fibrosis can become consistently colonized with A. fumigatus and develop allergic bronchopulmonary aspergillosis (ABPA).83 ABPA is characterized by eosinophilia, IgE antibody, and the development of Aspergillus‐specific Th2 cells.83 High levels of serum IgE, whose production from B cells is driven by Th2 cell‐derived IL‐4,84 are often found in patients suffering from ABPA.83 Experimental models using repeated exposures to A. fumigatus conidia have yielded insight into the mechanisms underpinning the development of anti‐A. fumigatus immune responses.9, 29, 31 In these models, mice repeatedly instilled with A. fumigatus conidia develop many of the hallmarks of allergic airway inflammation and ABPA, including robust eosinophil recruitment, arterial remodelling, and collagen deposition around airways.29, 31 Interestingly, this inflammatory response is associated with the development of Th1 and Th17 responses in addition to Th2 responses.29 Interleukin‐17A is required for full eosinophil recruitment at the peak of inflammation,9 underscoring the potential for non‐Type 2 cytokines in driving ‘allergic’ responses following fungal exposure. Recent work has shown that signalling via IL‐17RA and IL‐17RC may drive divergent allergic outcomes,85 with the IL‐17F–IL‐17RC axis favouring respiratory allergy in the proximal airways. Collectively, these studies demonstrate the potentially varied functions of T‐cell responses to fungal challenge, and how repeated exposure to fungi and their products can drive development of allergic airway disease.
Coccidioides posadasii and Coccidioides immitis are two closely related species of Coccidioides, a dimorphic primary fungal pathogen endemic to the American southwest and California, respectively, and the causative agents of Valley Fever.86 Infection is initiated when Coccidioides arthroconidia are inhaled into the lung, where these infectious particles undergo development and eventually grow into large spherules containing numerous endospores. When the spherule bursts, the newly freed endospores form new spherules, and the infection continues.86, 87, 88 The contribution of innate immunity to protection against Coccidioides remains incompletely understood. In experimental models of Coccidioides infection, the depletion of neutrophils does not result in increased lung or spleen fungal burden,89 suggesting that neutrophils may be dispensable in that model. Additionally, although the absence of functional TLR4 is not associated with any increase in lung burden, increased fungal dissemination to the spleen was reported.90
Experimental vaccination models have begun to elucidate the protective role of T‐cell responses against Coccidioides infection.89, 91 Subcutaneous vaccination with spores from an attenuated strain of Coccidioides engenders resistance against otherwise lethal pulmonary challenge with the wild‐type fungus, in association with robust Type 1, Type 2 and Type 17 responses.91 Th17 cells appear to be required for protective immunity following vaccination however, as IL‐17ra knockout mice exhibit a significant defect in survival during rechallenge after vaccination.91 Furthermore, MyD88 and CARD‐9 are required both for the development of vaccine‐induced resistance to infection and the development of robust Th17 responses in the lungs.89 Additionally, the depletion of neutrophils significantly impairs fungal clearance in vaccinated animals,89 offering further evidence that antifungal activity following vaccination is driven by Th17 cell activity.
Type 2 responses and pulmonary mycoses
Though Type 2 immunity is largely considered dispensable at best and detrimental at worst in response to pulmonary fungal challenge, numerous groups have reported beneficial facets of Type 2 immunity to a variety of fungal pathogens. Notably, although the absence of IL‐4 signalling is associated with improved control of C. neoformans lung burden at later time‐points post infection, IL‐4RaKO mice show increased pathogen burden early in infection,28 suggesting that IL‐4‐mediated responses are protective at this early time‐point. Additionally, whereas eosinophilia is associated with allergic airway inflammation following repeated exposure to Aspergillus conidia,9, 29, 31 defects in eosinophil activity are associated with impaired ability to clear Aspergillus conidia following installation into the lungs.92 Furthermore, eosinophils exhibit contact‐independent killing of Aspergillus conidia in vitro,92 suggesting that recruited eosinophils are capable of protecting the host by killing Aspergillus conidia after exposure.
