

Abstract
Background
Conotruncal heart defects (CTDs) are heterogeneous congenital heart malformations that result from outflow tract dysplasia; however, the genetic determinants underlying CTDs remain unclear. Increasing evidence demonstrates that dysfunctional TBX2 and TBX3 result in outflow tract malformations, implying that both of them are involved in CTD pathogenesis. We screened for TBX2 and TBX3 variants in a large cohort of CTD patients (n = 588) and population-matched healthy controls (n = 300) by target sequencing and genetically analyzed the expression and function of these variants.
Results
The probably damaging variants p.R608W, p.T249I, and p.R616Q of TBX2 and p.A192T, p.M65L, and p.A562V of TBX3 were identified in CTD patients, but none in controls. All altered amino acids were highly conserved evolutionarily. Moreover, our data suggested that mRNA and protein expressions of TBX2 and TBX3 variants were altered compared with those of the wild-type. We screened PEA3 and MEF2C as novel downstream genes of TBX2 and TBX3, respectively. Functional analysis revealed that TBX2R608W and TBX2R616Q variant proteins further activated HAS2 promoter but failed to activate PEA3 promoter and that TBX3A192T and TBX3A562V variant proteins showed a reduced transcriptional activity over MEF2C promoter.
Conclusions
Our results indicate that the R608W and R616Q variants of TBX2 as well as the A192T and A562V variants of TBX3 contribute to CTD etiology; this was the first association of variants of TBX2 and TBX3 to CTDs based on a large population.
Electronic supplementary material
The online version of this article (10.1186/s40246-018-0176-0) contains supplementary material, which is available to authorized users.
Keywords: TBX2, TBX3, Variant, Conotruncal heart defects, Target sequencing
Background
Conotruncal heart defects (CTDs) are a group of complex congenital heart malformations with an estimated prevalence of 0.1% of live births and roughly 10–25% in congenital heart defects (CHD) [1] and are caused by abnormal development of the outflow tract (OFT) in embryo or abnormal configuration and arrangement of the ventricle, septal tissue, and large vessels; CTDs include the tetralogy of Fallot (TOF), persistent truncus arteriosus (PTA), double outlet of right ventricle (DORV), transposition of the great arteries (TGA), single atrium (SA), single ventricle (SV), and others. CTDs commonly occur in infants and children and are chief causes of infant death and childhood disability. Most CTD patients require catheter-based or surgical interventions early in life because without intervention, the diseases could lead to poor quality of life with mental and physical retardation, severe cardiac arrhythmias, heart failure, and even sudden death. Although therapeutic regimens have increased survival into adulthood in patients with CTDs, the morbidity and mortality rates remain high in survivors [2, 3]. In addition, CTDs induce heavy economic burdens on society, especially as the survival rates and the number of adults living with CTDs increase [4].
The development of the OFT in the embryonic stage is an elaborate regulatory process, which includes the formation and development of the secondary heart field (SHF) and cardiac neural crest (CNC), during which any abnormal factor of inheritance or environment can lead to abnormal proliferation, differentiation, or migration of SHF and CNC cells, thereby causing CTDs [5–7]. Increasing studies demonstrated that genetic factors played primary roles in pathogenesis of CTDs, but the genetic determinants underlying CTDs remain unclear [8, 9].
In genetics, many transcription factors are recognized as major contributors to normal cardiac morphogenesis, including the T-box family of transcription factors [10]. TBX2 and TBX3 are members of the T-box family of transcription factors that are important for early cardiogenic lineage development as well as formation of chambers and the conduction system [11]. At embryonic day 8–10 mouse heart, Tbx2 was detected in the non-chamber myocardium, which included the atrioventricular canal (AVC), inner curvature, inflow tract, and OFT [12]. Tbx2 expression patterns during chick heart development are consistent with that of mouse [13]. Morphological defects of the heart were observed in Tbx2 knock-out mouse embryos, including abnormal atrioventricular morphology and outflow tract septation defects [14]. Mutations in TBX3 specifically cause ulnar-mammary syndrome [15]. TBX3 is most closely related to TBX2. Tbx3-null mouse embryos have atrioventricular alignment and OFT defects and various kinds of cardiac malformation, such as DORV and TGA. In addition, Tbx3 is involved in multiple signaling pathways that regulate OFT morphogenesis and SHF proliferation [16]. Both Tbx2 and Tbx3 express embryo mesoderm adjacent to CNC and the SHF [16–18]. Accordingly, overexpression/low expression or abnormal function of TBX2 and TBX3 causes heart defects and thus play an important role in heart development.
Current studies for TBX2 and TBX3 have typically focused on gene knock-out animal models to observe phenotype and explore mechanism; however, they have not investigated whether genetic variants were involved in pathogenesis in populations of CTD patients. Only Pang et al. reported variants (g.59477201C>T, g.59477347G>A, g.59477353delG, and g.59477371G>A) located at the TBX2 gene promoter in a cohort of 324 patients with ventricular septal defects, and the variants reduced the transcriptional activities of the TBX2 gene promoter [19]. Therefore, identifying rare variants of TBX2 and TBX3 in large CTD cohort is required urgently.
Here, we show several rare heterozygous variants of TBX2 and TBX3 by target sequencing in a cohort of 588 CTD patients without 22q11.2 deletion, but none in population-matched healthy controls. Our data shows that these variants alter mRNA and protein expression of TBX2 and TBX3. We screen PEA3 and MEF2C as novel downstream target genes of TBX2 and TBX3, respectively. Function analyses reveal that the variants of TBX2 or TBX3 may regulate PEA3, MEF2C, and HAS2 (the known downstream gene of TBX2) promoting CTD incidence, first defining the connection between TBX2/TBX3 variants and CTDs and further elucidating the genetic pathogenesis of CTDs.
Results
The variants of TBX2 and TBX3 identified in CTD patients
We found variants of TBX2 and TBX3 through target sequencing in 588 CTD patients and identified three variants of TBX2 in seven patients and three variants of TBX3 in six patients, including TOF, TGA, SA, and SV (Table 1). The variants of TBX2 were p.R608W, p.T249I, and p.R616Q (Fig. 1b, d, and f), and the variants of TBX3 were p.A192T, p.M65L, and p.A562V (Fig. 2b, d, and f). The p.R608W, p.T249I, and p.R616Q variants of TBX2 and p.A192T and p.M65L variants of TBX3 in both control and case groups were in HWE; the p.A562V variant of TBX3 in control group was in HWE, but in case group, it was not in HWE (Additional file 1: Table S1). However, these variants all led to amino acid substitutions and were predicted to be damaging as per SIFT, Polyphen-2, or Mutation Taster (Table 1).
