

Abstract
It is generally recognized that hepatic fibrogenesis is an end result of increased extracellular matrix (ECM) production from the activation and proliferation of hepatic stellate cells (HSCs). An in-depth understanding of the mechanisms of HSC necroptosis might provide a new therapeutic strategy for prevention and treatment of hepatic fibrosis. In this study, we attempted to investigate the effect of curcumol on necroptosis in HSCs, and further to explore the molecular mechanisms. We found that curcumol ameliorated the carbon tetrachloride (CCl4)-induced mice liver fibrosis and suppressed HSC proliferation and activation, which was associated with regulating HSC necroptosis through increasing the phosphorylation of receptor-interacting protein kinase 1 (RIPK1), receptor-interacting protein kinase 3 (RIPK3). Moreover, curcumol promoted the migration of RIPK1 and RIPK3 into necrosome in HSCs. RIPK3 depletion impaired the anti-fibrotic effect of curcumol. Importantly, we showed that curcumol-induced RIPK3 up-regulation significantly increased mitochondrial reactive oxygen species (ROS) production and mitochondrial depolarization. ROS scavenger, N-acetyl-L-cysteine (NAC) impaired RIPK3-mediated necroptosis. In addition, our study also identified that the activation of c-Jun N-terminal kinase1/2 (JNK1/2) was regulated by RIPK3, which mediated curcumol-induced ROS production. Down-regulation of RIPK3 expression, using siRIPK3, markedly abrogated JNK1/2 expression. The use of specific JNK1/2 inhibitor (SP600125) resulted in the suppression of curcumol-induced ROS production and mitochondrial depolarization, which in turn, contributed to the inhibition of curcumol-triggered necroptosis. In summary, our study results reveal the molecular mechanism of curcumol-induced HSC necroptosis, and suggest a potential clinical use of curcumol-targeted RIPK1/RIPK3 complex-dependent necroptosis via JNK1/2-ROS signaling for the treatment of hepatic fibrosis.
Keywords: Curcumol, Hepatic stellate cell, Liver fibrosis, Necroptosis, Receptor-interacting protein kinase, ROS
Graphical abstract
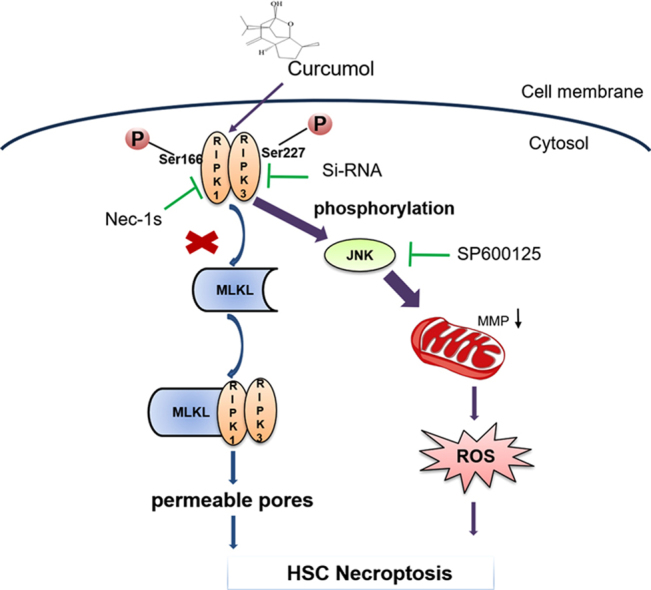
Highlights
-
•
Curcumol exerted anti-hepatic fibrogenesis effects in CCl4-treated mice.
-
•
Curcumol inhibited the activation of hepatic stellate cell in vitro.
-
•
Curcumol promoted the generation of RIPK1/RIPK3-complex to induce hepatic stellate cell necroptosis.
-
•
Curcumol modulated RIPK3/JNK/ROS signaling axis.
1. Introduction
Hepatic fibrosis caused by multiple chronic liver injuries, is a known contributor to cirrhosis, and even liver cancer [1], [2]. This scarring process starts with activation and proliferation of hepatic stellate cells (HSCs). Activated HSCs trans-differentiate into myofibroblasts during liver fibrosis, leading to the secretion and deposition of extracellular matrix (ECM) components [3], [4]. A growing evidence has shown that hepatic fibrosis is reversible [5], [6], [7]. The elimination of activated HSCs through cell death, including apoptosis, senescence, autophagy has been regarded as an effective antifibrogenic strategy [8], [9], [10]. We previously reported that HSC senescence could enhance immune surveillance, inhibit ECM components production, and consequently improve liver fibrosis [11]. Our recent study showed that the inhibition of autophagy in activated HSCs restored lipocyte phenotype, which was beneficial for the reverse of hepatic fibrosis [12]. Recent studies have highlighted a new model of programmed cell death, necroptosis, which is closely involved in liver disease including hepatocellular carcinoma (HCC), alcoholic fatty liver disease, and non-alcoholic fatty liver disease [13], [14], [15]. Investigations on necroptosis in liver fibrosis, however, are rarely performed. Until recently, only one published study showed that gallic acid could trigger necroptosis in activated HSCs [16]. In the current study, we intend to evaluate the role of necroptosis in liver fibrosis and further to explore the underlying molecular mechanisms.
Necroptosis is characterized as the cell death with the similar morphology as necrosis and the unique upstream signal pathway just as apoptosis [17]. Necroptosis may serve as an alternate pathway to enable cell death when apoptosis is restrained. Receptor-interacting protein kinase 1 and 3 (RIPK1 and RIPK3) are regarded as central regulators for initiating necroptosis [18], [19]. Activated RIPK1 binds to RIPK3, generating the necrosome complex. Necrosome could recruit and promote mixed lineage kinase domain-like (MLKL) phosphorylation [20]. Then, the activated MLKL oligomerizes and binds to membrane phospholipids, promoting the formation of pores that cause necroptotic cell death [21]. Recently, growing evidence has showed that reactive oxygen species (ROS) could change mitochondrial permeability, eventually leading to necroptosis [22]. However, it is still unknown whether the programmed necrosis ultimately result in cell death through the mitochondrial ROS pathway or the permeable pores induced by MLKL in some certain cells [23]. Moreover, the roles of RIPK1 and RIPK3 remain unclear in regulating ROS-mediated necroptosis. We previously reported that ROS-JNK1/2-induced autophagy in activated HSCs ameliorated inflammatory microenvironment [24]. It is interesting to explore whether ROS generation contributes to HSC necroptosis.
