

Summary
Type I interferons (IFNs) are a family of cytokines with a wide range of biological activities including anti‐viral and immune‐regulatory functions. Here, we focus on the protozoan parasitic disease malaria, and examine the effects of type I IFN‐signalling during Plasmodium infection of humans and experimental mice. Since the 1960s, there have been many studies in this area, but a simple explanation for the role of type I IFN has not emerged. Although epidemiological data are consistent with roles for type I IFN in influencing malaria disease severity, functional proof of this remains sparse in humans. Several different rodent‐infective Plasmodium species have been employed in in vivo studies of parasite‐sensing, experimental cerebral malaria, lethal malaria, liver‐stage infection, and adaptive T‐cell and B‐cell immunity. A range of different outcomes in these studies suggests a delicately balanced, multi‐faceted and highly complex role for type I IFN‐signalling in malaria. This is perhaps unsurprising given the multiple parasite‐sensing pathways that can trigger type I IFN production, the multiple isoforms of IFN‐α/β that can be produced by both immune and non‐immune cells, the differential effects of acute versus chronic type I IFN production, the role of low level ‘tonic’ type I IFN‐signalling, and that signalling can occur via homodimeric IFNAR1 or heterodimeric IFNAR1/2 receptors. Nevertheless, the data indicate that type I IFN‐signalling controls parasite numbers during liver‐stage infection, and depending on host–parasite genetics, can be either detrimental or beneficial to the host during blood‐stage infection. Furthermore, type I IFN can promote cytotoxic T lymphocyte immune pathology and hinder CD4+ T helper cell‐dependent immunity during blood‐stage infection. Hence, type I IFN‐signalling plays highly context‐dependent roles in malaria, which can be beneficial or detrimental to the host.
Keywords: cytokines, interferons, malaria, mouse models, parasitology
Introduction
Malaria remains an important public health problem, with 216 million cases and 445 000 deaths in 2016.1 The most life‐threatening forms of the disease are caused by Plasmodium falciparum, a protozoan parasite transmitted to human hosts by infected mosquitoes. During a blood meal, mosquitoes inject infectious sporozoites, some of which travel to the liver, infecting hepatocytes and initiating the clinically silent liver‐stage of infection. After a few days of rapid asexual replication, infected hepatocytes release thousands of parasites into the bloodstream, which rapidly invade red blood cells, initiating the cyclic and symptomatic blood‐stage of infection.2 Although anti‐malarial drugs and vector control approaches have contributed to a substantial global reduction in disease burden, eradication of malaria remains unlikely without the development of an efficacious vaccine or other interventions that improve anti‐parasitic immunity. A better understanding of immune responses that occur during Plasmodium infection may facilitate these goals. In this review we focus on the effects of type I interferon (IFN) signalling in malaria. Type I IFNs are a cytokine family that includes 13 different isoforms of IFN‐α in humans (14 in mice) and a single IFN‐β, as well as others, for example, IFN‐ε, which appears to function predominantly in the female reproductive tract.3 Although type I IFNs can play important roles in immunity to viral4, 5 and bacterial6, 7, 8 infections, excessive signalling may also contribute to autoimmune diseases such as systemic lupus erythematosus9 and rheumatoid arthritis.10 In addition, type I IFN‐signalling can impede immunity to pathogens such as Mycobacterium tuberculosis 11 and Leishmania.12 The effects of type I IFN‐signalling on host immune responses against viruses and bacteria have recently been reviewed in refs 13, 14, 15, 16.
Severe malaria remains a leading cause of death in children and pregnant mothers living in countries where malaria is endemic. In most patients, severe malaria manifests as cerebral malaria, severe anaemia or acidosis.2 Excessive pro‐inflammatory cytokine‐mediated pathways are thought to contribute to the immunopathogenesis of severe malaria.2, 17 Indeed, type I IFN‐associated responses have been reported in malaria patients,18, 19, 20 with single nucleotide polymorphisms in the Ifnar1 gene associating with greater risk of severe malaria.18, 19, 21 These single nucleotide polymorphisms exist in intronic or regulatory regions, and suggest that increased expression levels of the receptor might promote severe disease in malaria, whereas lower expression levels might be protective. Experimental evidence to support this hypothesis has been difficult to generate in humans, but has been the subject of study in mouse models. Here, we examine the effects of type I IFN‐signalling during Plasmodium infection, predominantly, though not exclusively, in experimental mice.
Type I IFN production during blood‐stage malaria
Innate immune sensing of Plasmodium parasites has been an important area of study over the past 15 years.22, 23, 24, 25 Researchers have employed a variety of in vitro and in vivo techniques to explore whether Plasmodium‐derived molecules, such as glycosylphosphatidylinositol anchors, CpG DNA motifs, AT‐rich DNA, haemozoin or Plasmodium RNA species might constitute pathogen‐associated molecular patterns that are recognized by cell surface, endosomal or cytoplasmic pattern recognition receptors.22, 23, 25, 26, 27 Such pattern recognition receptors include Toll‐like receptors (TLRs) and NOD‐like receptors, as well as cytosolic nucleic acid sensors such as retinoic acid inducible gene 1 protein, melanoma differentiation‐associated protein 5 (MDA5) and cyclic GMP‐AMP synthase (cGAS),28 which can signal via mitochondrial antiviral signalling protein (MAVS), and the stimulator of interferon genes (STING/MYPS, encoded by Tmem173).24, 29 The expression of TLR2 and TLR4 on macrophages was reported to detect Plasmodium‐associated glycosylphosphatidylinositol anchors,25 whereas TLR9 was implicated in the sensing of parasite DNA complexed with haemozoin.22 Haemozoin has also been associated with activation of NOD‐like receptor protein 3 inflammasomes in macrophages.23, 27 A common downstream readout for innate immune sensing of Plasmodium has been the detection of pro‐inflammatory cytokines in serum, plasma, or in tissue culture supernatants.25, 30, 31 This approach was driven technologically by sensitive (pg/ml level), multiplex bead‐based arrays for cytokine proteins.30 Unfortunately, such reagents have not been available for detection of IFN‐α/β at protein level. This has hampered research into the cellular sources of IFN‐α/β during malaria, with most studies resorting to mRNA‐based assessments, enzyme‐linked immunosorbent assays (ELISA) or, in the case of IFN‐β (but not the IFN‐α isoforms), reporter gene expression via yellow fluorescent protein (YFP) or luciferase (Fig. 1).
Figure 1.