Applications for antifungal T cells
One clinical application for antifungal T cells is the development of T‐cell‐based vaccines, especially for use in populations living in areas where fungal infections are endemic. Experimental models have identified candidate vaccination strategies against the endemic mycoses B. dermatitidis, 70 H. capsulatum,93 and Coccidioides posadasii.94 Wuthrich et al.24 recently demonstrated that T cells specific to an epitope found in fungal calnexin can respond to and expand following stimulation with the fungal pathogens mentioned above as well as A. fumigatus conidia. Vaccination with glucan particles loaded with calnexin peptide was capable of eliciting protective immunity against both B. dermatitidis and Coccidioides posadasii experimental pulmonary infection,24 demonstrating the potential for vaccination strategies promoting calnexin‐specific T‐cell responses to protect against multiple endemic fungal infections. Various strategies, including recombinant proteins in glucan particles, engineered attenuated strains and alkaline extracts have shown promise in vaccination against experimental murine Cryptococcus infections.95, 96, 97
An additional clinical application where one might leverage antifungal T‐cell responses is the development of immunotherapy treatments, especially in populations at high risk for fungal infections. Invasive aspergillosis is a severe fungal infection in immunocompromised individuals, especially those undergoing corticosteroid treatment, and mortality can be as high as 90%.79, 98 A therapeutic approach with promise is the transplantation of in vitro differentiated antifungal T cells to at‐risk patient populations.99 Preclinical models have demonstrated the efficacy of antifungal T‐cell responses in protecting mice from otherwise lethal doses of A. fumigatus.99, 100, 101 Recent studies by Kumaresan and colleagues have demonstrated the potential of CD8 T cells bioengineered to respond to β‐glucan via Dectin‐1 to impair A. fumigatus growth.102 Furthermore, human trials using the transplantation of in vitro stimulated donor Aspergillus‐specific T cells as a therapeutic intervention in response to evidence of invasive aspergillosis demonstrated a significant increase in survival compared with control patients,99, 103 so demonstrating the potential for this therapy in improving patient outcomes in an otherwise often intractable disease.
Similar immunotherapeutic strategies, where autologous antigen‐specific T cells are expanded in vitro and transferred to patients, have proven effective at protecting immunocompromised patients against cytomegalovirus (CMV) infection.104, 105 One difference between the two infections, and a technological challenge that must be met to develop a viable therapeutic, is the mechanisms by which the immunotherapy would kill the infectious agent. Anti‐CMV immunotherapeutic approaches use CMV‐specific cytotoxic T lymphocytes,104, 105 which can directly kill infected cells. In contrast, CD4 T helper cells will have to engage arms of innate immunity, such as neutrophils, monocytes, or macrophages,24 which may be absent or functionally impaired in immunosuppressed individuals, to protect against fungal infection. Hence, antifungal immunotherapeutic strategies may also involve the augmentation of effector myeloid cell responses in conjunction with antifungal T‐cell transfers.
Concluding remarks
The development of adaptive immunity and the engagement of CD4 T‐cell help is a key determinant of the outcome of numerous pulmonary fungal infections. In the case of H. capsulatum or C. neoformans infection, naturally developing Th1 responses drive clearance of fungal infections. In other cases, such as the Th2 responses that develop following repeated exposure to A. fumigatus conidia, CD4 T‐cell responses are dispensable to fungal killing and ultimately contribute to immune pathology. Furthermore in other contexts, including B. dermatitidis or Coccidioides infection, a failure of the development of robust CD4 T‐cell responses is associated with poor outcomes following infection. Hence, the phenotype and strength of antifungal T‐cell responses is a major factor in immunity and pathology during pulmonary fungal infections.
These insights are improving our understanding of the basic biology of fungal infections, and also informing the development of next‐generation antifungal therapies. As mentioned above, the transfer of antifungal CD4 T cells shows great promise as a therapy for difficult to treat fungal infections in immunocompromised hosts. Experimental vaccines against B. dermatitidis or Coccidioides are capable of eliciting protective immunity in preclinical models of fatal fungal infection. Future investigations of antifungal CD4 T‐cell responses should yield novel insights into the determinants of protective versus pathological host responses.