It is well-known that intracellular ROS could regulate mitogen activated protein kinases (MAPKs), including c-Jun N-terminal kinase1/2 (JNK1/2), extracellular regulated kinase1/2 (ERK1/2), and p38, which are the critical kinases that participate in numerous biological process, such as apoptosis, autophagy, and cell survival [25], [26], [27]. Meanwhile, ROS is vital for ferroptosis, a newly discovered type of regulated cell death [28]. Interestingly, recent study reported JNK activation could contribute to intracellular ROS production, promoting poly (ADP-ribose) polymerase-1 (PARP-1) dependent cell death (parthanatos) in glioma cells [29]. Besides, JNK could be phosphorylated by RIPK3, and activated JNK might contribute to necrosis via advancing the generation of intracellular ROS in hepatocytes [30]. These discoveries show that the JNK/ROS signaling pathway is important for cell survival. Thus, whether RIPK3/JNK/ROS signaling pathway involves in HSC necroptosis is worth further exploring.
Curcumol, a guaiane-type sesquiterpenoid hemiketal extracted from the roots of the herb Rhizoma Curcumae, exhibits multiple-pharmacological activities, including anti-inflammatory, and anti-tumor effect [31], [32]. A previous study reported that curcumol induced HSC-T6 cell death [33], but no major research of curcumol on liver fibrosis has been done. In the present study, we are the first to evaluated the effect of curcumol on protecting the liver from carbon tetrachloride (CCl4)-induced injury and fibrogenesis. Importantly, we verify that curcumol-induced RIPK1/RIPK3 complex promoted ROS production in HSCs via activating JNK1/2, thus triggering HSC necroptosis. Our study shows that curcumol can be a potential chemotherapeutic agent for the treatment of liver fibrosis.
2. Materials and methods
2.1. Chemicals and reagents
Curcumol, N-acetyl cysteine (NAC), N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (Z-VAD-FMK), necrotatin-1 (Nec-1), and SP600125 were brought from Sigma-Aldrich (St Louis, MO, USA). Dulbecco's modified essential medium (DMEM), fetal bovine serum (FBS), Opti MEM medium, phosphate buffered saline (PBS), and trypsin-EDTA were purchased from GIBCO BRL (Grand Island, NY, USA). Oxidation sensitive 2′,7′-dichlorodihydrofluorescin diacetate (DCFH-DA) and 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) were obtained by Beyotime Biotechnology (Shanghai, China). MitoSox Red was brought from Thermo Fisher Scientific (Waltham, MA, USA). Anti-rabbit IgG, anti-mouse IgG and antibodies against β-actin, and RIPK1 (17519-1-AP) were purchased from Proteintech Group, Inc. (Rosemont, IL, USA). Main antibodies against α-SMA, α1(I) procollagen, EGFR, fibronectin, MLKL, PDGF-βR, p-MLKL, p-RIPK3, RIPK3, and TGF-βRII were obtained from Abcam Technology (Abcam, Cambridge, UK). Primary antibodies against caspase3, cleaved-caspase3, caspase8, cleaved-caspase8, caspase9, cleaved-caspase9, ERK, JNK, p38, p-ERK, p-JNK, p-p38, p-RIPK1 and TIMP2 were procured by Cell Signaling Technology (Danvers, MA, USA). Lentivirus vectors encoding negative control shRNA (NC shRNA) and RIPK3 shRNA were designed by Hanbio (Shanghai, China).
2.2. Animal procedures and treatments
All experimental procedures received the approval of the institutional and local committee on the care and use of animals in Nanjing University of Chinese Medicine (Nanjing, China). The whole animals were given humane care according to the National Institutes of Health guidelines. Male ICR mice weighing about 18–22 g were purchased from Nanjing Medical University (Nanjing, China). A mixture of CCl4 (0.1 ml/20 g body weight) and olive oil (1:9 (v/v)) was utilized to trigger hepatic fibrosis in mice by intraperitoneal injection. Male ICR mice were randomly divided into seven groups (eight mice per group). Mice in groups 1–7 were correspondingly given the following treatments: group 1, negative control (NC) shRNA lentivirus and olive oil; group 2, NC shRNA lentivirus, CCl4, and olive oil; Mice in groups 3–5, NC shRNA lentivirus, CCl4, olive oil, and curcumol with 15, 30 and 60 mg kg−1, respectively, serving as treatment groups; group 6, RIPK3 shRNA lentivirus, CCl4, and olive oil; group 7, RIPK3 shRNA lentivirus, CCl4, olive oil and curcumol (30 mg kg−1). Mice in groups 2–7 were intraperitoneally injected with CCl4 every day for 8 weeks. Curcumol was dissolved in olive oil and given every other day via intraperitoneal injection during the 5–8 weeks period. Lentivirus with a titer of 2.5 × 107 pfu/g was injected into caudal vein of mice once per 2 weeks. At the end of the experiment, all mice were anesthetized with an injection of pentobarbital (50 mg kg−1), and then sacrificed. Blood was gathered for biochemical indicators and ELISA assay. A partial liver was removed for histopathological, immunofluorescence, and immunohistochemical studies by fixing with 10% formalin and subsequently embedded with paraffin. The rest of liver was cut into pieces and immediately frozen with liquid nitrogen for extracting total RNA and proteins.
2.3. Liver histopathology and immunohistochemistry
Hematoxylin and eosin (H&E), Masson and Sirius Red staining were conducted according to the previously established methods. Immunohistochemical staining was performed using antibodies against CD45, F4/80, and α-SMA as previously described [11]. Photographs were taken in a blinded fashion at random fields. Representative pictures of liver sections were displayed.
2.4. Cell culture
Primary HSCs were isolated from male ICR mice (Nanjing Medical University, Nanjing, China) as previously described [29]. Human HSC-LX2 cell line was brought from Cell Bank of Chinese Academy of Sciences (Shanghai, China). Isolated HSCs and HSC-LX2 cells were cultured in DMEM with 10% FBS, 1% antibiotics, and incubated in a 5% CO2 and 95% air humidified atmosphere at 37 °C.
2.5. Cell viability assay
Cellular viability was detected by the Cell Counting Kit-8 (Beyotime Biotechnology, Shanghai, China). Cells were cultured in 96-well plates at 100 μl volume per well. After drugs treatment, 10 μl CCK8 solution was added in the medium directly and incubated for 2 h at 37 °C. The absorbance of each well was determined at 450 nm by a Microplate Reader (Bio-Rad, Hercules, CA, USA).