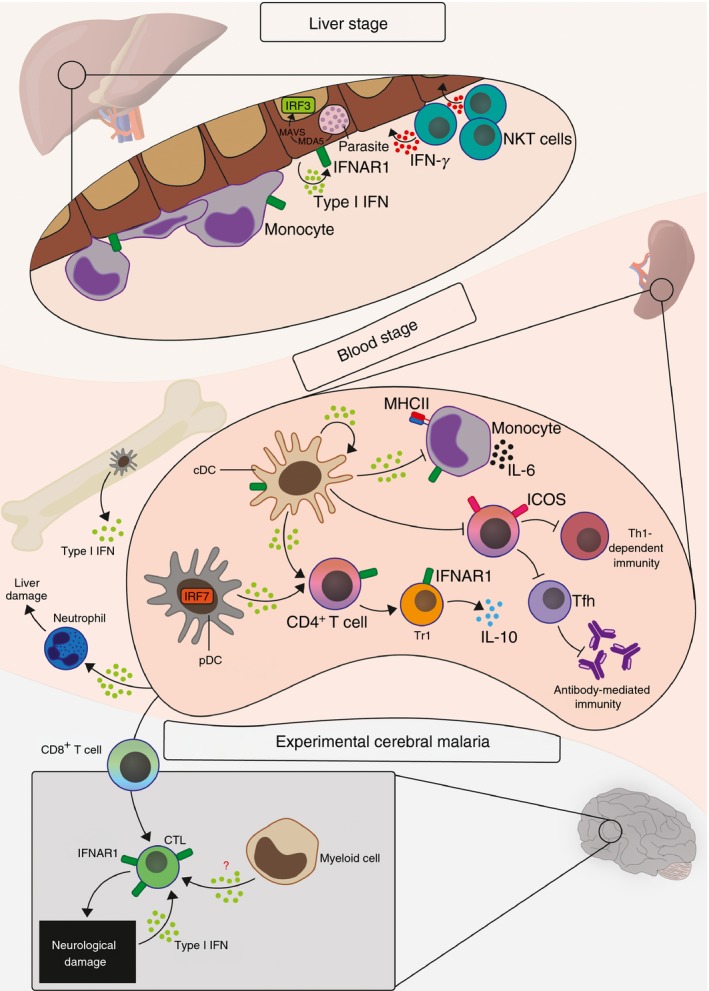
Effects of Type I interferon (IFN) during liver‐stage and blood‐stage Plasmodium infection. Liver‐stage of infection: parasite RNA is sensed partly via melanoma differentiation‐associated protein 5 (MDA5) and mitochondrial antiviral signalling protein (MAVS) in the cytosol of hepatocytes, triggering a type I IFN response dependent on IFN regulatory factor 3 (IRF3). This promotes anti‐parasitic immune responses that require involvement from monocytes and IFN‐γ‐producing natural killer T (NKT) cells. Blood‐stage of infection: Type I IFN can be produced by multiple myeloid cells including plasmacytoid dendritic cells (pDC) in the bone marrow, blood and spleen, and splenic conventional dendritic cells (cDC). This can impact on other myeloid cells, such as activating neutrophils to promote liver damage, or to suppress monocyte up‐regulation of major histocompatibility complex class II (MHCII) or interleukin‐6 (IL‐6) expression. Type I IFN can directly impede cDC from activating T helper type 1 (Th1) and follicular T helper (Tfh) differentiation in CD4+ T cells, with downstream negative impacts on cellular and antibody immunity. Type I IFN can also act directly on CD4+ T cells in the spleen to drive IL‐10 production by Type 1 regulatory (Tr1) cells. Finally, Type I IFN plays a major role in licensing cytotoxic T lymphocyte (CTL)‐mediated immune‐pathology in the brain during experimental cerebral malaria, via direct signalling to CD8+ T cells.
Early studies, examining splenic plasmacytoid dendritic cell (pDC) mRNA transcripts in a lethal mouse model (Plasmodium berghei ANKA infection),32 and a non‐lethal model (Plasmodium chabaudi chabaudi AS)33 or via IFN‐α ELISA of supernatants from in vitro culture of human pDC with P. falciparum schizonts,20 support the idea that pDC are important, and possibly dominant, sources of IFN‐α/β during blood‐stage infection. However, we have reported that other cells, in particular, splenic conventional dendritic cells (cDC), can also be a direct source of IFN‐α/β in mouse models of infection.32, 34 Despite the lack of research tools for detecting IFN‐α/β protein production by specific cells, several studies have attempted to determine the major cellular sources of these cytokines, principally during blood‐stage infection. A study in 2011 by Sharma et al. used ELISA and luciferase reporter expression to demonstrate that AT‐rich motifs from Plasmodium triggered IFN‐β production in vitro, via STING, TANK binding kinase 1, interferon regulatory factor 3 (IRF3) and IRF7.35 A more recent study from the same group assessed IFN‐β mRNA up‐regulation to examine a role for cGAS in promoting type I IFN production.36 Although important observations were made, formal in vivo evidence that cytoplasmic DNA sensing of Plasmodium triggers protein expression of IFN‐α isoforms or IFN‐β remains to be generated.
A recent study by Spaulding et al. examined IFN‐β production during infection with a lethal mouse strain, Plasmodium yoelii 17XYM (YM) using the YFP‐reporter mouse, and suggested that IFN‐β was rapidly made in the bone marrow and blood by pDC, with little contribution by other immune cells.37 It was also shown that STING‐mediated pathways were required, and that IFN‐α protein and IFN‐β‐driven YFP production were dependent on TLR7 and Myd88‐signalling. Interestingly, although systemic type I IFN production required STING in this study, pDC required only TLR7 and myeloid differentiation primary response 88 (MyD88) to produce IFN‐β. The authors then presented data to support that STING acting in macrophages served to boost type I IFN responses from pDC in vivo. This study, as well as another from our group34 suggest that co‐operation between different myeloid cell populations can promote a strong type I IFN response. Another study by Yu et al., published 1 month after the report by Spaulding et al., also examined IFN‐α/β production (by ELISA) during P. yoelii YM infection of mice.28 Consistent between both studies was the finding that systemic IFN‐α/β production was dependent on TLR7 and MyD88‐signalling, with evidence from Yu et al. that IRF7 was also essential for this process.28 Moreover, both studies implicate pDC over cDC or macrophages as the prime source of IFN‐α/β. However, Yu et al. found that mice deficient in MAVS or STING displayed an enormous increase in systemic IFN‐α/β protein levels, but Spaulding et al. found the exact opposite. It remains difficult to reconcile these two findings, published within a month of each other, particularly as both groups used the same parasite species and STING knockout mouse strain (Tmem173 gt/gt). However, the studies used different infectious doses and routes of infection. Hence, the effects of type I IFN during blood‐stage infection may be delicately balanced, so that variables such as infectious dose, route of infection, host genetic background, parasite genetic background and the microbiome, could all contribute to differences from one experimental system to another, and even from one laboratory to another. A major gap in our knowledge across all the common mouse models of blood‐stage infection, therefore, concerns the relative roles of STING versus MyD88‐dependent pathways in driving IFN‐α/β production from pDC and other myeloid cell populations. In addition, whether pDC‐ or cDC‐derived IFN‐α/β production occurs mainly in the bone marrow, blood or spleen during blood‐stage infection remains an important question. Finally, although some assessment of IFN‐α/β production during blood‐stage Plasmodium infection has been attempted in humans, it has been challenging to observe this occurring directly ex vivo.38, 39 It will be important to determine the cellular and molecular basis of IFN‐α/β production in human blood‐stage Plasmodium infection.