Table 1.
Clinical information and variant characteristics of TBX2 and TBX3 in patients with CTDs
| Patient | Gender | Age | Cardiac phenotype | Gene | Location in gene | Function | Nucleotide change | Amino acid change | dbSNP ID | SIFT | Mutation taster | PolyPhen-2 | ExAC allele frequency |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 4 months | TOF | TBX2 | exon7 | Missense | 1822C>T | R608W | rs764896880 | 0.01 | Disease causing | 1.000 | 1.726e−05 |
| 2 | F | 8 months | TOF | TBX2 | exon3 | Missense | 746C>T | T249I | rs778075071 | 0.02 | Disease causing | 0.967 | 6.633e−05 |
| 3 | F | 5 months | SV | TBX2 | exon3 | Missense | 746C>T | T249I | rs778075071 | 0.02 | Disease causing | 0.967 | 6.633e−05 |
| 4 | F | 5 years | SV | TBX2 | exon3 | Missense | 746C>T | T249I | rs778075071 | 0.02 | Disease causing | 0.967 | 6.633e−05 |
| 5 | M | 6 months | TOF | TBX2 | exon7 | Missense | 1847G>A | R616Q | rs191930922 | 0.01 | Disease causing | 0.865 | 0.0008162 |
| 6 | F | 3 years | SA | TBX2 | exon7 | Missense | 1847G>A | R616Q | rs191930922 | 0.01 | Disease causing | 0.865 | 0.0008162 |
| 7 | M | 1 year | TGA | TBX3 | exon6 | Missense | 574G>A | A192T | rs768160499 | 0.11 | Disease causing | 1.000 | 2.471e−05 |
| 8 | M | 1 year | TOF | TBX3 | exon1 | Missense | 193A>C | M65L | / | 0.56 | Disease causing | 0.734 | 8.675e−06 |
| 9 | M | 5 months | TOF | TBX3 | exon7 | Missense | 1685C>T | A562V | rs201325654 | 0.1 | Disease causing | 0.849 | 0.002761 |
| 10 | M | 6 months | TOF | TBX3 | exon7 | Missense | 1685C>T | A562V | rs201325654 | 0.1 | Disease causing | 0.849 | 0.002761 |
| 11 | F | 5 months | TOF | TBX3 | exon7 | Missense | 1685C>T | A562V | rs201325654 | 0.1 | Disease causing | 0.849 | 0.002761 |
| 12 | F | 6 months | TOF | TBX3 | exon7 | Missense | 1685C>T | A562V | rs201325654 | 0.1 | Disease causing | 0.849 | 0.002761 |
| 13 | M | 4 years | TGA | TBX3 | exon7 | Missense | 1685C>T | A562V | rs201325654 | 0.1 | Disease causing | 0.849 | 0.002761 |
F female, M male, CTDs conotruncal heart defects, TOF tetralogy of Fallot, SA single atrium, SV single ventricle, TGA transposition of the great arteries
Fig. 1.

Sequence chromatograms of TBX2 missense variants in patients and controls. a, c, and e Chromatograms of normal controls. b, d, and f Chromatograms of the three heterozygous variants. Arrows show heterozygous nucleotide changes
Fig. 2.

Sequence chromatograms of TBX3 missense variants in patients and controls. a, c, and e Chromatograms of normal controls. b, d, and f Chromatograms of the three heterozygous variants. Arrows show heterozygous nucleotide changes
Alignment of multiple TBX2 and TBX3 protein sequences and display of the structure of human TBX2 and TBX3 protein
All variation sites in this study were highly conserved in vertebrates, as shown in multiple TBX2 and TBX3 protein alignments (Fig. 3a, b), indicating that these variants were very important and might result in TBX2 and TBX3 gene function alterations. The human TBX2 spans 3396 bp, and has been mapped to chromosome 17q23, which is composed of seven exons and six introns (20). The T-box DNA-binding domain of TBX2 is located at amino acids 109–287 (Fig. 3c). The human TBX3 mapped to chromosome 12q24, spans 4814 bp and is composed of eight exons and seven introns. The T-box DNA-binding domain of TBX3 is located at amino acids 107–220 and 241–305 (Fig. 3d) (Uniprot: http://www.uniprot.org/).
Fig. 3.

Conservation and distribution of TBX2 and TBX3 variants. a, b Alignments of TBX2 and TBX3 protein among different species. All variants were highly conserved in vertebrates. c, d Diagram of the TBX2 and TBX3 gene exons and protein with location of variants identified in this study
Detection of TBX2 and TBX3 variant expression
To investigate whether the expression of the TBX2 and TBX3 variants were altered, we performed quantitative RT-PCR and Western blot. Quantitative RT-PCR analysis revealed that mRNA expression of R608W, T249I, and R616Q variants of TBX2 (Fig. 4a) and A192T and M65L variants of TBX3 (Fig. 4d) were greater than that of the group of the wild-type plasmid (P < 0.05). On Western blot, protein expression of R608W and R616Q variants (Fig. 4b, c) was distinctly greater than that of the wild-type TBX2 (P < 0.05), consistent with the mRNA expression of these two TBX2 variants; in contrast, protein expression of A192T and A562V variants (Fig. 4e, f) were notably lower than that of the wild-type TBX3 (P < 0.05), indicating that TBX3 variants might lead to protein degradation. Tbx3 is associated with SUMOylation (SUMO, small ubiquitin-related modifier) that may be a conserved mechanism controlling Tbx3 activity [21, 22]. Therefore, we observed the effect of ubiquitin-proteasome degradation and found that the reduction of A192T variant protein expression was rescued after adding the protease inhibitor, suggesting that the A192T variant decreased TBX3 protein stability by ubiquitin-proteasome degradation (Fig. 4g).
Fig. 4.