2.6. Analysis of cell death by Trypan Blue Staining
Cell death was determined by Trypan Blue Staining assay (Beyotime Biotechnology, Shanghai, China). In brief, after curcumol treatment, cells were digested using trypsin without EDTA, and gathered by centrifugation, then washed twice with PBS and resuspended in PBS. Cell suspension and trypan blue solution were mixed gently in 1:1 ratio and stained for 3 min. A few of stained cells were counted with a hemocytometer and photographs were taken in a blinded fashion at random fields. Stained blue cells represented dead cells.
2.7. SiRNA Transfection
RIPK3 siRNA (5′-GCAGUUGUAUAUGUUAACGAGCGGUCGdTdT-3′) was synthesized by GenScript (Nanjing, China). RIPK3 siRNA (4 μg/μl) and 10 μl lipofectamine 2000 reagent (Life Technologies, New York, NY) were mixed with 250 μl of Opti MEM medium, respectively. After incubating for 5 min at room temperature, the above two mixtures were lightly mixed and continuously cultured for 20 min at room temperature. Then the cells in 6-well plates were incubated with 500 μl mixed medium for 8 h at 37 °C, and were re-cultured in complete medium for an additional 16 h at 37 °C. Control siRNA was used as a negative control.
2.8. RNA isolation and real-time PCR
Total RNA was extracted from HSC-LX2 cells using Trizol reagent according to the protocol provided by manufacturer (Sigma-Aldrich, St. Louis, MO, USA) and then reversely transcribed to cDNA by PrimeScript RT reagent kit (TaKaRa Biotechnology, Beijing, China). Real-Time PCR was conducted by the SYBR Green I fluorescent dye (TaKaRa Biotechnology, Beijing, China), according to the manufacturer's guidelines. Glyceraldehyde phosphate dehydrogenase (GAPDH) was served as an invariant control, and mRNA levels were expressed as fold changes after normalizing to GAPDH. Results came from triplicate experiments. Table 1 listed out the primers (Genscript, Nanjing, China) used.
Table 1.
Primers used for determination of mRNA expression levels in human HSC-LX2 cells.
| Gene | Forward sequence | Reverse sequence |
|---|---|---|
| Fibronectin | 5′-AGCCGCCACGTGCCAGGATTAC-3′ | 5′-CTTATGGGGGTGGCCGTTGTGG-3′ |
| α1(I)-procollagen | 5′-AGAGGAAGGAAAGCGAGGAG-3′ | 5′-GGACCAGCAACACCATCTG-3′ |
| α-SMA | 5′-GACAATGGCTCTGGGCTCTGTAA-3′ | 5′-ATGCCATGTTCTATCGGGTACTTCA-3′ |
| GAPDH | 5′-CTTCTTTTGCGTCGCCAGCCGA-3′ | 5′-ACCAGGCGCCCAATACGACCAA-3′ |
2.9. Western blot analyses and immunoprecipitation assay
Tissue samples or cells were lysed by a mammalian lysis buffer (Sigma St. Louis, MO, USA) and western blot analysis was conduct according to the manufacturer's instructions (Bio/Rad, Hercules, CA, USA). After determining the concentrations of protein, the HSC-LX2 cell extraction was incubated with RIPK1 or RIPK3 antibody and followed by incubating with proteinA/G plus-agarose beads overnight at 4 °C. Then the complexes were washed 3 times with immunoprecipitation assay (RIPA) buffer on rice and boiled in SDS sample buffer for subsequent western blot analysis. β-actin was detected with anti-β-actin antibody (Danvers, MA, USA) for use as loading controls.
2.10. Immunofluorescence analysis
HSC-LX2 cells were planted on 24-well plates and cultured in DMEM with 10% FBS, then given corresponding reagents for 24 h. After that, cells were fixed with 4% PFA for 30 min at 37 °C. After permeabilized with PBS-T (0.1% Triton x-100 dissolved in PBS) and blocked with PBS-B (4% BSA dissolved in PBS). Cells were stained with the corresponding antibody (1:100 dilution) overnight at 4 ℃, followed by incubation with FITC-labeled Goat Anti-Rabbit IgG (1:2000 dilution) for 2 h. Finally, 4′,6-Diamidino-2-phenylindole (DAPI) was applied for staining the nucleus in dark for 5 min and the fluorescence was observed with fluorescence microscope (Nikon, Tokyo, Japan).
For liver tissues, sections including both the liver lobule and portal areas were cut into 5 µm in thickness, which were blocked with 1% bovine serum albumin after deparaffinization, and then followed by incubating with corresponding antibodies overnight at 4 ℃. After washing with PBS for 3 times, secondary antibodies were used to incubate with the sections for 1 h at room temperature. The sections incubated with secondary antibodies alone served as vertical controls. Sections were observed in a single plane under an MRC 1024 laser confocal microscope (Bio-Rad Laboratories).
2.11. Biochemical analysis
Sera were separated from whole blood by centrifugation. Serum levels of AST, ALT, ALP, LDH, TBIL, and IBIL were detected using commercial assay kits according to the protocols from manufacturer (Nanjing Jiancheng Bioengineering Institute, Nanjing, Jiangsu, China). The absorbance values were determined using a SpectraMax™ microplate spectrophotometer (Molecular Devices, Sunnyvale, CA, USA)
2.12. Enzyme-Linked Immunosorbent Assay (ELISA)
The serum levels of related factors, including IL-6, IL-8, IL-10, TNF-α, hyaluronic acid, laminin, type III procollagen, and type Ⅳ collagen were measured with ELISA kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) according to the manufacturer's protocols.
2.13. Transmission electron microscopy
HSC-LX2 cells (14,000 cells/well) of the same density were seeded onto 4-chambered coverglass (Lab-Tek Chambered Coverglass System) (Nalgene/Nunc, Rochester, NY, USA). Images were obtained from the Olympus EM208S transmission electron microscope.
2.14. Intracellular ROS levels assay
The level of intracellular ROS was determined by oxidation-sensitive fluorescent probe DCFH-DA as previously described [24]. To confirm that ROS involved in curcumol-induced HSC death, HSC-LX2 cells were pretreated with ROS scavenger, NAC (10 mM) for 1 h before being exposed to curcumol.
2.15. Determination of MDA, SOD levels and GSH/GSSG ratio
HSC-LX2 cells were plated into 6-well plates and treated with indicated reagents. After treatment, the levels of intracellular MDA, SOD, GSH and GSSG were quantified using commercially available kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China), according to the manufacturer's instructions.
2.16. Determination of mitochondrial superoxide
Mitochondrial superoxide was measured by MitoSox red staining as described by the manufacturer's protocol. After treatment with curcumol, HSC-LX2 cells were incubated with MitoSox reagent (5 μM) for 10 min at 37 °C, and washed with PBS. The red fluorescence density was detected by fluorescence microscope (Nikon, Tokyo, Japan) using the Rhodamine channel.