Type I IFN‐signalling during liver‐stage Plasmodium infection
Seminal studies from the laboratory of Ruth Nussenzweig, between 1968 and 1970, suggested that IFNs induced by viral infection could be harvested and used to completely prevent malaria in mice infected with liver‐infective P. berghei sporozoites.40, 41, 42 These studies revealed a short time window during the parasite replicative phase in hepatocytes, when exogenous IFNs, most likely type I IFNs based on their in vitro anti‐viral properties, potently controlled liver‐stage infection. However, a possible role for endogenously produced type I IFNs during liver‐stage infection remained unstudied for decades. Recent studies of type I IFN biology in malaria have tended to explore the blood‐stage of infection, possibly because of the relative ease of working with and detecting blood‐stage trophozoites and schizonts, over liver‐stage sporozoites and merozoites, and also because it had been generally thought that parasites invaded and replicated within hepatocytes without being detected or controlled by the innate immune system. However, two independent studies published in early 2014, by Liehl et al.43 and Miller et al.,44 revealed important roles for type I IFN‐signalling in controlling liver‐stage Plasmodium infection.
Liehl et al. and Miller et al. examined the liver transcriptome in mice infected with P. berghei ANKA sporozoites or P. yoelii 17XNL sporozoites respectively.43, 44 Liehl et al. found that all of the 89 genes up‐regulated more than twofold during infection of C57BL/6 mice were classified as IFN‐stimulated genes, and were linked to the type I IFN‐signalling pathway.43 Similarly, Miller et al. also found a strong type I IFN‐associated gene signature in BALB/c and C57BL/6 mice, and could confirm at protein level by ELISA that IFN‐β was produced in the liver.44 To examine the cellular and molecular basis of type I IFN responses in the liver, Liehl et al. assessed up‐regulation of five mRNA species, Ifit1, Ifi44, Usp18, ifit3 and irf7, whereas direct detection of IFN‐α/β at mRNA or protein level was not employed.43 Instead, the authors revealed that type I IFN‐associated transcriptional responses depended on hepatocyte expression of IFN‐α/β receptor subunit 1 (IFNAR1), as well as IRF3, MAVS, MDA5, but not TLR‐signalling via MyD88 or Toll/interleukin‐1‐receptor‐domain‐containing adaptor inducing IFN‐β. Crucially, liver parasite burden and the progression to blood‐stage infection was exacerbated when hepatocytes could not receive signals via IFNAR1. Using a slightly different system, an attenuated P. yoelii 17XNL that could not progress to blood‐stage infection, and which avoided any potential confounding immune responses due to blood‐stage sensing, Miller et al. also showed that IFNAR1 and IRF3 were crucial for liver‐stage protection against parasites.44 Liehl et al. provided important mechanistic insights by showing that in vitro stimulation of hepatocytes with the IRF3‐stimulator, DMXAA, was incapable of controlling parasite numbers, which suggested that type I IFN‐signalling was protective not through direct anti‐parasitic effects on hepatocytes, but that accessory cells, such as leucocytes, were needed.43 Taken together the studies showed that IFNAR1‐signalling was required in non‐hepatocytes for immune cell infiltration into the liver, with Miller et al. demonstrating that CD1d‐restricted natural killer T (NKT) cells were essential for type I IFN‐dependent parasite control, probably via IFN‐γ production. Together, these findings revealed that type I IFN‐signalling is protective against liver‐stage Plasmodium infection, by facilitating sensing of parasite RNA within hepatocytes via MDA5 and MAVS, as well as recruitment of anti‐parasitic immune cells such as NKT cells. These studies provide important first steps towards understanding type I IFN‐signalling networks within the liver during Plasmodium infection. However, we still lack a comprehensive description of how type I IFN‐dependent immune processes limit progression to the symptomatic blood‐stage of infection.
Effect of type I IFN‐signalling on parasite control during blood‐stage infection
The early studies from the Nussensweig laboratory indicated that exogenous type I IFN was highly protective during the liver stage of infection, but much less so during the blood‐stage of infection, providing the first clues that differences might exist in the effects of type I IFN between liver‐ and blood‐stages of infection.40, 41, 42 Studies from the Renia laboratory in 2001 and 2007 employed an engineered hybrid form of human IFN‐α (termed BDBB), comprising the N‐ and C‐terminal portions of human IFN‐α8, and a mid‐section from human IFN‐α1, that could bind and signal via mouse IFNAR receptors in vivo.45, 46 Using this as an exogenous form of type I IFN, the authors first showed that BDBB reduced parasitaemia during blood‐stage infection with P. yoelii 265 BY or P. yoelii 17XNL,45 but did not improve parasite control during liver‐stage infection. In 2007, Vigario et al., also showed that BDBB could prevent experimental cerebral malaria caused by P. berghei ANKA.46 The exact mechanisms underlying BDBB‐mediated protection were unclear, although parasitaemia, brain sequestration of parasites and pathogenic CD8+ T cells, as well as brain endothelial expression of the adhesion molecule, intercellular adhesion molecule 1, was reduced, while IFN‐γ levels in serum were elevated. Although it was not proven that BDBB signalled via IFNAR1 in these mouse malaria models, these papers suggested that exogenous type I IFN‐signalling could be beneficial during blood‐stage infection.
Two years later, Voisine et al., employed IFNAR‐deficient mice (129Sv background) to report that endogenous type I IFN‐signalling played no role in parasite control during non‐lethal infection with P. chabaudi chabaudi AS.33 However, subsequent studies from our group, using Ifnar1 −/− and Irf7 −/− mice (C57BL/6 background), have suggested that type I IFN‐signalling via IFNAR1/IRF7, but not IRF3, impairs early parasite control, not only in the P. chabaudi chabaudi AS model,47, 48 but also during non‐lethal infection with P. yoelii 17XNL.48, 49 Effects on parasite control during P. yoelii 17XNL infection were also observed in an independent study by Zander et al., which used the blocking antibody MAR1‐5A3 in C57BL/6 mice.50 Therefore, although it appears that in some host genetic backgrounds, type I IFN‐signalling via IFNAR1 can play little to no role during non‐lethal blood‐stage infection,33 in others, type I IFN‐signalling appears to hinder the control of primary blood‐stage infections, at least in non‐lethal models of blood‐stage infection.48, 49, 50 In contrast, a study employing another non‐lethal strain, P. yoelii N67, found a modest anti‐parasitic role for type I IFN (using both IFNAR1‐deficient mice and antibody blockade), which highlighted that Plasmodium genetics probably also influences the effect of type I IFN during non‐lethal blood‐stage infection.51 In the lethal P. yoelii YM model, Spaulding et al. employed IFNAR1‐deficient mice to reveal a clear role for type I IFN in promoting high parasitaemias and causing lethality.37 In contrast, Yu et al. employed an IFNAR1‐blocking antibody, the precise clone not being reported, and saw no effect on lethality or parasite control.28 These two studies are difficult to reconcile, and so further independent studies will be required to confirm the role of type I IFN‐signalling via IFNAR1 during lethal P. yoelii YM infection.
One of the most commonly employed, lethal mouse models of malaria is infection of C57BL/6 mice with P. berghei ANKA, which triggers multi‐organ, severe malaria symptoms, including parasite‐dependent, cytotoxic T‐lymphocyte (CTL) ‐mediated neurological damage that is usually fatal within 6–10 days. This model is known as ‘experimental cerebral malaria’ (ECM). The effect of type I IFN in this model will be discussed separately in the section below.