mRNA abundance, protein expression level, and subcellular localization of TBX2 and TBX3. Plasmids were transfected into HEK 293T cells and harvested. a, d Relative mRNA expression of the blank vector, wild-type plasmid, and variants of TBX2 and TBX3 (n = 3). GAPDH was used as an internal control. b, e Western blot analysis of the blank vector, wild-type plasmid, and variants of TBX2 and TBX3. Actin was used as an internal control. c, f Density quantitation of TBX2 and TBX3 variant protein expression as shown in b and e (n = 3). g Western blot analysis of TBX3 variant protein degradation through the ubiquitin-proteasome pathway. MG-132 was the proteinase inhibitor and DMSO was used as control. h, i Representative images of immunofluorescence staining of TBX2 and TBX3 variants and wild-type proteins. *P < 0.05, **P < 0.01 versus WT; data represented here are obtained from three biological replicates
Nuclear localization of TBX2 and TBX3 variants
To detect the cellular distribution of TBX2 and TBX3 variant proteins, we carried out immunofluorescence assays that demonstrated that all the TBX2 and TBX3 variant proteins were expressed in the nucleus, as were wild-type TBX2 and TBX3 proteins (Fig. 4h, i). The result suggested that these variant proteins might affect TBX2 or TBX3 gene function through other mechanisms.
Screening of downstream target genes of TBX2 and TBX3
We selected several genes related to CTDs or OFT development from previous studies (BMP2 [13], BMP4 [13], FGF8 [23, 24], HAS2 [25], PEA3 [16]), and the MalaCards database (CRELD1, DKK1, FOG2, GATA4, GATA6, HAND2, MEF2C, PLXND1) and then screened the possible downstream genes of TBX2/TBX3 by observing mRNA expression alterations of these target genes after TBX2/TBX3 overexpression (Fig. 5a, b). The results showed that TBX2 activated HAS2 and PEA3, while TBX3 activated MEF2C (P < 0.05), indicating that these were downstream genes of TBX2/TBX3 in CTD incidence.
Fig. 5.
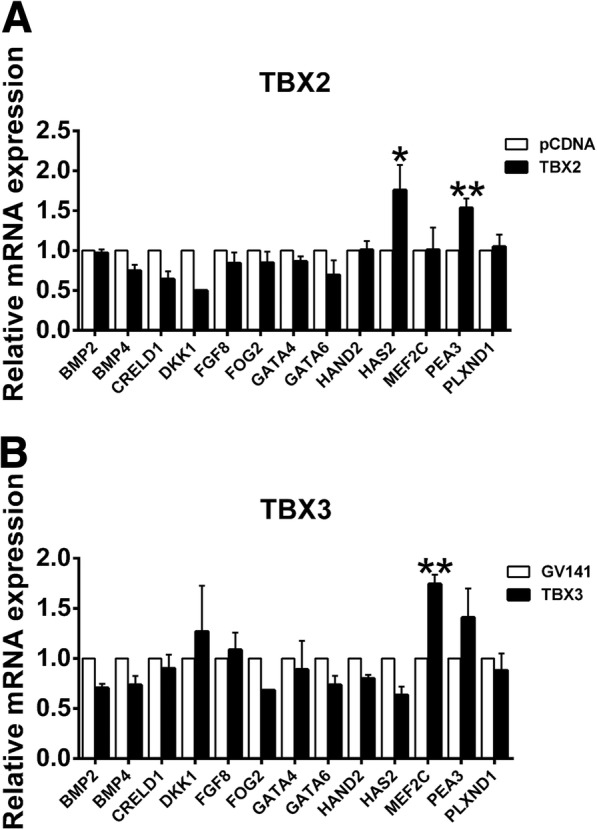
Screening of downstream target genes related to CTDs of TBX2 or TBX3. a, b Relative mRNA expression of candidate downstream target genes affected by wild-type TBX2 or TBX3 transfection (n = 3). GAPDH was used as an internal control. *P < 0.05, **P < 0.01 versus pCDNA or GV141; data represented here are obtained from three biological replicates
Transcriptional activity of TBX2 and TBX3 variant proteins
To evaluate the ability of the TBX2/TBX3 variants to regulate downstream genes, we constructed luciferase reporter genes for human HAS2, MEF2C, and PEA3 promoters selected from the mRNA expression screening and co-transfected them with wild-type or variants of TBX2/TBX3. Compared with wild-type TBX2, R608W and R616Q variant proteins were able to activate HAS2 promoter up to approximately 1.7 times (P < 0.05) and 2.6 times (P < 0.01), respectively (Fig. 6a), whereas the transcriptional activity of R608W and R616Q variant proteins to activate PEA3 promoter reduced by 45% (P < 0.05) and 55% (P < 0.01), respectively (Fig. 6b). Compared with wild-type TBX3, A192T, and A562V variant proteins showed a reduced transcriptional activity over MEF2C promoter (Fig. 6c). Therefore, R608W and R616Q variants of TBX2 and A192T and A562V variants of TBX3 may affect TBX2/TBX3 regulation on downstream target genes through transcriptional activity alteration, thereby promoting CTD incidence.
Fig. 6.

The activity of HAS2, PEA3, and MEF2C luciferase reporter genes regulated by TBX2 or TBX3. HEK 293T cells that were co-transfected with vector or wild-type or variant plasmid and a luciferase reporter; transcriptional activity was measured by a luciferase reporter gene detection system. pRL-TK was used as an internal control. a, b Luciferase activity of HAS2, PEA3 promoter regulated by blank vector, and wild-type and variants of TBX2 (n = 3). c Luciferase activity of MEF2C promoter regulated by blank vector, wild-type, and variants of TBX3 (n = 3). #P < 0.01 versus pCDNA or GV141 and *P < 0.05, **P < 0.01 versus WT; data represented here are obtained from three biological replicates
Expression of TBX3 protein in the human embryo
TBX2 has been reported as being expressed at the AVC and OFT of the heart in animals and human embryos [12]. However, TBX3 expression has not been identified in the human embryo; therefore, we selected human embryos in Carnegie 13 stage, which is the crucial period of the OFT formation to carry out immunohistochemistry. The results showed that TBX3 was expressed in the nucleus in the OFT (Fig. 7c, d), indicating that TBX3 might have a function in the development of the OFT.