2.17. Mitochondrial membrane potential assay
The mitochondrial membrane potential (MMP) was determined by JC-1 staining, following the manufacturer's guidelines. In brief, the cells were washed with PBS and incubated with JC-1 work solution at 37 °C in the dark for 20 min. After the removal of JC-1 work solution, the cells were washed with PBS, and images were taken using a fluorescence microscope (Nikon, TiE, Japan) with both red and green channels. The MMP is represented by the average red/green fluorescent intensity ratio.
2.18. Statistical analysis
Results were expressed as mean ± S.D. or mean ± S.E.M, and the differences between groups were analyzed using Prism 5.0 (Graph Pad Software Inc., San Diego, CA). The statistical analysis was determined by Student's t-test (two-group comparison) or one-way analysis of variance with the Student-Newman-Keuls test (more than two groups). P < 0.05 was considered a significance.
3. Results
3.1. Curcumol protects mice liver from CCl4-induced injury and ameliorates murine hepatic fibrogenesis
To investigate the effect of curcumol on liver injury and hepatic fibrosis, a classical murine liver fibrosis model was built through intraperitoneal injection of CCl4 [34]. CCl4 causes hepatocyte steatosis, mild thickening of the central venous wall, fibro-plastic proliferation, inflammatory cell infiltration and collagen deposition. The result showed that curcumol treatment markedly ameliorated pathological features of liver fibrosis in a dose-dependent manner (Fig. 1A). There was obvious increase in levels of serum ALT, AST, ALP, LDH, TBIL, and IBIL in CCl4-treated mice. Treatment with curcumol significantly decreased these serum markers of hepatic injury (Fig. 1B). Inflammation is a vital precursor to hepatic fibrogenesis among chronic liver diseases [35]. The result of ELISA showed that the serum levels of pro-inflammatory factors, including IL-6, IL-8, and TNF-α, were dose-dependently decreased by curcumol. By contrast, the serum level of anti-inflammatory factor IL-10 was dramatically upregulated (Supplementary Fig. 1A). Furthermore, immunohistochemistry (IHC) revealed that curcumol reduced inflammatory cell infiltration in CCl4-treated liver, illustrated by decreased intrahepatic CD45-, and F4/80-positive cells (Supplementary Fig. 1B).
Fig. 1.

Curcumol protects mice liver from CCl4-induced injury and ameliorates murine hepatic fibrogenesis. Mice were randomly divided into the following 5 groups (eight mice per group): vehicle control, CCl4, CCl4 + curcumol 15 mg kg−1, CCl4 + curcumol 30 mg kg−1 and CCl4 + curcumol 60 mg kg−1. (A) Representative photograph of liver tissues, and microphotograph of H&E-stained, Masson-stained and Sirius Red-stained paraffin-embedded sections of liver tissues (Scale bars, 100 µm). (B) Determination of serum ALT, AST, ALP, LDH, TBIL and IBIL levels. (C) ELISA analysis of serum hyaluronic acid, laminin, type III procollagen, and Ⅳ collagen. (D) Western blot analysis of α-SMA, α1(I) procollagen, fibronectin and TGF-βRⅡ expression from mouse liver. (E) Immunofluorescence staining of liver sections using antibodies against EGFR, PPAR-γ. Scale bar, 100 µm. Data are expressed as mean ± SD (n = 6); *P < 0.05 versus CCl4, **P < 0.01 versus CCl4, and ***P < 0.001 versus CCl4, ##P < 0.01 versus vehicle control, ###P < 0.001 versus vehicle control.
As shown in Fig. 1C, CCl4 apparently induced liver fibrogenesis, whereas curcumol could markedly counteract the increase in serum levels of hyaluronic acid, laminin, type III procollagen, and type Ⅳ collagen in CCl4-treated mice. In addition, we examined the protein abundances of α-smooth muscle actin (α-SMA), α1(I) procollagen, fibronectin, key markers of liver fibrosis and transforming growth factor-β receptor II (TGF-βRII), the key receptor of transmitting profibrogenic pathways. Treatment with curcumol markedly reduced these protein expressions dose-dependently (Fig. 1D). Similarly, immunofluorescence analyses of epidermal growth factor receptor (EGFR) with α-SMA (a characteristic HSC marker) indicated that curcumol abrogated EGFR expression in CCl4-induced fibrotic liver of mice dose-dependently (Fig. 1E). Given that peroxisome proliferator activated receptor-γ (PPARγ) could control the profibrogenic biology of HSCs [36], we then evaluated the effect of curcumol on the expression of PPARγ. Immunofluorescence double staining indicated that curcumol inhibited the activation of HSC in fibrotic liver of mice by upregulating the expression of PPARγ (Fig. 1F). Collectively, curcumol protects mice liver from CCl4-caused injury and reduces inflammation, ultimately ameliorating hepatic fibrogenesis in CCl4-treated mice.
3.2. Curcumol inhibits HSC activation in vitro
It is generally accepted that HSC activation is the main cause of subsequent excessive collagen synthesis and deposition in the fibrotic liver [37]. We used cultured human cell line HSC-LX2 cells to verify whether curcumol could directly affect HSC activation. We measured the markers for signaling transduction of HSC activation in vitro. Our results showed that curcumol dose-dependently inhibited the transcriptional and translational regulation of α-SMA, α1 (I) collagen and fibronectin in HSC-LX2 cells (Fig. 2A, B). Immunofluorescence staining for α-SMA, α1 (I) collagen and fibronectin further demonstrated that HSC activation was dramatically suppressed under curcumol treatment (Fig. 2C). Meanwhile, we found that curcumol downregulated the protein expression of TGF-βRI, TGF-βRII, and EGFR in HSCs (Fig. 2D). Moreover, the protein level of platelet derived growth factor β receptor (PDGF-βR), a mitogenic factor, was also reduced by curcumol, as proved by the immunofluorescence staining (Fig. 2E). In addition, curcumol significantly reduced tissue inhibitor of metalloproteinase 2 (TIMP-2) protein expression in HSCs (Fig. 2F), which was partially responsible for increased ECM degradation [38]. These data suggest that curcumol can impair HSC activation via regulating several fibrotic markers.
Fig. 2.