Effect of type I IFN‐signalling during experimental cerebral malaria
In 2011, Sharma et al. and our group independently published, within 1 month of each other, that C57BL/6 mice deficient in IFNAR1 were completely resistant to ECM, so demonstrating for the first time a detrimental role for endogenous type I IFN‐signalling during blood‐stage Plasmodium infection.35, 52 Sharma et al. also showed that mice deficient in both IRF3 and IRF7 survived this lethal infection, providing further evidence of a detrimental role for type I IFN‐signalling.35 Our group provided early characterization of IFNAR1‐deficient mice during P. berghei ANKA infection, specifically showing that brain pathology was reduced, and that parasite burden in the whole bodies and brain were substantially reduced.52 Our data suggested that neurological damage, known in this system to be dependent upon parasite burden and the action of CD8+ T cells in the brain, was type I IFN‐dependent. We also found that CD8+ T‐cell recruitment to the brain was unaffected by IFNAR1 deficiency. Subsequent studies by Ball et al. and Palomo et al., reported very similar outcomes – that IFNAR1‐deficient mice were less susceptible to brain pathology and ECM compared with wild‐type controls.19, 53 However, minor differences were apparent between our three independent studies: Palomo et al. showed that IFNAR1‐deficient mice were not completely resistant to ECM, but did exhibit reduced CTL numbers in the brain,53 whereas Ball et al. reported complete resistance to ECM associated with activation, though not recruitment,19 of CTL in the brain. Moreover, whereas our group used parasite‐derived bioluminescence to show that parasite burdens in the brain were reduced,52 Ball et al. employed quantitative polymerase chain reaction of parasite RNA to show that parasite burdens were unaffected by IFNAR1 deficiency.19 It is perhaps difficult to reconcile differences in parasite burden and CTL responses in the brain between these three studies, which could again be due to minor changes in infectious dose, route of infection or the microbiome across different laboratories. Instead, it is useful to conclude that all three independent studies revealed a substantial deleterious role for type I IFN‐signalling in mediating severe and lethal disease during P. berghei ANKA infection. This conclusion was also supported by our observation that the canonical type I IFN‐associated transcription factor IRF7 promotes lethal symptoms in this model.47
Mechanistic details of type I IFN‐signalling during P. berghei ANKA infection have recently been explored by our group,34 and others.19 Ball et al. employed a pre‐sensitization approach to adoptively transfer splenocytes from donor mice that had received irradiated parasites, into IFNAR1‐deficient mice, in an effort to re‐capitulate ECM symptoms.19 Using this technique, the authors showed that IFNAR1‐expression on CD8+ T cells was sufficient to induce ECM in IFNAR1‐deficient recipients. Hence, T‐cell intrinsic IFNAR1 was implicated in the pathogenic effects of type IFN‐signalling in this model. In contrast, our group used a combination of mixed bone marrow chimeras and CD11c‐Cre ifnar1 f/f mice to implicate a role for cDC in mediating the deleterious effects of type I IFN‐signalling.34 It remains difficult to determine whether pathogenic type I IFN‐signalling during P. berghei ANKA infection proceeds exclusively through T cells or myeloid cells, particularly as both techniques have potential caveats. For instance in our study, IFNAR1 deficiency, although profound in cDC populations, was also observed to a limited degree in NK cells and T cells. Hence the fidelity of the CD11c promoter may not be absolute. A possible complication of studying CD8+ T cells from Ifnar1 −/− mice, is that tonic levels of type I IFN‐signalling have been suggested to play a role in the final stages of T‐cell development in the thymus.54 Therefore, while T cells still develop in the absence of type I IFN‐signalling, it is possible that their precise phenotype is affected by a total absence of IFNAR1. Given potential caveats with genetic manipulation of IFNAR1, our group's use of the efficacious IFNAR1‐blocking monoclonal antibody MAR1‐5A3 provides perhaps less confounded evidence that type I IFN‐signalling through IFNAR1 promotes severe and lethal neurological symptoms in experimental cerebral malaria.34, 47 Futhermore, late blockade of IFNAR1 in partially ECM‐susceptible IRF7‐deficient mice, provided evidence that type I IFN‐signalling also acted late, perhaps on brain‐recruited CD8+ T cells, as originally suggested by Ball et al.19 In a more recent study, activation of type I IFN responses in neurological tissue via the deubiquitinase enzyme USP15 and the E3 Ubiquitin ligase TRIM25 promoted ECM, further supporting the concept that type I IFN‐signalling in the brain may be deleterious during P. berghei ANKA infection.55 Therefore, although type I IFN‐signalling clearly induces ECM symptoms in this model, the precise mechanisms by which this occurs remain to be fully determined. In particular, the effects of type I IFN‐signalling in the brain have not been fully defined in this model. Nevertheless, a working model, based on all recent studies, is that type I IFN‐signalling via the IFNAR1 receptor acts on numerous cell populations during blood‐stage P. berghei ANKA infection, including an early signal via cDC to impair the onset of CD4+ T‐cell immunity (as discussed below), and a later signal via CD8+ T cells, to support not their priming in the spleen, but their recruitment, activation and function in the brain.
Immune‐regulation via type I IFN‐signalling
Although type I IFNs were discovered for their potent anti‐viral properties, it is established now that signalling via IFNAR1 constitutes a major immunomodulatory pathway in a wide range of immune cells. This has been increasingly reported in a number of studies in mice and humans infected with Plasmodium.
Effect of type I IFN‐signalling on myeloid cells
Type I IFN‐signalling has been implicated in regulating the function of myeloid cells during infection with viruses,56, 57 bacteria58, 59 and parasites,60, 61 including during liver‐stage and blood‐stage Plasmodium infection.34, 39, 43, 48, 62 Previously, blood‐stage Plasmodium parasites were reported to trigger systemic cDC activation and dysfunction in human and mouse experimental systems;60, 63, 64, 65 but for some time, host‐derived factors mediating this process had remained unclear. Our group recently employed bone marrow chimeric mice and CD11c‐Cre ifnar1 f/f mice during P. berghei ANKA and P. chabaudi chabaudi AS infection to identify a role for type I IFN‐signalling via IFNAR1 in promoting excessive activation and dysfunction of cDC.34, 48 Specifically, we noted that CD4+ T‐cell activation and differentiation was impaired by type I IFN.34, 48 We also reported a stronger effect of type I IFN suppression in CD8α − CD11b+ splenic cDC compared with CD8+ CD11b− cDC. From a mechanistic perspective, we did not determine what molecular pathways in cDC were functionally impaired by type I IFN‐signalling during CD4+ T‐cell priming, although we noted that il10 transcription by cDC, and the ratio of programmed cell death protein ligand 1 (PDL1)/PDL2 were controlled by type I IFN‐signalling. In another recent study, it was revealed that type I IFN‐signalling affected cDC survival, as IFNAR1 blockade reduced caspase‐3‐associated apoptosis in splenic cDC during P. berghei ANKA and P. yoelii YM infection.62 Together, these studies suggest that at least one aspect of type I IFN‐mediated immune‐modulation in malaria is enacted via signalling in cDC.