Disclosures
The authors have no competing financial, professional or personal interests that might have influenced the performance or presentation of the work described in the manuscript.
Acknowledgements
The authors thank members of the Klein Laboratory and Hull laboratory for helpful discussions. AJM received support from T32AI007635 (NIH‐NIAID). BSK is supported by NIH R01 AI035681.
References
- 1. Wiesner DL, Klein BS. Lung epithelium: barrier immunity to inhaled fungi and driver of fungal‐associated allergic asthma. Curr Opin Microbiol 2017; 40:8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Frohlich‐Nowoisky J, Pickersgill DA, Despres VR, Poschl U. High diversity of fungi in air particulate matter. Proc Natl Acad Sci USA 2009; 106:12814–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue‐specific context. Nat Rev Immunol 2014; 14:81–93. [DOI] [PubMed] [Google Scholar]
- 4. Plato A, Willment JA, Brown GD. C‐type lectin‐like receptors of the dectin‐1 cluster: ligands and signaling pathways. Int Rev Immunol 2013; 32:134–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Inoue M, Shinohara ML. Clustering of pattern recognition receptors for fungal detection. PLoS Pathog 2014; 10:e1003873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kerrigan AM, Brown GD. C‐type lectins and phagocytosis. Immunobiology 2009; 214:562–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang H, Lee TJ, Fites SJ, Merkhofer R, Zarnowski R, Brandhorst T et al Ligation of Dectin‐2 with a novel microbial ligand promotes adjuvant activity for vaccination. PLoS Pathog 2017; 13:e1006568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stappers MHT, Clark AE, Aimanianda V, Bidula S, Reid DM, Asamaphan P et al Recognition of DHN‐melanin by a C‐type lectin receptor is required for immunity to Aspergillus . Nature 2018; 555:382–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Murdock BJ, Falkowski NR, Shreiner AB, Sadighi Akha AA, McDonald RA, White ES et al Interleukin‐17 drives pulmonary eosinophilia following repeated exposure to Aspergillus fumigatus conidia. Infect Immun 2012; 80:1424–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wiesner DL, Specht CA, Lee CK, Smith KD, Mukaremera L, Lee ST et al Chitin recognition via chitotriosidase promotes pathologic type‐2 helper T cell responses to cryptococcal infection. PLoS Pathog 2015; 11:e1004701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Horwath MC, Fecher RA, Deepe GS Jr. Histoplasma capsulatum, lung infection and immunity. Future Microbiol 2015; 10:967–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mircescu MM, Lipuma L, van Rooijen N, Pamer EG, Hohl TM. Essential role for neutrophils but not alveolar macrophages at early time points following Aspergillus fumigatus infection. J Infect Dis 2009; 200:647–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Espinosa V, Jhingran A, Dutta O, Kasahara S, Donnelly R, Du P et al Inflammatory monocytes orchestrate innate antifungal immunity in the lung. PLoS Pathog 2014; 10:e1003940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McDermott AJ, Tumey TA, Huang M, Hull CM, Klein BS. Inhaled Cryptococcus neoformans elicits allergic airway inflammation independent of Nuclear Factor κB signalling in lung epithelial cells. Immunology 2018; 153:513–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Amarsaikhan N, O'Dea EM, Tsoggerel A, Templeton SP. Lung eosinophil recruitment in response to Aspergillus fumigatus is correlated with fungal cell wall composition and requires γδ T cells. Microbes Infect 2017; 19:422–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Obst R. The timing of T cell priming and cycling. Front Immunol 2015; 6:563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Plunkett CH, Nagler CR. The influence of the microbiome on allergic sensitization to food. J Immunol 2017; 198:581–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mempel TR, Henrickson SE, Von Andrian UH. T‐cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 2004; 427:154–9. [DOI] [PubMed] [Google Scholar]