Fig. 7.

Immunohistochemistry of TBX3 in human embryos in Carnegie 13 stage. a, b Negative control. c, d Wild-type TBX3. Scale bar 20 μm
We show the illustration summarizing our results and claims (Fig. 8).
Fig. 8.

The diagram of the regulation TBX2 and TBX3 variants involved in CTD pathogenesis
Discussion
Identifying rare variants of TBX2 and TBX3 in a large CTD cohort is required urgently; our study found six rare heterozygous variants of TBX2 and TBX3 in 13 patients from 588 sporadic patients with CTDs using target sequencing that included p.R608W, p.T249I, and p.R616Q variants of TBX2 and p.A192T, p.M65L, and p.A562V variants of TBX3, all of which were all highly conserved based on multiple sequence alignment, suggesting that these variants might have important biological functions. Among these variants, TBX3M65L was the novel variant first reported, and TBX2R608W had not been reported formerly in East Asians. The other four variants were reported in Ensemble (http://ensemblgenomes.org/); their functions had never been studied before. All these variants were predicted as damaging as per SIFT, Mutation Taster or Polyphen-2. Moreover, TBX2R608W, TBX2R616Q, TBX3A192T, and TBX3A562V showed altered expression and function compared with wild-type TBX2/TBX3. Nevertheless, none of the six variants affected the subcellular expression of TBX2 and TBX3 in nucleus, and it is possible that these variants were not located in the critical region that influenced the nuclear distribution of TBX2 and TBX3 [26].
Tbx2 and Tbx3 have common downstream target genes that have been identified as chamber myocardium-specific genes, including connexin 40 (Cx40), connexin 43 (Cx43), and natriuretic precursor peptide type A (Nppa) [27, 28]. Locally repressing these chamber specific genes is required for the formation of non-chamber myocardium and induction of the development of the AVC, inflow tract, and OFT [29, 30]. However, mRNA expression of Cx40, Cx43, and Nppa in HEK293 cells showed no difference after TBX2 or TBX3 overexpression (data not shown) probably due to the limitations of the cell line or the experimental model.
We screened 12 genes selected from previous studies and the MalaCards database (http://www.malacards.org/) as candidates for TBX2 or TBX3 downstream genes and found that mRNA expression of HAS2 and PEA3 in HEK293 cells were elevated substantially after TBX2 overexpression, while TBX3 transfection upregulated MEF2C abundance significantly. The activity of HAS2, MEF2C, and PEA3 luciferase reporter genes was dramatically increased by wild-type TBX2/TBX3 compared with that of blank vectors, providing further evidence that the expression of HAS2, PEA3, and MEF2C was regulated by TBX2 or TBX3. HAS2 has conserved T-box binding sites among promoter regions and encodes hyaluronan synthases 2, the major enzyme responsible for hyaluronan (HA) production in the heart [31], and it is expressed in the OFT [20]. However, excess HA deposition may cause hemodynamic alterations and may block cardiomyocyte differentiation. Tbx2 contributes to the expansion of the extracellular matrix (ECM) and epithelial-mesenchymal transformation (EMT) by inducing Has2 myocardial expression and increasing HA deposition to drive endocardial cushion formation and altered cardiogenic lineage specification in embryonic hearts [25]. Overexpression of HAS2 leading to HA deposition may hinder cardiomyocyte differentiation. We found that TBX2 variants activated HAS2 and caused CTDs and were similar to this report. PEA3 (polyomavirus enhancer activator 3, currently called ETV4, ETS variant 4) is a transcription factor belonging to the PEA3 family and is involved in chromosomal translocation associated with Ewing tumors, whose overexpression promotes cell proliferation, motility, and invasion. The evidences suggest that PEA3 plays a role in cellular proliferation, differentiation, and migration [32, 33]. A previous study reported that Pea3 was associated with Tbx3 involved in regulating OFT morphogenesis [16]. However, in the present study, TBX3 did not affect mRNA expression of PEA3, whereas TBX2 markedly increased PEA3 mRNA abundance and promoter activity; meanwhile, the wild-type TBX2 protein was able to significantly activate its promoter and the TBX2 variant proteins failed to activate its promoter, indicating that PEA3 was a novel downstream gene of TBX2 causing CTDs. TBX2 contributes to oncogenesis and cell cycle regulation [34], analogous to the functions of PEA3 [32, 33]. Therefore, we inferred that TBX2 and PEA3 may engage in crosstalk during cell cycle regulation and that more studies are needed to determine whether they take part in cardiac development through the regulation that these pathways require. MEF2C (MEF2 polypeptide C) is a member of the MADS box transcription enhancer factor 2 (MEF2) family of proteins, which play roles in myogenesis. Previous studies demonstrated that MEF2C is required for proper OFT alignment [35]. Moreover, MEF2C has also been involved in congenital OFT defects in humans [36]. Our results showed that MEF2C was a novel downstream gene of TBX3 and that variants of TBX3 might regulate MEF2C to cause CTDs.
Previous studies reported Tbx2 and Tbx3 were transcriptional repressors [22, 23]. In contrast, TBX2 and TBX3 in our study were activators; we showed that wild-type TBX2 activated the HAS2 gene promoter and wild-type TBX3 activated the MEF2C gene promoter. On the one hand, TBX2 has the capacity to activate a promoter including multiple T-box elements by a weak activation domain located within the T-box [37]. We presumed that the regulating domain of TBX3 resembles that of TBX2. On the other hand, it can also be explained by the presumption that transcriptional repression of TBX2 and TBX3 may depend on the cell line and the primary cardiomyocyte; therefore, our results are limited by the single cell line we used.
Dysfunction of Tbx2 and Tbx3 leading to heart defects has been verified in animals, providing strong evidence that TBX2 and TBX3 are significant for human cardiac morphogenesis and the underlying etiology of CTDs. There were some limitations to our study; for example, the lack of parental samples limited our ability to study the genetic background of these variants. Nevertheless, this study provided the first genetic evidence of an association between malfunctioning TBX2 and TBX3 and CTDs based on a large population, contributing to prenatal diagnosis and prenatal consultation in favor of the early prophylaxis and allele-specific therapy of CTDs.