Curcumol inhibits HSC activation in vitro. Human HSC-LX2 cells were treated with DMSO (0.02%, w/v), or curcumol at indicated concentrations for 24 h. (A, D and F) Western blot analysis of protein expression of profibrogenic genes. (B) Real-time PCR analyses of genes related to fibrogenesis, including a-SMA, α1(I) procollagen, fibronectin. (C, E) Immunofluorescence staining of a-SMA, α1(I) procollagen, fibronectin, and PDGF-βR. Scale bar, 50 µm. Data are expressed as mean ± SD (n = 3); *P < 0.05 versus DMSO, **P < 0.01 versus DMSO, and ***P < 0.001 versus DMSO.
3.3. Curcumol induces necroptosis but not apoptosis in HSCs
The inhibition of HSC viability is crucial for attenuating HSC activation. Cell Counting Kit-8 analysis showed that curcumol could dose-dependently affect HSC-LX2 cell viability (Fig. 3A), having no cytotoxicity on human hepatocytes at doses lower than 80 μM (Fig. 3B). Light microscopy indicated that adherent and long spindle-shaped HSCs shrunk and changed to be semi-suspended with increased dose gradually (Fig. 3C). It is widely known that apoptosis is important for clearing activated HSCs [39]. We first examined that whether curcumol induced HSC apoptosis. Pretreatment with apoptosis inhibitor Z-VAD-FMK had no influence on curcumol-induced anti-proliferation (Fig. 3D). In addition, the protein levels of some key caspases did not change under curcumol treatment (Fig. 3E). Therefore, curcumol-induced cell death was independent on apoptosis. Interestingly, the LDH release in HSCs increased dramatically in a dose-dependent manner in response to curcumol, and the levels of ATP decreased significantly at a dose of 45 μM (Fig. 3F, G). At the same time, membrane permeability was concentration-dependently changed by curcumol, which was observable as Trypan blue-positive staining 24 h after curcumol treatment (Fig. 3H). Furthermore, no obvious apoptotic features in curcumol-treated cells could be observed through transmission electron microscopy (TEM) analysis (Fig. 3I). Instead, the cells exhibited a typical necrotic cell death morphology, including swelling of organelles (especially mitochondria), condensation of chromatin into small, irregular patches, and chromatin margination [17]. Along with its effect on the loss of plasma membrane integrity and the decrease of ATP production, we demonstrate that curcumol induces HSC necroptosis.
Fig. 3.

Curcumol induces necroptosis but not apoptosis in HSCs. Human HSC-LX2 cells were treated with DMSO (0.02%, w/v) or curcumol at indicated concentrations for 24 h. (A) and (B) Cell Counting Kit-8 analysis of the cell viability. (C) Cell morphology assessment. Scale bar, 200 µm. (D) Human HSC-LX2 cells were pretreated with 30 μM pan-caspase inhibitor z-vad-fmk for 1 h, followed by curcumol treatment for 24 h. Cell viability was measured by Cell Counting Kit-8 assay. (E) Western blot analysis of the protein expression of caspase 3, 8, 9 in the curcumol-treated human HSC-LX2 cells (F) LDH release assay. (G) CellTiter-Glo ATP-based luminescence assays for the detection of ATP levels. (H) Transmission electron microscopy (TEM)of cells. Black arrows represent swollen mitochondria (M), while white arrows represent condensed and marginated chromatins in nuclei (N). (I) Human HSC-LX2 cells were treated with curcumol treatment for 24 h. Trypan blue staining was used to detect cell death. Scale bar, 100 µm. Data are expressed as mean ± SD (n = 3); *P < 0.05 versus DMSO, **P < 0.01 versus DMSO, ***P < 0.001 versus DMSO.
3.4. The RIPK1/RIPK3-complex is required for curcumol to induce HSC necroptosis in vitro
It has been reported that the RIPK1-RIPK3-MLKL signaling pathway was involved in the induction of necroptosis [40]. Thus, we investigated that whether curcumol changed the protein expression or phosphorylation of RIPK1, RIPK3 and MLKL by western blot analysis. As shown in Fig. 4A, curcumol treatment upregulated the total protein expression of RIPK1 and RIPK3 in dose-dependent manner. Besides, the phosphorylation of RIPK1 and RIPK3 increased after curcumol treatment. Interestingly, the up-regulated phosphorylation level of RIPK1 was sustained, whereas the phosphorylation of RIPK3 began to decrease at 16 h. However, curcumol had no significant influence on the expression of phospho-MLKL (Fig. 4A, B). Immunofluorescence staining of RIPK1 and RIPK3 further validated the results of western blot (Fig. 4C, D). We further investigated the interaction between RIPK1 and RIPK3 in curcumol-treated HSCs by co-immunoprecipitation. When RIPK1 was immunoprecipitated with its antibody, the level of co-immunoprecipitated RIPK3 rose significantly in the curcumol-treated cells, compared to the control cells, which was impaired by RIPK1 inhibitor Nec-1. Similarly, immunoprecipitation of RIPK3 with its antibody caused co-immunoprecipitation of much more RIPK1 in the curcumol-treated cells. which was also abrogated by Nec-1 (Fig. 4E). Overall, these results demonstrate that curcumol not only upregulates the phosphorylation levels of RIPK1 and RIPK3, but also promotes their aggregation to form necrosome in HSCs, and eventually induces HSC necroptosis.
Fig. 4.

The RIPK1/RIPK3-complex is required for curcumol to induce HSC necroptosis in vitro. (A, B) Human HSC-LX2 cells were treated with DMSO (0.02%, w/v) and/or curcumol at the indicated doses for 24 h, or curcumol at 30 μM for various hours. Western blot analyses of phosphorylated RIPK1 (Ser166), RIPK3 (Ser227), MLKL(Ser358) and corresponding total protein expression in activated HSCs treated with curcumol at different concentrations for 24 h or curcumol at 30 μM for various hours. (C, D) Human HSC-LX2 cells were treated with DMSO (0.02%, w/v) and/or curcumol at the indicated doses for 24 h. Immunofluorescence staining for RIPK1, RIPK3 in HSC-LX2 cells. Scale bar, 50 µm. (E) Human HSC-LX2 cells were pretreated with 50 μM Nec-1 for 1 h, followed by curcumol treatment for 24 h. RIPK1 was immunoprecipitated with its antibody and resulted in co-immunoprecipitation of RIPK3. Immunoprecipitation of RIPK3 with its antibody caused co-immunoprecipitation of RIPK1 in HSC-LX2 cells. Data are expressed as mean ± SD (n = 3); *P < 0.05 versus DMSO, **P < 0.01 versus DMSO, ***P < 0.001 versus DMSO.