- 19. Chen L, Flies DB. Molecular mechanisms of T cell co‐stimulation and co‐inhibition. Nat Rev Immunol 2013; 13:227–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luckheeram RV, Zhou R, Verma AD, Xia B. CD4+ T cells: differentiation and functions. Clin Dev Immunol 2012; 2012:925135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Edwards JA, Rappleye CA. Histoplasma mechanisms of pathogenesis–one portfolio doesn't fit all. FEMS Microbiol Lett 2011; 324:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Flaczyk A, Duerr CU, Shourian M, Lafferty EI, Fritz JH, Qureshi ST. IL‐33 signaling regulates innate and adaptive immunity to Cryptococcus neoformans . J Immunol 2013; 191:2503–13. [DOI] [PubMed] [Google Scholar]
- 23. Myers RC, Dunaway CW, Nelson MP, Trevor JL, Morris A, Steele C. STAT4‐dependent and ‐independent Th2 responses correlate with protective immunity against lung infection with Pneumocystis murina . J Immunol 2013; 190:6287–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wuthrich M, Brandhorst TT, Sullivan TD, Filutowicz H, Sterkel A, Stewart D et al Calnexin induces expansion of antigen‐specific CD4+ T cells that confer immunity to fungal ascomycetes via conserved epitopes. Cell Host Microbe 2015; 17:452–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ziegler SF. Division of labour by CD4+ T helper cells. Nat Rev Immunol 2016; 16:403. [DOI] [PubMed] [Google Scholar]
- 26. Romagnani S. T‐cell subsets (Th1 versus Th2). Ann Allergy Asthma Immunol 2000; 85:9–18; quiz, 21. [DOI] [PubMed] [Google Scholar]
- 27. Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev 2008; 223:87–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grahnert A, Richter T, Piehler D, Eschke M, Schulze B, Muller U et al IL‐4 receptor‐α‐dependent control of Cryptococcus neoformans in the early phase of pulmonary infection. PLoS ONE 2014; 9:e87341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Murdock BJ, Shreiner AB, McDonald RA, Osterholzer JJ, White ES, Toews GB et al Coevolution of TH1, TH2, and TH17 responses during repeated pulmonary exposure to Aspergillus fumigatus conidia. Infect Immun 2011; 79:125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Perez‐Nazario N, Rangel‐Moreno J, O'Reilly MA, Pasparakis M, Gigliotti F, Wright TW. Selective ablation of lung epithelial IKK2 impairs pulmonary Th17 responses and delays the clearance of Pneumocystis . J Immunol 2013; 191:4720–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shreiner AB, Murdock BJ, Sadighi Akha AA, Falkowski NR, Christensen PJ, White ES et al Repeated exposure to Aspergillus fumigatus conidia results in CD4+ T cell‐dependent and ‐independent pulmonary arterial remodeling in a mixed Th1/Th2/Th17 microenvironment that requires interleukin‐4 (IL‐4) and IL‐10. Infect Immun 2012; 80:388–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pottratz ST. Pneumocystis carinii interactions with respiratory epithelium. Semin Respir Infect 1998; 13:323–9. [PubMed] [Google Scholar]
- 33. Limper AH. Parasitic adherence and host responses in the development of Pneumocystis carinii pneumonia. Semin Respir Infect 1991; 6:19–26. [PubMed] [Google Scholar]
- 34. Bello‐Irizarry SN, Wang J, Olsen K, Gigliotti F, Wright TW. The alveolar epithelial cell chemokine response to pneumocystis requires adaptor molecule MyD88 and interleukin‐1 receptor but not toll‐like receptor 2 or 4. Infect Immun 2012; 80:3912–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang J, Gigliotti F, Bhagwat SP, Maggirwar SB, Wright TW. Pneumocystis stimulates MCP‐1 production by alveolar epithelial cells through a JNK‐dependent mechanism. Am J Physiol Lung Cell Mol Physiol 2007; 292:L1495–505. [DOI] [PubMed] [Google Scholar]