Type I interferon‐signalling may also impact the function of other myeloid cells, including monocytes and neutrophils. In one of the only human studies to be conducted in this field, Montes de Oca et al. employed controlled human malaria infection with blood‐stage P. falciparum parasites to reveal that monocytes produced more IL‐6 protein on a per cell basis when type I IFN‐signalling was blocked in vitro using an IFNAR2‐blocking antibody.39 Similar effects were noted for IL‐1β and IL‐17, although the cellular sources of these were not examined. IFNAR1‐mediated suppression of monocyte activation in the spleen and blood was also observed in our studies in mice infected with P. berghei ANKA.34 It will be important in the future, when reagents permit, to examine in humans whether immune‐suppressive effects on monocytes are seen equally by IFNAR2 and IFNAR1 blockade, because IFN‐β can signal via IFNAR1, independently of IFNAR2.66
Another recent study implicated type I IFN‐signalling in the activation and function of neutrophils during infection, a process that potentially contributed to pathology.67 In this report, up‐regulation of a type I IFN‐associated transcriptional signature was observed in neutrophils from Plasmodium vivax‐infected patients compared with healthy controls. This associated with elevated levels of liver enzymes, alanine aminotransferase and aspartate aminotransferase, suggesting a type I IFN‐dependent neutrophil‐mediated mechanism in inducing liver damage during Plasmodium infection. Further experiments using P. chabaudi chabaudi AS infection in IFN‐αβR−/− (129Sv background) mice showed that type I IFN‐signalling partially supported the recruitment of pathogenic neutrophils into the liver and was absolutely essential for elevation in alanine aminotransferase.67 Collectively, these studies indicate that type I IFN‐signalling can profoundly influence the responses of cDC, monoyctes and neutrophils during blood‐stage Plasmodium infection. Although there are some data to suggest that type I IFN‐signalling can proceed directly via cDC, exactly which myeloid cells receive type I IFN signals, and moreover, which isoforms, and which forms of the IFNAR receptors are involved in modulating cell function remain to be elucidated. Finally, it will be important to determine cellular pathways that mediate type I IFN‐dependent leucocyte responses during liver‐stage infection.
Effect of type I IFN‐signalling on adaptive T helper and B‐cell responses
Given that adaptive T‐cell and B‐cell responses can influence the outcome of both liver‐stage and blood‐stage Plasmodium infections, several studies have explored how they might be influenced by type I IFN‐signalling. Here, we first briefly summarize current knowledge of T‐cell effector responses in malaria. During liver‐stage infection, cytotoxic CD8+ T‐cell responses elicited by sporozoites or other vaccination strategies strongly protect against primary or secondary challenge.68, 69 During blood‐stage infection, there is little evidence that CD8+ T cells are protective during primary infection.70 Rather, CD8+ T cells in mice infected with P. berghei ANKA, though not in other mouse models, promote neurological damage, high parasite burdens and lethality.71, 72 In contrast, CD4+ helper T‐cell responses are crucial for inducing cellular and humoral immunity during blood‐stage Plasmodium infection.48 In particular, as recently reviewed elsewhere by our group,73 IFN‐γ‐producing, T helper type 1 (Th1) cells, and high‐affinity antibody‐inducing T follicular helper (Tfh) cells play particularly important roles in controlling and resolving blood‐stage infections. In addition, certain types of regulatory CD4+ T cells, in particular Type 1 regulatory (Tr1) cells that express the canonical immune‐suppressive cytokine IL‐10, are required to mitigate the possible deleterious effects of T‐cell‐mediated inflammation.74, 75, 76
During liver‐stage infection, Miller et al. found that type I IFN‐signalling was necessary for increases in CD8+ T‐cell proportions (as well as NKT‐cell responses). This are perhaps the first data to suggest that type I IFN responses might be beneficial in the liver for driving protective CTL responses. Given that much of the lethality in the P. berghei ANKA model is due to pathogen burden and brain‐recruited CTL, it was also shown by several groups that CD8+ T‐cell numbers and/or activation within the brain were dependent upon type I IFN signals.53 Although our earlier study had found little requirement for type I IFN‐signalling in promoting T‐cell activation in the spleen, a subsequent study blocked lethality with late treatment with anti‐IFNAR1 blocking antibodies.47 Taken together, we propose that type I IFN‐signalling probably promotes CTL responses during blood‐stage Plasmodium infection, which could result in immune pathology in the brain, but might conceivably protect other organs, such as the liver.70 It is intriguing that in a study of patients with malaria, type I IFN‐dependent Flt3L production correlated positively with the number of activated CD8+ T cells detectable in peripheral blood.77 This suggests that type I IFN‐signalling may also support parasite‐specific CD8+ T‐cell responses in human malaria.
Given that CD4+ T‐cell subsets, particularly Th1, Tfh and Tr1 cells, are crucial for immunity and immune‐regulation in blood‐stage malaria, several studies, including our own, have explored a possible role for type I IFN in influencing T helper cells during blood‐stage infection. Our initial studies using P. berghei ANKA infection, which exhibits a relatively weak Th1 response and minimal Tfh responses before the onset of lethality,78 demonstrated, through the employment of IFNAR1‐deficient mice or IFNAR1‐blocking antibodies, that type I IFN‐signalling limited Th1 responses, although Tfh differentiation was not assessed.34, 52 Our subsequent experiments employed ifnar1 −/− and irf7 −/− mice during P. chabaudi chabaudi AS infection, and similarly found that Th1 responses were suppressed in these mice.47 Finally, using P. chabaudi chabaudi AS and P. yoelii 17XNL infection, we found that Th1 and Tfh cell responses, as well as germinal centre B‐cell development and high‐affinity IgG production, all increased in the absence of type I IFN‐signalling.48 One month earlier, an independent study by Zander et al., had also employed P. yoelii 17XNL infection, and similarly found that Tfh, B‐cell and antibody responses were restricted by type I IFN‐signalling.50 Therefore together, our data suggest that type I IFN‐signalling serves to limit Th1, Tfh and antibody responses during blood‐stage infection. One major difference between studies by our group, and that of Zander et al. has been in the proposed cellular target of IFN‐α/β signalling. Our studies have implicated a role for signalling in splenic cDC, while Zander et al. revealed that signalling occurred in a CD4+ T‐cell intrinsic manner. As these two scenarios are not mutually exclusive, the relative contribution of signalling through myeloid cells versus T cells remains to be determined. Furthermore, it will be of great interest to examine any possible effects of direct IFNAR1‐signalling in B cells. Finally, in human studies by Montes de Oca et al., in vitro blockade of IFNAR2 also increased IFN‐γ production by CD4+ T cells during controlled human malaria infection.39 Hence, taken together, data from several studies including our own show that type I IFN‐signalling limits Th1‐ and Tfh‐dependent immune responses during blood‐stage malaria. The exact pathways mediating such T‐cell immune‐suppression are unclear, but could be related to a number of different mechanisms. First, our study and that of Zander et al. revealed that inducible co‐stimulator expression by CD4+ T cells was restrained by type I IFN‐signalling.48, 50 This might limit their capacity to interact with inducible co‐stimulator ligand‐expressing B cells or myeloid cells. Second, primary effects of type I IFN‐signalling on myeloid cells, such as IL‐10 expression or PDL1/PDL2 expression by cDC and inflammatory monocytes, might be responsible for reduced activation of T cells. Third, it was shown by Zander et al., that type I IFN‐signalling promoted the emergence of IL‐10‐producing Tr1 cells, which in turn acted alongside IFN‐γ to suppress Tfh‐dependent immune responses.50 Furthermore, in vitro culture of peripheral blood mononuclear cells from controlled human malaria infection studies in the presence of IFNAR2‐blocking antibodies reduced Tr1 formation and IL‐10 production compared with isotype control‐treated cultures.39 Together, these reports in humans and mice suggest that multiple cellular and molecular mechanisms, both T‐cell intrinsic and extrinsic, may mediate type I IFN‐dependent immune‐regulation of CD4+ T‐cell‐dependent immunity during blood‐stage Plasmodium infection, while also acting to support CD8+ T‐cell responses.