Conclusions
We found that the variants of TBX2 and TBX3 contributed to the occurrence of CTDs, and we explored the interesting downstream genes of TBX2 and TBX3 to illuminate the mechanisms of CTD etiology. Meanwhile, our findings open new fields of investigation into CTD genetic pathogenesis.
Methods
Study population
Our study population included 588 sporadic nonsyndromic CTD patients diagnosed by echocardiogram, cardiac catheterization, or surgery from Shanghai Xin Hua Hospital. The participants are from the Chinese Han population and included 388 males and 200 females, with ages ranging from 0.1 to 17 years (Table 2). Patients with known syndromes or chromosomal abnormalities, such as 22q11.2 deletion, were excluded from our study. The controls were 300 healthy children without heart disease. Both patient and control groups gave informed consent for inclusion, and then, peripheral blood was collected for DNA extraction. The genomic DNA of participants was extracted by using the QIAamp DNA Blood Mini Kit (QIAGEN, Germany) following the manufacturer’s instructions and was then stored at − 80 °C.
Table 2.
Cardiac diagnoses for study population of patients with CTDs
| Diagnoses | Number | Gender/number | Age |
|---|---|---|---|
| TGA | 90 | F18M72 | 1 day–16 years |
| TOF | 234 | F83M151 | 1 month–13 years |
| DORV | 98 | F32M66 | 1 month–17 years |
| PA/VSD | 97 | F36M61 | 3 months–12 years |
| IAA | 13 | F7M6 | 7 days–1 year |
| PTA | 10 | F5M5 | 3 days–2 years |
| SA/SV | 46 | F19M27 | 1 month–13 years |
| Total | 588 | F200M388 | 1 day–17 years |
TGA transposition of the great arteries, TOF tetralogy of Fallot, DORV double outlet of right ventricle, PA/VSD pulmonary atresia with ventricular septal defect, IAA interruption of aortic arch, PTA persistent truncus arteriosus, SA single atrium, SV single ventricle, F female, M male
Target sequencing and variant analysis
Target sequencing was performed using the Illumina HiSeq 2000 platform for variants in TBX2 (GenBank accession number NC_000017.11, NM_005994.3) and TBX3 (GenBank accession number NC_000012.12, NM_016569.3). The candidate variants were validated by Sanger sequencing, and the primers were designed for PCR amplification of TBX2 and TBX3. To predict the effects of nonsynonymous variants, we used several bioinformatics criteria including SIFT (http://sift.jcvi.org/www/SIFT_enst_submit.html), Mutation Taster (http://www.mutationtaster.org/), and Polyphen-2 (http://genetics.bwh.havard.edu/pph2/). Amino acid substitutions were predicted as damaging when the score was ≤ 0.05 in SIFT or ≥ 0.85 in Polyphen-2. In our study, variants with a minor allele frequency (MAF) < 0.5% were defined as rare [38].
Multiple TBX2 and TBX3 protein sequence alignment
TBX2 and TBX3 protein sequences from Homo sapiens (human), Mus musculus (house mouse), Gallus gallus (chicken), Bos taurus (cattle), Canis lupus familiaris (dog), Pan troglodytes (chimpanzee), and Sus scrofa (pig) were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/protein/) and were aligned with ClustalX software to confirm the conservation of TBX2 and TBX3 sequences.
Plasmid construction and site-directed mutagenesis
The TBX2 and TBX3 cDNA plasmid was purchased from Genomeditech. Mutated primers were designed to amplify human TBX2 and TBX3 cDNA according to the protocol provided by the QuikChange SiteDirected Mutagenesis Kit (Stratagene, USA), and then, variant TBX2 cDNAs were cloned into a pCDNA3.1-3xFlag vectors while variant TBX3 cDNAs were cloned into GV141-3xFlag vectors. For recombining luciferase reporter plasmid, a 5′-flanking region of downstream gene promoter was subcloned into Kpn I and Bgl II sites of the pGL3 luciferase reporter-basic vector (Promega, USA).
Cell cultures and transfection
HEK 293T cells (Human embryonic kidney cells) were maintained in Dulbecco’s modified Eagle’s medium (HyClone, USA) with 10% fetal bovine serum (MP Biomedicals, USA) and 1% penicillin-streptomycin (Gibco, USA). pcDNA3.1-3xFlag-TBX2 and GV141-3xFlag-TBX3 including wild-type and variants were transfected into 293T cells with FuGene HD (Promega, USA) according to the manufacturer’s protocol after seeding 24 h.
Quantitative RT-PCR
Plasmids were transfected into HEK 293T cells that were seeded in 12-well plates. Cells were harvested 36 h after transfection. Total RNA was extracted with TRIzol reagent (Invitrogen, USA), and then, reverse transcription of cDNA was performed using Prime Script RT Master Mix (Takara, Japan) and was followed by quantitative RT-PCR using SYBR Premix Ex Taq (Takara, Japan) on an Applied Biosystems 7500 system (Applied Biosystems, USA). The relative quantification of expression was determined using the 2^-△△Ct method [39], and glyceraldehyde-3-phosphate dehydrogenase (GAPDH, human) was used as an internal control. Primer sequences of TBX2, TBX3, GAPDH, and candidate downstream genes are listed in Table 3.
Table 3.