- 36. Wang J, Gigliotti F, Maggirwar S, Johnston C, Finkelstein JN, Wright TW. Pneumocystis carinii activates the NF‐κB signaling pathway in alveolar epithelial cells. Infect Immun 2005; 73:2766–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Beck JM, Newbury RL, Palmer BE, Warnock ML, Byrd PK, Kaltreider HB. Role of CD8+ lymphocytes in host defense against Pneumocystis carinii in mice. J Lab Clin Med 1996; 128:477–87. [DOI] [PubMed] [Google Scholar]
- 38. Lidman C, Berglund O, Tynell E, Lindback S. CD4+ cells and CD4+ percent as risk markers for Pneumocystis carinii pneumonia (PCP): implications for primary PCP prophylaxis. Scand J Infect Dis 1992; 24:157–60. [DOI] [PubMed] [Google Scholar]
- 39. Ripamonti C, Bishop LR, Kovacs JA. Pulmonary interleukin‐17‐positive lymphocytes increase during Pneumocystis murina infection but are not required for clearance of Pneumocystis. Infect Immun 2017; 85:e00434‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rudner XL, Happel KI, Young EA, Shellito JE. Interleukin‐23 (IL‐23)‐IL‐17 cytokine axis in murine Pneumocystis carinii infection. Infect Immun 2007; 75:3055–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nandakumar V, Hebrink D, Jenson P, Kottom T, Limper AH. Differential macrophage polarization from pneumocystis in immunocompetent and immunosuppressed hosts: potential adjunctive therapy during pneumonia. Infect Immun 2017; 85:e00939‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kolls JK, Habetz S, Shean MK, Vazquez C, Brown JA, Lei D et al IFN‐γ and CD8+ T cells restore host defenses against Pneumocystis carinii in mice depleted of CD4+ T cells. J Immunol 1999; 162:2890–4. [PubMed] [Google Scholar]
- 43. Kauffman CA. Histoplasmosis: a clinical and laboratory update. Clin Microbiol Rev 2007; 20:115–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Deepe GS Jr, Gibbons RS, Smulian AG. Histoplasma capsulatum manifests preferential invasion of phagocytic subpopulations in murine lungs. J Leukoc Biol 2008; 84:669–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Van Prooyen N, Henderson CA, Hocking Murray D, Sil A. CD103+ conventional dendritic cells are critical for TLR7/9‐dependent host defense against Histoplasma capsulatum, an endemic fungal pathogen of humans. PLoS Pathog 2016; 12:e1005749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Deepe GS Jr, Gibbons R, Woodward E. Neutralization of endogenous granulocyte‐macrophage colony‐stimulating factor subverts the protective immune response to Histoplasma capsulatum . J Immunol 1999; 163:4985–93. [PubMed] [Google Scholar]
- 47. Allendoerfer R, Deepe GS Jr. Intrapulmonary response to Histoplasma capsulatum in γ interferon knockout mice. Infect Immun 1997; 65:2564–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Allendoerfer R, Deepe GS Jr. Blockade of endogenous TNF‐α exacerbates primary and secondary pulmonary histoplasmosis by differential mechanisms. J Immunol 1998; 160:6072–82. [PubMed] [Google Scholar]
- 49. Szymczak WA, Deepe GS Jr. The CCL7‐CCL2‐CCR2 axis regulates IL‐4 production in lungs and fungal immunity. J Immunol 2009; 183:1964–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Allendorfer R, Brunner GD, Deepe GS Jr. Complex requirements for nascent and memory immunity in pulmonary histoplasmosis. J Immunol 1999; 162:7389–96. [PubMed] [Google Scholar]
- 51. Kroetz DN, Deepe GS. The role of cytokines and chemokines in Histoplasma capsulatum infection. Cytokine 2012; 58:112–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lin JS, Wu‐Hsieh BA. Functional T cells in primary immune response to histoplasmosis. Int Immunol 2004; 16:1663–73. [DOI] [PubMed] [Google Scholar]