Conclusions
Seminal studies from the 1960s revealed that interferons induced by viral infection could provide excellent protection against liver‐stage Plasmodium parasites in mouse models. This seeded the concept that type I IFN‐signalling could be beneficial in malaria. Since then, recombinant protein technologies and knockout mice have permitted more detailed assessment of the roles of IFN‐α/β and signalling via the IFNAR1 receptor in influencing the liver‐ and blood‐stages of Plasmodium infection. Although it is clear that type I IFN‐associated transcriptional responses occur in both humans and mice, during liver‐ and blood‐stage infections, due to a lack of reagents it remains difficult to ascertain which IFN isoforms are being generated by specific cells during infection. Nevertheless, studies in mice reveal that type I IFN‐signalling both in hepatocytes and in leucocytes is essential for promoting immune responses that can control liver‐stage infection. Type I IFN‐signalling during the blood‐stage of infection also appears to promote CD8+ T‐cell responses in humans and mice, and in some cases, this may induce CTL‐mediated immune‐pathology. In stark contrast, however, type I IFN‐signalling during blood‐stage infection restricts the development of CD4+ T‐cell‐mediated immunity to parasites, probably through multiple cellular mechanisms. Hence, an emerging picture in malaria is that type I IFN‐signalling can be either detrimental or beneficial, depending on the context in which it is being studied. Therefore, based on the current literature it is difficult to present a simple, unified picture of the effects of type I IFN in malaria, not only because consensus has been difficult to reach in experimental animal studies, but also because there remains a paucity of data in humans. In addition, effects of type I IFN in malaria may be more complicated than currently reported, as studies of different IFN‐α isoforms, or homodimeric versus heterodimeric cytokine receptors have yet to be conducted during either blood‐ or liver‐stage infection. One possible path through this complexity may be to understand in greater detail the basic biology of how different IFN‐α/β isoforms signal through homodimeric versus heterodimeric IFNAR1 receptors, and then to determine how these different signalling pathways are employed by a variety of cells in vivo. Although it is perhaps too early to herald type I IFN‐signalling as a viable molecular target for improving clinical outcomes in malaria, reports from the past 10 years reveal that endogenous type I IFN‐signalling probably plays a crucial role in shaping the host response to Plasmodium infection.
Disclosures
The authors declare that there are no competing interests.
References
- 1. WHO . 2016 World Malaria report, 2016. 1–186.
- 2. Cunnington AJ, Walther M, Riley EM. Piecing together the puzzle of severe malaria. Sci Transl Med 2013; 5:211 ps18. [DOI] [PubMed] [Google Scholar]
- 3. Fung KY, Mangan NE, Cumming H, Horvat JC, Mayall JR, Stifter SA et al Interferon‐ε protects the female reproductive tract from viral and bacterial infection. Science 2013; 339:1088–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Versteeg GA, Garcia‐Sastre A. Viral tricks to grid‐lock the type I interferon system. Curr Opin Microbiol 2010; 13:508–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yan N, Chen ZJ. Intrinsic antiviral immunity. Nat Immunol 2012; 13:214–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ishihara T, Aga M, Hino K, Ushio C, Taniguchi M, Iwaki K et al Inhibition of Chlamydia trachomatis growth by human interferon‐α: mechanisms and synergistic effect with interferon‐γ and tumor necrosis factor‐α . Biomed Res 2005; 26:179–85. [DOI] [PubMed] [Google Scholar]
- 7. Parker D, Prince A. Staphylococcus aureus induces type I IFN signaling in dendritic cells via TLR9. J Immunol 2012; 189:4040–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rothfuchs AG, Trumstedt C, Mattei F, Schiavoni G, Hidmark A, Wigzell H et al STAT1 regulates IFN‐α/β‐ and IFN‐γ‐dependent control of infection with Chlamydia pneumoniae by nonhemopoietic cells. J Immunol 2006; 176:6982–90. [DOI] [PubMed] [Google Scholar]
- 9. Mauri C, Menon M. The many faces of type I interferon in systemic lupus erythematosus. J Clin Investig 2015; 125:2562–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Conigliaro P, Perricone C, Benson RA, Garside P, Brewer JM, Perricone R et al The type I IFN system in rheumatoid arthritis. Autoimmunity 2010; 43:220–5. [DOI] [PubMed] [Google Scholar]
- 11. Berry MPR, Graham CM, Xu Z, McNab FW, Bloch SAA, Oni T et al An interferon‐inducible neutrophil‐driven blood transcriptional signature in human tuberculosis. Nature 2010; 466:973–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xin L, Vargas‐Inchaustegui DA, Raimer SS, Kelly BC, Hu J, Zhu L et al Type I IFN receptor regulates neutrophil functions and innate immunity to Leishmania parasites. J Immunol 2010; 184:7047–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McNab F, Mayer‐Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol 2015; 15:87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boxx GM, Cheng G. The roles of type I interferon in bacterial infection. Cell Host Microbe 2016; 19:760–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eshleman EM, Lenz LL. Type I interferons in bacterial infections: taming of myeloid cells and possible implications for autoimmunity. Front Immunol 2014; 5:431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moreira‐Teixeira L, Mayer‐Barber K, Sher A, O'Garra A. Type I interferons in tuberculosis: foe and occasionally friend. J Exp Med 2018; 215:1273–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cunnington AJ, Riley EM, Walther M. Stuck in a rut? Reconsidering the role of parasite sequestration in severe malaria syndromes. Trends Parasitol 2013; 29:585–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aucan C, Walley AJ, Hennig BJW, Fitness J, Frodsham A, Zhang L et al Interferon‐α receptor‐1 (IFNAR1) variants are associated with protection against cerebral malaria in the Gambia. Genes Immun 2003; 4:275–82. [DOI] [PubMed] [Google Scholar]
- 19. Ball EA, Sambo MR, Martins M, Trovoada MJ, Benchimol C, Costa J et al IFNAR1 controls progression to cerebral malaria in children and CD8+ T cell brain pathology in Plasmodium berghei‐infected mice. J Immunol 2013; 190:5118–27. [DOI] [PubMed] [Google Scholar]
- 20. Pichyangkul S, Yongvanitchit K, Kum‐arb U, Hemmi H, Akira S, Krieg AM et al Malaria blood stage parasites activate human plasmacytoid dendritic cells and murine dendritic cells through a Toll‐like receptor 9‐dependent pathway. J Immunol 2004; 172:4926–33. [DOI] [PubMed] [Google Scholar]