Sequences of the primers used for real-time quantitative PCR
| Gene | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| GAPDH | GGAGCGAGATCCCTCCAAAAT | GGCTGTTGTCATACTTCTCATGG |
| TBX2 | CACGGCTTCACCATCCTAAAC | TGCGGAAGGTGCTGTAAGG |
| TBX3 | GAGGCTAAAGAACTTTGGGATCA | CATTTCGGGGTCGGCCTTA |
| BMP2 | GAGGTCCTGAGCGAGTTCGA | ACCTGAGTGCCTGCGATACA |
| BMP4 | ATGATTCCTGGTAACCGAATGC | CCCCGTCTCAGGTATCAAACT |
| CRELD1 | GCTCCTATGAGTGCCGAGAC | CTACACTTCTTACAGCGACCTG |
| DKK1 | ATAGCACCTTGGATGGGTATTCC | CTGATGACCGGAGACAAACAG |
| FGF8 | GACCCCTTCGCAAAGCTCAT | CCGTTGCTCTTGGCGATCA |
| FOG2 | GGCCTGAAAATCTGAGCTGC | CAGTCGTCTGTCTCAACTCCA |
| GATA4 | CGACACCCCAATCTCGATATG | GTTGCACAGATAGTGACCCGT |
| GATA6 | CTCAGTTCCTACGCTTCGCAT | GTCGAGGTCAGTGAACAGCA |
| HAND2 | CGCCGACACCAAACTCTCC | TCGCCATTCTGGTCGTCCT |
| HAS2 | CTCATCATCCAAAGCCTGTT | GCTGGGTCAAGCATAGTGTC |
| MEF2C | CCAACTTCGAGATGCCAGTCT | GTCGATGTGTTACACCAGGAG |
| PLXND1 | AATGGGCGGAACATCGTCAAG | CGAGACTGGTTGGAAACACAG |
| PEA3 | GAGAAACCTCTGCGACCATT | GCCCGTCCAGGCAATGAAAT |
Western blot
HEK 293T cells were transfected with 1 μg of wild-type and variant plasmid DNA. Cells were harvested 48 h after transfection. For protein degradation experiments, HEK 293T cells were cultured in the presence of 10 mM MG132 solution dissolved in DMSO for 8–10 h before harvest. Then, cells were lysed in RIPA lysis buffer (Beyotime, China) with PMSF (1:100). The proteins were subjected to 10% SDS-PAGE and were then transferred onto nitrocellulose membranes (Millipore, USA) and immunostained with rabbit anti-FLAG antibody (1:1000, Sigma-Aldrich, USA) and mouse anti-β-actin antibody (1:5000, Sigma-Aldrich, USA) at 4 °C overnight. The membranes were incubated with horseradish peroxidase-conjugated anti-rabbit secondary antibody (1:10000) and anti-mouse secondary antibody (1:10000). Immobilon Western Chemiluminescent HRP Substrate (Millipore, USA) was used for chemiluminescent immunodetection.
Luciferase assays
HEK 293T cells were transfected with 200 ng of wild-type or variant plasmid DNA, 200 ng of luciferase reporter plasmid, and 8 ng of an internal control reporter plasmid (pRL-TK) (Promega, USA) in a 48-well plate. The luciferase activity was measured on the Dual-Glo luciferase assay system (Promega, USA) following the manufacturer’s protocol after 48 h.
Immunofluorescence assay
HEK 293T cells were seeded onto a 24-well plate covered with slips coated with poly-L-lysine (0.1 mg/mL) for 24 h and were then transfected with wild-type or variant plasmid DNA. Cells were harvested 24 h after transfection. Cells were incubated with rabbit anti-Flag antibody (1:100, Sigma-Aldrich, USA) diluted in PBS containing 5% BSA and 0.1% Triton X-100 at 4 °C overnight and followed by incubation with Cy3-conjugated goat anti-rabbit secondary antibody (1:250). Cell nuclei were stained by 4,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, USA). A Leica SP8 microscope was used for image analysis.
Tissue collection and immunohistochemistry
Human embryos of Carnegie 13 stage were acquired after medical termination of pregnancy at Shanghai Xin Hua Hospital. The medical ethics committee of Xin Hua Hospital approved the study. Embryos were fixed overnight in 4% paraformaldehyde in PBS, embedded in paraffin, and sectioned at a thickness of 7 μm. For immunolocalization of TBX3, paraffin sections were incubated with a primary rabbit anti-TBX3 antibody (1:50, Protein-tech), followed by horseradish peroxidase-conjugated secondary anti-rabbit antibody and DAB (Abcam, UK).
Statistical analysis
Each assay was performed for three independent biological replicates. The data are shown as the mean ± standard deviation (SD). Statistical differences were evaluated by one-way ANOVA and two-tailed unpaired t test. A P value < 0.05 was considered statistically significant.
Additional file
Table S1. Frequencies of alleles and genotypes of TBX2 and TBX3 variants in CTD patients and controls. (DOCX 19 kb)
Acknowledgements
We are grateful to all subjects who participated in this study.
Funding
This study received financial supports from the National Natural Science Foundation of China (81670285), the key international (regional) cooperation projects of the National Natural Science Foundation of China (81720108003), the multi-center clinical research project of Shanghai Jiao Tong University (DLY201609), and the public health system construction of Shanghai Municipal Commission of Health and Family Planning (GWIV-23), the Experts Recruitment Program of Xinhua Hospital (Re-013).
Availability of data and materials
The datasets analyzed during the current study are available from the corresponding author with a reasonable request.
Abbreviations
- AVC
Atrioventricular canal
- CNC
Cardiac neural crest
- CTDs
Conotruncal heart defects
- HA
Hyaluronan
- OFT
Outflow tract
- SHF
Secondary heart field
- TBX2/3
T-box 2/3 [Homo sapiens (human)]
- Tbx2/3
T-box 2/3 [Mus musculus (house mouse)]
Authors’ contributions
KS, YY, and HX conceived and designed the experiments; HX, EZ, and NH performed the experiments; HX, EZ, YY, and KS analyzed the data; QF, FL, and SC collected the blood samples from all subjects. HX wrote the first draft of the manuscript. YY and KS revised the manuscript. All authors contributed to writing this paper and approved the final manuscript.
Ethics approval and consent to participate
Written informed consent was obtained from both patient and control group participants. The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of Xin Hua Hospital.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Huilin Xie, Email: xiehuilin_irene@foxmail.com.
Erge Zhang, Email: iriserge@sjtu.edu.cn.
Nanchao Hong, Email: hncbaogu@126.com.
Qihua Fu, Email: qfu@shsmu.edu.cn.
Fen Li, Email: lifen_88@aliyun.com.
Sun Chen, Email: chensun@xinhuamed.com.cn.
Yu Yu, Email: yuyu@xinhuamed.com.cn.
Kun Sun, Email: sunkun@xinhuamed.com.cn.