- 53. Deepe GS Jr, Gibbons RS. T cells require tumor necrosis factor‐α to provide protective immunity in mice infected with Histoplasma capsulatum . J Infect Dis 2006; 193:322–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gildea LA, Gibbons R, Finkelman FD, Deepe GS Jr. Overexpression of interleukin‐4 in lungs of mice impairs elimination of Histoplasma capsulatum . Infect Immun 2003; 71:3787–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kozubowski L, Heitman J. Profiling a killer, the development of Cryptococcus neoformans . FEMS Microbiol Rev 2012; 36:78–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Osterholzer JJ, Milam JE, Chen GH, Toews GB, Huffnagle GB, Olszewski MA. Role of dendritic cells and alveolar macrophages in regulating early host defense against pulmonary infection with Cryptococcus neoformans . Infect Immun 2009; 77:3749–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Herring AC, Falkowski NR, Chen GH, McDonald RA, Toews GB, Huffnagle GB. Transient neutralization of tumor necrosis factor α can produce a chronic fungal infection in an immunocompetent host: potential role of immature dendritic cells. Infect Immun 2005; 73:39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chen GH, McDonald RA, Wells JC, Huffnagle GB, Lukacs NW, Toews GB. The γ interferon receptor is required for the protective pulmonary inflammatory response to Cryptococcus neoformans . Infect Immun 2005; 73:1788–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wormley FL Jr, Perfect JR, Steele C, Cox GM. Protection against cryptococcosis by using a murine γ interferon‐producing Cryptococcus neoformans strain. Infect Immun 2007; 75:1453–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Traynor TR, Kuziel WA, Toews GB, Huffnagle GB. CCR2 expression determines T1 versus T2 polarization during pulmonary Cryptococcus neoformans infection. J Immunol 2000; 164:2021–7. [DOI] [PubMed] [Google Scholar]
- 61. Chen GH, McNamara DA, Hernandez Y, Huffnagle GB, Toews GB, Olszewski MA. Inheritance of immune polarization patterns is linked to resistance versus susceptibility to Cryptococcus neoformans in a mouse model. Infect Immun 2008; 76:2379–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. McDonough ES. Blastomycosis‐epidemiology and biology of its etiologic agent Ajellomyces dermatitidis . Mycopathol Mycol Appl 1970; 41:195–201. [DOI] [PubMed] [Google Scholar]
- 63. McBride JA, Gauthier GM, Klein BS. Clinical manifestations and treatment of Blastomycosis. Clin Chest Med 2017; 38:435–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Klein BS, Vergeront JM, Weeks RJ, Kumar UN, Mathai G, Varkey B et al Isolation of Blastomyces dermatitidis in soil associated with a large outbreak of blastomycosis in Wisconsin. N Engl J Med 1986; 314:529–34. [DOI] [PubMed] [Google Scholar]
- 65. Lemos LB, Baliga M, Guo M. Blastomycosis: the great pretender can also be an opportunist. Initial clinical diagnosis and underlying diseases in 123 patients. Ann Diagn Pathol 2002; 6:194–203. [DOI] [PubMed] [Google Scholar]
- 66. Sterkel AK, Mettelman R, Wuthrich M, Klein BS. The unappreciated intracellular lifestyle of Blastomyces dermatitidis . J Immunol 2015; 194:1796–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hernandez‐Santos N, Wiesner DL, Fites JS, McDermott AJ, Warner T, Wuthrich M et al Lung epithelial cells coordinate innate lymphocytes and immunity against pulmonary fungal infection. Cell Host Microbe 2018; 23:511–22.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Inglis DO, Berkes CA, Hocking Murray DR, Sil A. Conidia but not yeast cells of the fungal pathogen Histoplasma capsulatum trigger a type I interferon innate immune response in murine macrophages. Infect Immun 2010; 78:3871–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Narra HP, Shubitz LF, Mandel MA, Trinh HT, Griffin K, Buntzman AS et al A Coccidioides posadasii CPS1 deletion mutant is avirulent and protects mice from lethal infection. Infect Immun 2016; 84:3007–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wuthrich M, Filutowicz HI, Klein BS. Mutation of the WI‐1 gene yields an attenuated Blastomyces dermatitidis strain that induces host resistance. J Clin Invest 2000; 106:1381–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Brandhorst TT, Wuthrich M, Warner T, Klein B. Targeted gene disruption reveals an adhesin indispensable for pathogenicity of Blastomyces dermatitidis . J Exp Med 1999; 189:1207–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wuthrich M, Filutowicz HI, Warner T, Klein BS. Requisite elements in vaccine immunity to Blastomyces dermatitidis: plasticity uncovers vaccine potential in immune‐deficient hosts. J Immunol 2002; 169:6969–76. [DOI] [PubMed] [Google Scholar]