3.5. Curcumol reduces the expression of pro-fibrotic proteins by inducing RIPK1/RIPK3-mediated necroptosis in HSCs
Recent study has shown that gallic acid could trigger necroptosis in HSCs, indicating that HSC necroptosis might have a beneficial effect on ameliorating animal hepatic fibrosis [16]. We proceeded to investigate whether curcumol could inhibit profibrotic response in HSCs by regulating necroptosis. At first, we used selective RIPK1 inhibitor Nec-1 to verify that Nec-1 decreased curcumol-induced RIPK1 expression (Fig. 5A). Cell Counting Kit-8 analysis showed that the inhibition of HSC viability under curcumol treatment was unexpectedly enhanced by Nec-1 (Fig. 5B). Besides, Nec-1 did not impair the effect of curcumol on decreasing the markers of HSC activation (Fig. 5C). These results suggested that the deficiency of RIPK1 protein might switch the cell death from necroptosis to apoptosis when HSCs were treated with curcumol. In fact, we found that Nec-1 promoted curcumol-induced apoptosis of HSCs, which was consistent with prior published studies [41], [42] (Supplementary Fig. 2). Next, we explored the role of RIPK3 in the activation of HSCs. RIPK3 siRNA significantly inhibited cellular RIPK3 expression, suggesting a high transfection efficiency (Fig. 5D). Results of Cell Counting Kit-8 analysis indicated that HSC viability was restrained by curcumol, but rescued by administration of RIPK3 siRNA (Fig. 5E). We further investigated the role of RIPK3 in inhibiting HSC activation provided by curcumol. SiRNA-mediated knockdown of RIPK3 significantly ameliorated the inhibitory effect of curcumol on fibrotic makers (Fig. 5F, G). Taken together, these findings show that curcumol abolishes HSC activation, which is associated with RIPK1/RIPK3-mediated necroptosis in activated HSCs.
Fig. 5.

Curcumol reduces the expression of pro-fibrotic proteins by inducing RIPK1/RIPK3-mediated necroptosis in HSCs. Human HSC-LX2 cells were pretreated with 50 μM Nec-1 for 1 h or stably transfected with RIPK3 siRNA construction for 6 h, and then treated with the indicated concentration of curcumol for 24 h. (A) Detection of RIPK1 was done by immunoblot analysis. (B, E) Cell Counting Kit-8 assayed for detecting the cell viability. (C, F) Immunoblot analysis of α-SMA, α1(I) procollagen, and fibronectin was performed. (D) Western blot analysis of the transfection efficiency. (G) The expression of α-SMA and α1(I) procollagen were assessed by immunofluorescence. Scale bar, 50 µm. Data are expressed as mean ± SD (n = 3); *P < 0.05 versus DMSO, **P < 0.01 versus DMSO, and ***P < 0.001 versus DMSO. & P < 0.05 versus si-Ctrl. #P < 0.05 versus curcumol, ##P < 0.01 versus curcumol.
3.6. Curcumol induces mitochondrial ROS generation and depolarization in HSCs via a RIPK3-dependent mechanism
Numerous studies have shown that ROS production is necessary for necroptosis in several cell lines such as macrophages [43], MEFs and L929 cells [44]. To investigate whether ROS participated in curcumol-induced cell death in HSCs, we first analyzed the effect of curcumol on the intracellular ROS levels. Observation using fluorescence microscopy showed that curcumol treatment dose-dependently increased ROS level in HSCs (Fig. 6A). The overproduction of ROS triggers serious damages in various cells, related to increased MDA levels and decreased SOD and GSH content. Consistently, we found that curcumol treatment concentration-dependently promoted MDA formation, and conversely reduced SOD cellular vitality and GSH-to-GSSG ratio (Fig. 6E). Further study with Cell Counting kit-8 analysis showed that curcumol-induced cell death was greatly suppressed when cells were pretreated with the ROS scavenger, NAC (Fig. 6B), suggesting the critical role of ROS production in curcumol-triggered cell death in HSCs.
Fig. 6.

Curcumol induces mitochondrial ROS generation and depolarization in HSCs via a RIPK3-dependent mechanism. Human HSC-LX2 cells were treated with DMSO (0.02%, w/v), or curcumol at indicated concentrations for 24 h. Following the treatment, (A) ROS production was assessed by DCFH-DA staining, Scale bar, 50 µm. (C) Mitochondrial superoxide was detected by immunofluorescence using MitoSox Red staining. Scale bar, 50 µm. (F) The JC-1 fluorescence ratio evaluated mitochondrial membrane potential. Scale bar, 50 µm. (B) HSC-LX2 cells were pretreated with NAC (10 mM) for 1 h and then were treated with the indicated concentration of curcumol for 24 h. Cell viability was evaluated by Cell Counting Kit-8 analysis. HSC-LX2 cells were steadily transfected with RIPK3 siRNA, and then treated with the indicated concentration of curcumol for 24 h. Following the treatment, (D) MitoSOX Red immunofluorescence staining evaluated mitochondrial superoxide. Scale bar, 50 µm. (E) MDA levels, SOD cellular viability, and the GSH/GSSG ratio were measured by corresponding test kit. (G) Mitochondrial membrane potential was detected by the JC-1 fluorescence ratio. Scale bar, 50 µm. Data are expressed as mean ± SD (n = 3); *P < 0.05 versus DMSO, **P < 0.01 versus DMSO, and ***P < 0.001 versus DMSO. && P < 0.01 versus control. #P < 0.05 versus curcumol (30 μM), ##P < 0.01 versus curcumol (30 μM), ###P < 0.001 versus curcumol (30 μM).
Necrosomes could disturb ROS homeostasis, induce ATP depletion and mitochondrial depolarization, eventually impairing mitochondrial metabolism and even resulting in cell death [45]. Next, we investigated whether mitochondrial ROS production and loss of mitochondrial membrane potential (MMP) in necroptosis were dependent on RIPK3 expression. MitoSox Red staining was used to evaluate the mitochondrial ROS production. As shown in Fig. 6C, curcumol induced significant increase of mitochondrial superoxide, which was attenuated by RIPK3 siRNA (Fig. 6D). In addition, we found that pretreatment of cells with administration of RIPK3 siRNA significantly reversed curcumol-induced reduction of intracellular GSH and SOD levels (Fig. 6E). The effect of curcumol on MMP was assessed using the JC-1 fluorescence ratio, where a low ratio of red/green means a dissipation of MMP [46]. The result indicated that curcumol led to the dissipation of MMP, whereas RIPK3 inhibition via siRNA dramatically ameliorated mitochondrial depolarization (Fig. 6F, G). Collectively, these discoveries suggest that RIPK3-dependent mitochondrial ROS production and loss of MMP contribute to curcumol-induced necroptosis in HSCs.