- 21. Feintuch CM, Benayoun J, Laufer M, Ryu S, Sixpence A, Suh Y et al Type I interferon receptor variants in gene regulatory regions are associated with susceptibility to cerebral malaria in Malawi. Am J Trop Med Hyg 2018; 98:1692–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Coban C, Ishii KJ, Kawai T, Hemmi H, Sato S, Uematsu S et al Toll‐like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J Exp Med 2005; 201:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Griffith JW, Sun T, McIntosh MT, Bucala R. Pure Hemozoin is inflammatory in vivo and activates the NALP3 inflammasome via release of uric acid. J Immunol 2009; 183:5208–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kalantari P, DeOliveira RB, Chan J, Corbett Y, Rathinam V, Stutz A et al Dual engagement of the NLRP3 and AIM2 inflammasomes by Plasmodium‐derived hemozoin and DNA during malaria. Cell Rep 2014; 6:196–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu J, Krishnegowda G, Gowda DC. Induction of proinflammatory responses in macrophages by the glycosylphosphatidylinositols of Plasmodium falciparum: the requirement of extracellular signal‐regulated kinase, p38, c‐Jun N‐terminal kinase and NF‐κB pathways for the expression of proinflammatory cytokines and nitric oxide. J Biol Chem 2005; 280:8617–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sherry BA, Alava G, Tracey KJ, Martiney J, Cerami A, Slater AF. Malaria‐specific metabolite hemozoin mediates the release of several potent endogenous pyrogens (TNF, MIP‐1α, and MIP‐1β) in vitro, and altered thermoregulation in vivo . J Inflamm 1995; 45:85–96. [PubMed] [Google Scholar]
- 27. Tiemi Shio M, Eisenbarth SC, Savaria M, Vinet AF, Bellemare M‐J, Harder KW et al Malarial hemozoin activates the NLRP3 inflammasome through Lyn and Syk kinases. PLoS Pathog 2009; 5:e1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu X, Cai B, Wang M, Tan P, Ding X, Wu J et al Cross‐regulation of two type I interferon signaling pathways in plasmacytoid dendritic cells controls anti‐malaria immunity and host mortality. Immunity 2016; 45:1093–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Parroche P, Lauw FN, Goutagny N, Latz E, Monks BG, Visintin A et al Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll‐like receptor 9. Proc Natl Acad Sci USA 2007; 104:1919–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lyke KE, Burges R, Cissoko Y, Sangare L, Dao M, Diarra I et al Serum levels of the proinflammatory cytokines interleukin‐1 beta (IL‐1β), IL‐6, IL‐8, IL‐10, tumor necrosis factor α, and IL‐12(p70) in Malian children with severe Plasmodium falciparum malaria and matched uncomplicated malaria or healthy controls. Infect Immun 2004; 72:5630–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Prakash D, Fesel C, Jain R, Cazenave P, Mishra GC, Pied S. Clusters of cytokines determine malaria severity in Plasmodium falciparum‐infected patients from endemic areas of Central India. J Infect Dis 2006; 194:198–207. [DOI] [PubMed] [Google Scholar]
- 32. de Walick S, Amante FH, McSweeney KA, Randall LM, Stanley AC, Haque A et al Cutting edge: conventional dendritic cells are the critical APC required for the induction of experimental cerebral malaria. J Immunol 2007; 178:6033–7. [DOI] [PubMed] [Google Scholar]
- 33. Voisine C, Mastelic B, Sponaas AM, Langhorne J. Classical CD11c+ dendritic cells, not plasmacytoid dendritic cells, induce T cell responses to Plasmodium chabaudi malaria. Int J Parasitol 2010; 40:711–9. [DOI] [PubMed] [Google Scholar]
- 34. Haque A, Best SE, Montes de Oca M, James KR, Ammerdorffer A, Edwards CL et al Type I IFN signaling in CD8– DCs impairs Th1‐dependent malaria immunity. J Clin Invest 2014; 124:2483–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sharma S, DeOliveira RB, Kalantari P, Parroche P, Goutagny N, Jiang Z et al Innate immune recognition of an AT‐rich stem‐loop DNA motif in the Plasmodium falciparum genome. Immunity 2011; 35:194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gallego‐Marin C, Schrum JE, Andrade WA, Shaffer SA, Giraldo LF, Lasso AM et al Cyclic GMP‐AMP synthase is the cytosolic sensor of Plasmodium falciparum genomic DNA and activates type I IFN in malaria. J Immunol 2018; 200:768–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Spaulding E, Fooksman D, Moore JM, Saidi A, Feintuch CM, Reizis B et al STING‐licensed macrophages prime type I IFN production by plasmacytoid dendritic cells in the bone marrow during severe Plasmodium yoelii malaria. PLoS Pathog 2016; 12:e1005975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Loughland JR, Minigo G, Sarovich DS, Field M, Tipping PE, Montes de Oca M et al Plasmacytoid dendritic cells appear inactive during sub‐microscopic Plasmodium falciparum blood‐stage infection, yet retain their ability to respond to TLR stimulation. Sci Rep 2017; 7:2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Montes de Oca M, Kumar R, Rivera FL, Amante FH, Sheel M, Faleiro RJ et al Type I interferons regulate immune responses in humans with blood‐stage Plasmodium falciparum infection. Cell Rep 2016; 17:399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jahiel RI, Nussenzweig RS, Vanderberg J, Vilcek J. Anti‐malarial effect of interferon inducers at different stages of development of Plasmodium berghei in the mouse. Nature 1968; 220:710–1. [DOI] [PubMed] [Google Scholar]
- 41. Jahiel RI, Vilcek J, Nussenzweig R, Vanderberg J. Interferon inducers protect mice against Plasmodium berghei malaria. Science 1968; 161:802–4. [DOI] [PubMed] [Google Scholar]
- 42. Jahiel RI, Vilcek J, Nussenzweig RS. Exogenous interferon protects mice against Plasmodium berghei malaria. Nature 1970; 227:1350–1. [DOI] [PubMed] [Google Scholar]
- 43. Liehl P, Zuzarte‐Luís V, Chan J, Zillinger T, Baptista F, Carapau D et al Host‐cell sensors for Plasmodium activate innate immunity against liver‐stage infection. Nat Med 2014; 20:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Miller JL, Sack BK, Baldwin M, Vaughan AM, Kappe SH. Interferon‐mediated innate immune responses against malaria parasite liver stages. Cell Rep 2014; 7:436–47. [DOI] [PubMed] [Google Scholar]
- 45. Vigario AM. Inhibition of Plasmodium yoelii blood‐stage malaria by interferon α through the inhibition of the production of its target cell, the reticulocyte. Blood 2001; 97:3966–71. [DOI] [PubMed] [Google Scholar]
- 46. Vigario AM, Belnoue E, Gruner AC, Mauduit M, Kayibanda M, Deschemin J‐C et al Recombinant human IFN‐α inhibits cerebral malaria and reduces parasite burden in mice. J Immunol 2007; 178:6416–25. [DOI] [PubMed] [Google Scholar]
- 47. Edwards CL, Best SE, Gun SY, Claser C, James KR, de Oca MM et al Spatiotemporal requirements for IRF7 in mediating type I IFN‐dependent susceptibility to blood‐stage Plasmodium infection. Eur J Immunol 2015; 45:130–41. [DOI] [PubMed] [Google Scholar]
- 48. Sebina I, James KR, Soon MSF, Fogg LG, Best SE, Labastida Rivera F et al IFNAR1‐signalling obstructs ICOS‐mediated humoral immunity during non‐lethal blood‐stage Plasmodium infection. PLoS Pathog 2016; 12:e1005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. James KR, Soon MSF, Sebina I, Fernandez‐Ruiz D, Davey G, Liligeto UN et al IFN regulatory factor 3 balances Th1 and T follicular helper immunity during nonlethal blood‐stage Plasmodium infection. J Immunol 2018; 200:1443–56. [DOI] [PubMed] [Google Scholar]