- 73. Wuthrich M, Gern B, Hung CY, Ersland K, Rocco N, Pick‐Jacobs J et al Vaccine‐induced protection against 3 systemic mycoses endemic to North America requires Th17 cells in mice. J Clin Invest 2011; 121:554–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wuthrich M, LeBert V, Galles K, Hu‐Li J, Ben‐Sasson SZ, Paul WE et al Interleukin 1 enhances vaccine‐induced antifungal T‐helper 17 cells and resistance against Blastomyces dermatitidis infection. J Infect Dis 2013; 208:1175–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wuthrich M, Filutowicz HI, Warner T, Deepe GS Jr, Klein BS. Vaccine immunity to pathogenic fungi overcomes the requirement for CD4 help in exogenous antigen presentation to CD8+ T cells: implications for vaccine development in immune‐deficient hosts. J Exp Med 2003; 197:1405–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Nanjappa SG, McDermott AJ, Fites JS, Galles K, Wuthrich M, Deepe GS Jr et al Antifungal Tc17 cells are durable and stable, persisting as long‐lasting vaccine memory without plasticity towards IFNγ cells. PLoS Pathog 2017; 13:e1006356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nanjappa SG, Heninger E, Wuthrich M, Gasper DJ, Klein BS. Tc17 cells mediate vaccine immunity against lethal fungal pneumonia in immune deficient hosts lacking CD4+ T cells. PLoS Pathog 2012; 8:e1002771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nanjappa SG, Hernandez‐Santos N, Galles K, Wuthrich M, Suresh M, Klein BS. Intrinsic MyD88‐Akt1‐mTOR signaling coordinates disparate Tc17 and Tc1 responses during vaccine immunity against fungal pneumonia. PLoS Pathog 2015; 11:e1005161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Dagenais TR, Keller NP. Pathogenesis of Aspergillus fumigatus in invasive aspergillosis. Clin Microbiol Rev 2009; 22:447–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bonnett CR, Cornish EJ, Harmsen AG, Burritt JB. Early neutrophil recruitment and aggregation in the murine lung inhibit germination of Aspergillus fumigatus conidia. Infect Immun 2006; 74:6528–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Jhingran A, Kasahara S, Shepardson KM, Junecko BA, Heung LJ, Kumasaka DK et al Compartment‐specific and sequential role of MyD88 and CARD9 in chemokine induction and innate defense during respiratory fungal infection. PLoS Pathog 2015; 11:e1004589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Caffrey AK, Lehmann MM, Zickovich JM, Espinosa V, Shepardson KM, Watschke CP et al IL‐1α signaling is critical for leukocyte recruitment after pulmonary Aspergillus fumigatus challenge. PLoS Pathog 2015; 11:e1004625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Knutsen AP, Slavin RG. Allergic bronchopulmonary aspergillosis in asthma and cystic fibrosis. Clin Dev Immunol 2011; 2011:843763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Walker JA, McKenzie ANJ. TH2 cell development and function. Nat Rev Immunol 2018; 18:121–33. [DOI] [PubMed] [Google Scholar]
- 85. De Luca A, Pariano M, Cellini B, Costantini C, Villella VR, Jose SS et al The IL‐17F/IL‐17RC axis promotes respiratory allergy in the proximal airways. Cell Rep 2017; 20:1667–80. [DOI] [PubMed] [Google Scholar]
- 86. Castro‐Lopez N, Hung CY. Immune response to Coccidioidomycosis and the development of a vaccine. Microorganisms 2017; 5:E13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hector RF, Laniado‐Laborin R. Coccidioidomycosis – a fungal disease of the Americas. PLoS Med 2005; 2:e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Erwig LP, Gow NA. Interactions of fungal pathogens with phagocytes. Nat Rev Microbiol 2016; 14:163–76. [DOI] [PubMed] [Google Scholar]