3.7. RIPK3 promotes curcumol-induced mitochondrial ROS generation and depolarization via activating JNK
Accumulating evidence report that JNK activation promotes the increase of intracellular ROS generation, and contributes to the oxidative stress [27], [28]. Another report suggests that RIPK3 is great importance for JNK activation [29]. We hypothesized that JNK could mediate curcumol-induced RIPK3/ROS-dependent necroptosis in HSCs. To verify this hypothesis, we measured the levels of entirely phosphorylated MAPK signaling proteins in HSCs under curcumol treatment. Curcumol herein selectively increased JNK phosphorylation in dose and time-dependent manners (Fig. 7A, B). Cell Counting Kit-8 analysis showed that curcumol inhibited HSC viability, but regained by selective JNK1/2 inhibitor (SP600125), suggesting JNK was vital for curcumol-induced HSC death (Fig. 7C). Besides, the pretreatment with RIPK3 siRNA obviously impaired the curcumol-induced JNK1/2 activation, proving that JNK1/2 phosphorylation was a RIPK3-dependent process (Fig. 7D, E). Given that JNK activation was essential for curcumol-induced necroptosis, we investigated whether mitochondrial ROS imbalance and depolarization were regulated by JNK1/2. The inhibition of phosphorylated-JNK by SP600125 could markedly protect HSCs from suffering the imbalance of mitochondrial homeostasis, indicated by the increase of MitoSox Red fluorescence (Fig. 7F). These results confirm that JNK activation, as the upstream mechanism of mitochondrial dysfunction, is essential for RIPK3-dependent necroptosis in HSCs under curcumol treatment.
Fig. 7.

RIPK3 promotes curcumol-induced mitochondrial ROS generation and depolarization via activating JNK. (A) Human HSC-LX2 cells were treated with DMSO (0.02%, w/v), or curcumol at indicated concentrations for 24 h. Western blot analyses of the protein expression of phospho-ERK1/2, ERK1/2, phospho-JNK1/2, JNK1/2, phospho-p38, and p38. (B) Human HSC-LX2 cells were treated with 30 μM curcumol for the indicated time period. The ratio of phospho-JNK1/2 and JNK1/2 were detected by western blot analysis. (C) Human HSC-LX2 cells were pretreated with SP600125 (20 μM) for 1 h and then treated with the indicated concentration of curcumol for 24 h. Cell Counting Kit-8 assayed for evaluating cell viability. Human HSC-LX2 cells were stably transfected with RIPK3 siRNA, and then treated with the indicated concentration of curcumol for 24 h. Following the treatment, (D) the ratio of phospho-JNK1/2 and JNK1/2 was measured by western blot analysis, (E) the expression of phospho-JNK1/2 were detected by immunofluorescence. Scale bar, 50 µm. (F) MitoSox Red staining was use to evaluate mitochondrial superoxide. Scale bar, 50 µm. *P < 0.05 versus DMSO, **P < 0.01 versus DMSO, and ***P < 0.001 versus DMSO. & P < 0.05 versus control. ##P < 0.01 versus curcumol (30 μM).
3.8. RIPK3 knockdown in HSCs impairs the effect of curcumol on attenuating hepatic fibrosis
To address the effect of necroptosis on liver fibrosis, we firstly examined the protein levels of RIPK1 and RIPK3 in mice liver tissues. Results from double immunofluorescence staining of murine liver tissues displayed that curcumol increased the expression of RIPK1 and RIPK3 in mice fibrotic livers. Dual immunofluorescence staining of murine liver tissues clearly showed a close co-localization of RIPK1, and RIPK3 with HSC marker α-SMA (Fig. 8A, B). To further define the role of necroptosis in hepatic fibrosis, RIPK3-deficient mice were treated with CCl4 to trigger liver fibrosis. H&E, Masson and picrosirius red staining demonstrated that CCl4 aggravated liver bridging fibrosis and caused excessive collagen deposition in RIPK3-knockdown mice, but improving by curcumol (Fig. 8C). Besides, increased α-SMA expression by immunohistochemistry analysis indicated that RIPK3 shRNA significantly enhanced HSC activation, whereas the up-regulation of RIPK3 by curcumol inhibited HSC activation in CCl4-treated mice (Fig. 8D).
Fig. 8.

RIPK3 knockdown in HSCs impairs the effect of curcumol on attenuating hepatic fibrosis. (A, B) Mice were randomly divided into the following 5 groups (eight mice every group): vehicle control, CCl4, CCl4 + curcumol 15 mg kg−1, CCl4 + curcumol 30 mg kg−1 and CCl4 + curcumol 60 mg kg−1. The expression of RIPK1, RIPK3 in liver sections were detected by immunofluorescence staining. Scale bar, 100 µm. Mice were randomly divided into 5 groups (eight mice every group): vehicle control (no CCl4, no treatment); CCl4 + NC shRNA lentivirus; CCl4 + curcumol + NC shRNA lentivirus; CCl4 + RIPK3 shRNA lentivirus; and CCl4 + RIPK3 shRNA lentivirus + curcumol. (C) Representative photograph of liver tissues, and microphotograph of H&E-stained, Masson-stained and Sirius Red-stained paraffin-embedded sections of liver tissues. Scale bar, 100 µm. (D) Immunochemical staining for α-SMA of liver sections. Scale bar, 100 µm. (E, F) Western blot analyses of RIPK1, p-RIPK1, RIPK3, p-RIPK3, α-SMA, and α1(I)-procollagen protein levels in isolated HSCs. Data are expressed as mean ± SD (n = 6); *P < 0.05 versus CCl4, **P < 0.01 versus CCl4, and ***P < 0.001 versus CCl4, ##P < 0.01, ###P < 0.001 versus vehicle control. & P < 0.05 versus curcumol, && P < 0.01 versus curcumol.
Importantly, the primary HSCs and hepatocytes were isolated from fibrotic livers of mice treated with CCl4 and/or curcumol, and the effect of curcumol on necroptosis in isolated primary HSCs and hepatocytes was investigated. Interestingly, curcumol did not induce cell death in primary hepatocytes, but could resulted in a significant decrease of living primary HSCs. (Supplementary Fig. 3A). Moreover, treatment with curcumol could not trigger ROS overproduction in primary hepatocytes, whereas curcumol markedly induced ROS generation in CCl4-treated primary HSCs (Supplementary Fig. 3B). Furthermore, curcumol could significantly reduce the activities of supernatant ALT, AST and LDH in primary hepatocytes from CCl4-treated mice liver (Supplementary Fig. 3C). Attractively, curcumol could bring the phosphorylation of RIPK1 and RIPK3 to approximately normal levels in primary HSCs in a concentration-dependent manner (Fig. 8E). As expected, the role of curcumol in rescuing the protein expression of RIPK3 in primary HSCs extracted from CCl4-treated mice liver, was partially abrogated by RIPK3 shRNA (Fig. 8F). In addition, curcumol treatment reduced the translation of the major profibrotic genes, including α-SMA and α1(I)-collagen, whereas RIPK3 shRNA significantly increased the protein expression of these profibrotic makers (Fig. 8F). Therefore, our data demonstrate that curcumol attenuates liver fibrosis dependent on RIPK3-mediated HSC necroptosis.