- 50. Zander RA, Guthmiller JJ, Graham AC, Pope RL, Burke BE, Carr DJJ et al Type I interferons induce T regulatory 1 responses and restrict humoral immunity during experimental malaria. PLoS Pathog 2016; 12:e1005945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wu J, Tian L, Yu X, Pattaradilokrat S, Li J, Wang M et al Strain‐specific innate immune signaling pathways determine malaria parasitemia dynamics and host mortality. Proc Natl Acad Sci U S A 2014; 111:E511–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Haque A, Best SE, Ammerdorffer A, Haque A, Best SE, Ammerdorffer A et al Type I interferons suppress CD4+ T‐cell‐dependent parasite control during blood‐stage Plasmodium infection. Eur J Immunol 2011; 41:2688–98. [DOI] [PubMed] [Google Scholar]
- 53. Palomo J, Fauconnier M, Coquard L, Gilles M, Meme S, Szeremeta F et al Type I interferons contribute to experimental cerebral malaria development in response to sporozoite or blood‐stage Plasmodium berghei ANKA. Eur J Immunol 2013; 43:2683–95. [DOI] [PubMed] [Google Scholar]
- 54. Xing Y, Wang X, Jameson SC, Hogquist KA. Late stages of T cell maturation in the thymus involve NF‐κB and tonic type I interferon signaling. Nat Immunol 2016; 17:565–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Torre S, Polyak MJ, Langlais D, Fodil N, Kennedy JM, Radovanovic I et al USP15 regulates type I interferon response and is required for pathogenesis of neuroinflammation. Nat Immunol 2017; 18:54–63. [DOI] [PubMed] [Google Scholar]
- 56. Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KCF, Welch M et al Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013; 340:207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G et al Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013; 340:202–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rayamajhi M, Humann J, Kearney S, Hill KK, Lenz LL. Antagonistic crosstalk between type I and II interferons and increased host susceptibility to bacterial infections. Virulence 2010; 1:418–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rayamajhi M, Humann J, Penheiter K, Andreasen K, Lenz LL. Induction of IFN‐α/β enables Listeria monocytogenes to suppress macrophage activation by IFN‐γ . J Exp Med 2010; 207:327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wilson NS, Behrens GMN, Lundie RJ, Smith SM, Waithman J, Young L et al Systemic activation of dendritic cells by Toll‐like receptor ligands or malaria infection impairs cross‐presentation and antiviral immunity. Nat Immunol 2006; 7:165–72. [DOI] [PubMed] [Google Scholar]
- 61. Silva‐Barrios S, Stager S. Protozoan parasites and type I IFNs. Front Immunol 2017; 8:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tamura T, Kimura K, Yui K, Yoshida S. Reduction of conventional dendritic cells during Plasmodium infection is dependent on activation induced cell death by type I and II interferons. Exp Parasitol 2015; 159:127–35. [DOI] [PubMed] [Google Scholar]
- 63. Elliott SR, Spurck TP, Dodin JM, Maier AG, Voss TS, Yosaatmadja F et al Inhibition of dendritic cell maturation by malaria is dose dependent and does not require Plasmodium falciparum erythrocyte membrane protein 1. Infect Immun 2007; 75:3621–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Urban BC, Ferguson DJ, Pain A, Willcox N, Plebanski M, Austyn JM et al Plasmodium falciparum‐infected erythrocytes modulate the maturation of dendritic cells. Nature 1999; 400:73–7. [DOI] [PubMed] [Google Scholar]
- 65. Wykes MN, Good MF. What really happens to dendritic cells during malaria? Nat Rev Microbiol 2008; 6:864–70. [DOI] [PubMed] [Google Scholar]
- 66. De Weerd NA, Vivian JP, Nguyen TK, Mangan NE, Gould JA, Braniff S‐J et al Structural basis of a unique interferon‐beta signaling axis mediated via the receptor IFNAR1. Nat Immunol 2013; 14:901–7. [DOI] [PubMed] [Google Scholar]
- 67. Rocha BC, Marques PE, Leoratti FM, Junqueira C, Pereira DB, Antonelli LR et al Type I interferon transcriptional signature in neutrophils and low‐density granulocytes are associated with tissue damage in malaria. Cell Rep 2015; 13:2829–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schofield L, Villaquiran J, Ferreira A, Schellekens H, Nussenzweig R, Nussenzweig V. γ Interferon, CD8+ T cells and antibodies required for immunity to malaria sporozoites. Nature 1987; 330:664–6. [DOI] [PubMed] [Google Scholar]
- 69. Zavala F, Gwadz RW, Collins FH, Nussenzweig RS, Nussenzweig V. Monoclonal antibodies to circumsporozoite proteins identify the species of malaria parasite in infected mosquitoes. Nature 1982; 299:737–8. [DOI] [PubMed] [Google Scholar]
- 70. Haque A, Best SE, Amante FH, Ammerdorffer A, de Labastida F, Pereira T et al High parasite burdens cause liver damage in mice following Plasmodium berghei ANKA infection independently of CD8+ T cell‐mediated immune pathology. Infect Immun 2011; 79:1882–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Swanson PA, Hart GT, Russo MV, Nayak D, Yazew T, Peña M et al CD8+ T cells induce fatal brainstem pathology during cerebral malaria via luminal antigen‐specific engagement of brain vasculature. PLoS Pathog 2016; 12:e1006022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Shaw TN, Stewart‐Hutchinson PJ, Strangward P, Dandamudi DB, Coles JA, Villegas‐Mendez A et al Perivascular arrest of CD8+ T cells is a signature of experimental cerebral malaria. PLoS Pathog 2015; 11:e1005210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Soon MSF, Haque A. Recent insights into CD4+ Th cell differentiation in malaria. J Immunol 2018; 200:1965–75. [DOI] [PubMed] [Google Scholar]
- 74. Montes de Oca M, Kumar R, de Labastida Rivera F, Amante FH, Sheel M, Faleiro RJ et al Blimp‐1‐dependent IL‐10 production by Tr1 cells regulates TNF‐mediated tissue pathology. PLoS Pathog 2016; 12:e1005398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Couper KN, Blount DG, Riley EM. IL‐10: the master regulator of immunity to infection. J Immunol 2008; 180:5771–7. [DOI] [PubMed] [Google Scholar]
- 76. Couper KN, Blount DG, Wilson MS, Hafalla JC, Belkaid Y, Kamanaka M et al IL‐10 from CD4CD25Foxp3CD127 adaptive regulatory T cells modulates parasite clearance and pathology during malaria infection. PLoS Pathog 2008; 4:e1000004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Guermonprez P, Helft J, Claser C, Deroubaix S, Karanje H, Gazumyan A et al Inflammatory Flt3 l is essential to mobilize dendritic cells and for T cell responses during Plasmodium infection. Nat Med 2013; 19:730–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ryg‐Cornejo V, Ioannidis LJ, Ly A, Chiu CY, Tellier J, Hill DL et al Severe malaria infections impair germinal center responses by inhibiting t follicular helper cell differentiation. Cell Rep 2016; 14:68–81. [DOI] [PubMed] [Google Scholar]