- 89. Hung CY, Jimenez‐Alzate Mdel P, Gonzalez A, Wuthrich M, Klein BS, Cole GT. Interleukin‐1 receptor but not Toll‐like receptor 2 is essential for MyD88‐dependent Th17 immunity to Coccidioides infection. Infect Immun 2014; 82:2106–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Awasthi S. Susceptibility of TLR4‐defective C3H/HeJ mice to Coccidioides posadasii infection. Med Mycol 2010; 48:470–5. [DOI] [PubMed] [Google Scholar]
- 91. Hung CY, Gonzalez A, Wuthrich M, Klein BS, Cole GT. Vaccine immunity to coccidioidomycosis occurs by early activation of three signal pathways of T helper cell response (Th1, Th2, and Th17). Infect Immun 2011; 79:4511–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lilly LM, Scopel M, Nelson MP, Burg AR, Dunaway CW, Steele C. Eosinophil deficiency compromises lung defense against Aspergillus fumigatus . Infect Immun 2014; 82:1315–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Gomez FJ, Allendoerfer R, Deepe GS Jr. Vaccination with recombinant heat shock protein 60 from Histoplasma capsulatum protects mice against pulmonary histoplasmosis. Infect Immun 1995; 63:2587–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Xue J, Chen X, Selby D, Hung CY, Yu JJ, Cole GT. A genetically engineered live attenuated vaccine of Coccidioides posadasii protects BALB/c mice against coccidioidomycosis. Infect Immun 2009; 77:3196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Specht CA, Lee CK, Huang H, Hester MM, Liu J, Luckie BA et al Vaccination with recombinant Cryptococcus proteins in glucan particles protects mice against cryptococcosis in a manner dependent upon mouse strain and cryptococcal species. MBio 2017; 8:e01872‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Specht CA, Lee CK, Huang H, Tipper DJ, Shen ZT, Lodge JK et al Protection against experimental cryptococcosis following vaccination with glucan particles containing Cryptococcus alkaline extracts. MBio 2015; 6:e01905–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Upadhya R, Lam WC, Maybruck B, Specht CA, Levitz SM, Lodge JK. Induction of protective immunity to Cryptococcal infection in mice by a heat‐killed, chitosan‐deficient strain of Cryptococcus neoformans . MBio 2016; 7:e00547‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Lin SJ, Schranz J, Teutsch SM. Aspergillosis case‐fatality rate: systematic review of the literature. Clin Infect Dis 2001; 32:358–66. [DOI] [PubMed] [Google Scholar]
- 99. Deo SS, Gottlieb DJ. Adoptive T‐cell therapy for fungal infections in haematology patients. Clin Transl Immunology 2015; 4:e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Cenci E, Mencacci A, Bacci A, Bistoni F, Kurup VP, Romani L. T cell vaccination in mice with invasive pulmonary aspergillosis. J Immunol 2000; 165:381–8. [DOI] [PubMed] [Google Scholar]
- 101. Bozza S, Perruccio K, Montagnoli C, Gaziano R, Bellocchio S, Burchielli E et al A dendritic cell vaccine against invasive aspergillosis in allogeneic hematopoietic transplantation. Blood 2003; 102:3807–14. [DOI] [PubMed] [Google Scholar]
- 102. Kumaresan PR, Manuri PR, Albert ND, Maiti S, Singh H, Mi T et al Bioengineering T cells to target carbohydrate to treat opportunistic fungal infection. Proc Natl Acad Sci USA 2014; 111:10660–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Perruccio K, Tosti A, Burchielli E, Topini F, Ruggeri L, Carotti A et al Transferring functional immune responses to pathogens after haploidentical hematopoietic transplantation. Blood 2005; 106:4397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Blyth E, Clancy L, Simms R, Ma CK, Burgess J, Deo S et al Donor‐derived CMV‐specific T cells reduce the requirement for CMV‐directed pharmacotherapy after allogeneic stem cell transplantation. Blood 2013; 121:3745–58. [DOI] [PubMed] [Google Scholar]
- 105. Peggs KS, Thomson K, Samuel E, Dyer G, Armoogum J, Chakraverty R et al Directly selected cytomegalovirus‐reactive donor T cells confer rapid and safe systemic reconstitution of virus‐specific immunity following stem cell transplantation. Clin Infect Dis 2011; 52:49–57. [DOI] [PubMed] [Google Scholar]