References
- 1.Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39(12):1890–1900. doi: 10.1016/S0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- 2.van der Bom T, Zomer AC, Zwinderman AH, Meijboom FJ, Bouma BJ, Mulder BJ. The changing epidemiology of congenital heart disease. Nat Rev Cardiol. 2011;8(1):50–60. doi: 10.1038/nrcardio.2010.166. [DOI] [PubMed] [Google Scholar]
- 3.Tutarel O, Kempny A, Alonso-Gonzalez R, Jabbour R, Li W, Uebing A, Dimopoulos K, Swan L, Gatzoulis MA, Diller GP. Congenital heart disease beyond the age of 60: emergence of a new population with high resource utilization, high morbidity, and high mortality. Eur Heart J. 2014;35(11):725–732. doi: 10.1093/eurheartj/eht257. [DOI] [PubMed] [Google Scholar]
- 4.Verheugt CL, Uiterwaal CS, van der Velde ET, Meijboom FJ, Pieper PG, Sieswerda GT, Plokker HW, Grobbee DE, Mulder BJ. The emerging burden of hospital admissions of adults with congenital heart disease. Heart. 2010;96(11):872–878. doi: 10.1136/hrt.2009.185595. [DOI] [PubMed] [Google Scholar]
- 5.Hutson MR, Kirby ML. Model systems for the study of heart development and disease. Cardiac neural crest and conotruncal malformations. Semin Cell Dev Biol. 2007;18(1):101–110. doi: 10.1016/j.semcdb.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ward C, Stadt H, Hutson M, Kirby ML. Ablation of the secondary heart field leads to tetralogy of Fallot and pulmonary atresia. Dev Biol. 2005;284(1):72–83. doi: 10.1016/j.ydbio.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 7.Waldo KL, Hutson MR, Ward CC, Zdanowicz M, Stadt HA, Kumiski D, Abu-Issa R, Kirby ML. Secondary heart field contributes myocardium and smooth muscle to the arterial pole of the developing heart. Dev Biol. 2005;281(1):78–90. doi: 10.1016/j.ydbio.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 8.Jenkins KJ, Correa A, Feinstein JA, Botto L, Britt AE, Daniels SR, Elixson M, Warnes CA, Webb CL. Noninherited risk factors and congenital cardiovascular defects: current knowledge: a scientific statement from the American Heart Association Council on Cardiovascular Disease in the Yong: endorsed by the American Academy of Pediatrics. Circulation. 2007;115(23):2995–3014. doi: 10.1161/CIRCULATIONAHA.106.183216. [DOI] [PubMed] [Google Scholar]
- 9.Cooper WO, Hernandez-Diaz S, Arboast PG, Dudley JA, Deyer S, Giddeon PS, Hall K, Ray WA. Major congenital malformations after first-trimester exposure to ACE inhibitors. N Engl J Med. 2006;354(23):2443–2451. doi: 10.1056/NEJMoa055202. [DOI] [PubMed] [Google Scholar]
- 10.Meganathan K, Sotiriadou I, Natarajan K, Hescheler J, Sachinidis A. Signaling molecules, transcription growth factors and other regulators revealed from in-vivo and in-vitro models for the regulation of cardiac development. Int J Cardiol. 2015;183:117–128. doi: 10.1016/j.ijcard.2015.01.049. [DOI] [PubMed] [Google Scholar]
- 11.Plageman TF, Jr, Yutzey KE. T-box genes and heart development: putting the “T” in heart. Dev Dyn. 2005;232(1):11–20. doi: 10.1002/dvdy.20201. [DOI] [PubMed] [Google Scholar]
- 12.Christoffels VM, Hoogaars WM, Tessari A, Clout DE, Moorman AF, Campione M. T-box transcription factor Tbx2 represses differentiation and formation of the cardiac chambers. Dev Dyn. 2004;229(4):763–770. doi: 10.1002/dvdy.10487. [DOI] [PubMed] [Google Scholar]
- 13.Yamada M, Revelli JP, Eichele G, Barron M, Schwartz RJ. Expression of chick Tbx-2, Tbx-3, and Tbx-5 genes during early heart development: evidence for BMP2 induction of Tbx2. Dev Biol. 2000;228(1):95–105. doi: 10.1006/dbio.2000.9927. [DOI] [PubMed] [Google Scholar]
- 14.Harrelson Z, Kelly RG, Goldin SN, Gibson-Brown JJ, Bollag RJ, Silver LM, Papaioannou VE. Tbx2 is essential for patterning the atrioventricular canal and for morphogenesis of the outflow tract during heart development. Development. 2004;131(20):5041–5052. doi: 10.1242/dev.01378. [DOI] [PubMed] [Google Scholar]
- 15.Bamshad M, Lin RC, Law DJ, Watkins WC, Krakowiak PA, Moore ME, Franceschini P, Lala R, Holmes LB, Gebuhr TC, Bruneau BG, Schinzel A, Seidman JG, Seidman CE, Jorde LB. Mutations in human TBX3 alter limb, apocrine and genital development in ulnar-mammary syndrome. Nat Genet. 1997;16(3):311–315. doi: 10.1038/ng0797-311. [DOI] [PubMed] [Google Scholar]
- 16.Mesbah K, Harrelson Z, Theveniau-Ruissy M, Papaioannou VE, Kelly RG. Tbx3 is required for outflow tract development. Circ Res. 2008;103(7):743–750. doi: 10.1161/CIRCRESAHA.108.172858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dupays L, Kotecha S, Angst B, Mohun TJ. Tbx2 misexpression impairs deployment of second heart field derived progenitor cells to the arterial pole of the embryonic heart. Dev Biol. 2009;333(1):121–131. doi: 10.1016/j.ydbio.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 18.Mesbah K, Rana MS, Francou A, van Duijvenboden K, Papaioannou VE, Moorman AF, Kelly RG, Christoffels VM. Identification of a Tbx1/Tbx2/Tbx3 genetic pathway governing pharyngeal and arterial pole morphogenesis. Hum Mol Genet. 2012;21(6):1217–1229. doi: 10.1093/hmg/ddr553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pang S, Liu Y, Zhao Z, Huang W, Chen D, Yan B. Novel and functional sequence variants within the TBX2 gene promoter in ventricular septal defects. Biochimie. 2013;95(9):1087–1089. doi: 10.1016/j.biochi.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 20.Campbell C, Goodrich K, Casey G, Beatty B. Cloning and mapping of a human gene (TBX2) sharing a highly conserved protein motif with the Drosophila omb gene. Genomics. 1995;28(2):255–260. doi: 10.1006/geno.1995.1139. [DOI] [PubMed] [Google Scholar]
- 21.Crum TL, Okkema PG. SUMOylation-dependent function of a T-box transcriptional repressor in Caenorhabditis elegans. Biochem Soc Trans. 2007;35(6):1424–1426. doi: 10.1042/BST0351424. [DOI] [PubMed] [Google Scholar]
- 22.Huber P, Crum T, Clary LM, Ronan T, Packard AV, Okkema PG. Function of the C. elegans T-box factor TBX-2 depends on SUMOylation. Cell Mol Life Sci. 2013;70(21):4157–4168. doi: 10.1007/s00018-013-1336-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Firnberg N, Neubuser A. FGF signaling regulates expression of Tbx2, Erm, Pea3, and Pax3 in the early nasal region. Dev Biol. 2002;247(2):237–250. doi: 10.1006/dbio.2002.0696. [DOI] [PubMed] [Google Scholar]
- 24.Ilagan R, Abu-Issa R, Brown D, Yang YP, Jiao K, Schwartz RJ, Klingensmith J, Meyers EN. Fgf8 is required for anterior heart field development. Development. 2006;133(12):2435–2445. doi: 10.1242/dev.02408. [DOI] [PubMed] [Google Scholar]
- 25.Shirai M, Imanaka-Yoshida K, Schneider MD, Schwartz RJ, Morisaki T. T-box 2, a mediator of Bmp-Smad signaling, induced hyaluronan synthase 2 and Tgfbeta2 expression and endocardial cushion formation. Proc Natl Acad Sci U S A. 2009;106(44):18604–18609. doi: 10.1073/pnas.0900635106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang YQ, Gharibeh L, Li RG, Xin YF, Wang J, Liu ZM, Qiu XB, Xu YJ, Xu L, Qu XK, Liu X, Fang WY, Huang RT, Xue S, Nemer G. GATA4 loss-of-function mutations underlie familial tetralogy of Fallot. Hum Mutat. 2013;34(12):1662–1671. doi: 10.1002/humu.22434. [DOI] [PubMed] [Google Scholar]
- 27.Hoogaars WM, Tessari A, Moorman AF, de Boer PA, Hagoort J, Soufan AT, Campione M, Christoffels VM. The transcriptional repressor Tbx3 delineates the developing central conduction system of the heart. Cardiovasc Res. 2004;62(3):489–499. doi: 10.1016/j.cardiores.2004.01.030. [DOI] [PubMed] [Google Scholar]
- 28.Habets PE, Moorman AF, Clout DE, van Roon MA, Lingbeek M, van Lohuizen M, Campione M, Christoffels VM. Cooperative action of Tbx2 and Nkx2.5 inhibits ANF expression in the atrioventricular canal: implications for cardiac chamber formation. Genes Dev. 2002;16(10):1234–1246. doi: 10.1101/gad.222902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ribeiro I, Kawakami Y, Büscher D, Raya A, Rodríguez-León J, Morita M, Rodríguez Esteban C, Izpisúa Belmonte JC. Tbx2 and Tbx3 regulate the dynamics of cell proliferation during heart remodeling. PLoS One. 2007;2(4):e398. doi: 10.1371/journal.pone.0000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh R, Hoogaars WM, Barnett P, Grieskamp T, Rana MS, Buermans H, Farin HF, Petry M, Heallen T, Martin JF, Moorman AF, ‘t Hoen PA, Kispert A, Christoffels VM. Tbx2 and Tbx3 induce atrioventricular myocardial development and endocardial cushion formation. Cell Mol Life Sci. 2012;69(8):1377–1389. doi: 10.1007/s00018-011-0884-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Camenisch TD, Spicer AP, Brehm-Gibson T, Biesterfeldt J, Augustine ML, Calabro A, Jr, Kubalak S, Klewer SE, McDonald JA. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J Clin Invest. 2000;106(3):349–360. doi: 10.1172/JCI10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akagi T, Kuure S, Uranishi K, Koide H, Costantini F, Yokota T. ETS-related transcription factors ETV4 and ETV5 are involved in proliferation and induction of differentiation-associated genes in embryonic stem (ES) cells. J Biol Chem. 2015;290(37):22460–22473. doi: 10.1074/jbc.M115.675595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oh S, Shin S, Janknecht R. ETV1, 4 and 5: an oncogenic subfamily of ETS transcription factors. Biochim Biophys Acta. 2012;1826(1):1–12. doi: 10.1016/j.bbcan.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sinclair CS, Adem C, Naderi A, Soderberg CL, Johnson M, Wu K, Wadum L, Couch VL, Sellers TA, Schaid D, Slezak J, Fredericksen Z, Ingle JN, Hartmann L, Jenkins RB, Couch FJ. TBX2 is preferentially amplified in BRCA1- and BRCA2-related breast tumors. Cancer Res. 2002;62(13):3587–3591. [PubMed] [Google Scholar]
- 35.Barnes RM, Harris IS, Jaehnig EJ, Sauls K, Sinha T, Rojas A, Schachterle W, McCulley DJ, Norris RA, Black BL. MEF2C regulates outflow tract alignment and transcriptional control of Tdgf1. Development. 2016;143(5):774–779. doi: 10.1242/dev.126383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kodo K, Nishizawa T, Furutani M, Arai S, Ishihara K, Oda M, Makino S, Fukuda K, Takahashi T, Matsuoka R, Nakanishi T, Yamagishi H. Genetic analysis of essential cardiac transcription factors in 256 patients with non-syndromic congenital heart defects. Circ J. 2012;76(7):1703–1711. doi: 10.1253/circj.CJ-11-1389. [DOI] [PubMed] [Google Scholar]
- 37.Paxton C, Zhao H, Chin Y, Langner K, Reecy J. Murine Tbx2 contains domains that activate and repress gene transcription. Gene. 2002;283(1–2):117–124. doi: 10.1016/S0378-1119(01)00878-2. [DOI] [PubMed] [Google Scholar]
- 38.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Frequencies of alleles and genotypes of TBX2 and TBX3 variants in CTD patients and controls. (DOCX 19 kb)
Data Availability Statement
The datasets analyzed during the current study are available from the corresponding author with a reasonable request.