4. Discussion
It is well known that the imbalanced synthesis and degradation of ECM ultimately result in liver fibrosis and later cirrhosis. ECM-producing myofibroblasts in liver are predominantly derive from HSCs. Once liver suffers injury, HSCs become activated [47]. Pharmacological induction of HSC death, such as apoptosis, senescence, and autophagy are one of the effective strategies to treat liver fibrosis [8], [9], [10]. In this study, we first confirmed the protective effect of curcumol on CCl4-triggered liver injury, inflammation, and hepatic fibrosis. Besides, we demonstrated that curcumol-induced HSC necroptosis could play a vital role in reversing hepatic fibrosis process, and also discovered that the RIPK1/RIPK3 complex-dependent necroptosis via JNK1/2-ROS signaling in HSCs was a novel approach to treat liver fibrosis.
Previous studies have identified the anti-tumor and anti-inflammatory role of curcumol [30], [31]. However, there are few studies on assessing the effect of curcumol on hepatic fibrosis. Chen et al. reported that curcumol induced HSC-T6 cell death through inhibiting of PI3K/NF-κB pathway, which suggested that curcumol might ameliorate hepatic fibrosis [33]. Herein, we investigated the effect of curcumol on liver fibrogenesis. The CCl4 murine model was established as our previous description [11]. For the first time, we found that curcumol not only could ameliorate CCl4-induced liver injury and inflammatory effect, as serum levels of hepatic ALT, AST, ALP, and LDH all decreased, but also inhibit the release of inflammatory cytokines as well as hepatic inflammatory infiltration. Histopathological analysis showed that curcumol dramatically improved multiple features linked to liver fibrosis, such as adipose degeneration of hepatocytes, fibrous hyperplasia, and collagen deposition. In addition, serum hyaluronic acid, laminin, type III procollagen and type Ⅳ collagen were also effectively reduced by curcumol in the fibrotic murine liver. Furthermore, curcumol down-regulated the expression of α-SMA, α1(I) procollagen, and fibronectin to approximately normal levels, and inhibited the pro-fibrogenic pathway transmission, including TGF, EGF pathways, which consisted with our vitro experiments. These data clearly showed that curcumol protected against liver fibrosis.
Accumulating evidence identified that the proliferation and growth of tumor cells were inhibited through the induction of necroptosis. Salvatore et al. found that S-nitrosoglutathione reductase-deficient HepG2 cells and tumors were more sensitive to succinate dehydrogenase (SDH) inhibitors, and SDH inhibitor suppressed the growth of HCC by inducing RIPK1/PARP1-mediated necroptosis [48]. Besides, Jing et al. showed cisplatin triggered lung cancer A549 cells death via MLKL-PITP alpha signaling-mediated necroptosis [49]. Moreover, Jing et al. identified the transcription factor specific protein-1 (SP1) and the epigenetic regulator ubiquitin-like, containing PHD and RING finger domains 1 (UHRF1) promoted RIPK3-mediated necroptosis in colon cancer cells [50]. Despite of all these, the function of necroptosis in non-tumor conditions has not been well understood. Interestingly, Dai et al. reported that curcumin, an effective ingredient isolated from the rhizome of the plant turmeric (Curcuma longa Linn), protected against iron induced neurotoxicity by modulating necroptosis [51]. Prior study has also discovered the existence of necroptosis in activated HSCs [16]. In the current study, we demonstrated that the other active component of curcuma, curcumol, could inhibit HSC proliferation in vitro. We also found that curcumol had no effect on the activation of caspases. Therefore, curcumol-induced cell vitality inhibition did not result from apoptosis. Typically, necroptosis is initiated when caspases are in the state of insufficient activation or their activities are interdicted [17]. Trypan blue staining provided the evidence that curcumol induced cell death of HSCs. Further study showed curcumol upregulated the totalprotein expression and phosphorylation of RIPK1 and RIPK3, as well as promoting the formation of necrosome. Necrosome stimulates the phosphorylation of MLKL, further resulting in the generation of autologous oligomers that is a core machinery for triggering necroptosis [21]. Attractively, our study showed that curcumol had no significant effect on the expression of phospho-MLKL. Therefore, phosphorylated MLKL inducing pores did not trigger necroptotic cell death. Obviously, curcumol induced RIPK1/RIPK3-complex dependent necroptosis in human HSC-LX2 cells. As expected, curcumol-induced inhibition of cellular viability and HSC activation were resumed when RIPK3 was genetically reduced by siRNA. However, down-regulation of RIPK1 activity by pharmacological inhibitor Nec-1 enhanced the effect of curcumol. Notably, the positive expression of RIPK1 was further found to be a molecular switch for curcumol to induce necroptosis or apoptosis, which was consistent with previous studies [41], [42]. Interestingly, recent studies have reported that catalytically active RIPK3 induces necroptosis, whereas catalytically inactive platform RIPK3 favors apoptosis [52]. Besides, RIPK3 inhibitors at higher concentrations induce RIPK3-scaffold-dependent apoptosis [53]. In our study, siRIPK3 did not switch cell death from necroptosis to apoptosis as Nec-1 did, in that siRIPK3 mainly affected the expression of RIPK3, having little effect on its kinase activity or conformation. At the same time, our experiments in vivo demonstrated that RIPK3 was co-localized with α-SMA, the marker of activated HSCs in murine fibrotic liver. The induction of RIPK3-necroptosis in HSCs showed negative correlation with liver fibrosis. For the first time, we demonstrated the potential of curcumol to induce RIPK3-dependent necroptosis in HSCs, in turn, could reduce the fibrotic response.
ROS accumulation is gradually regarded as the executioner and mediator of necroptosis. Wang et al. reported that AMPK protected against myocardial ischemia and reperfusion (IR) injury resulting from ROS-induced necroptosis. Similarly, the increase of ROS generation in endothelial cells induced necroptosis, providing new insights into targeting organelle-derived ROS for improving endothelial death and related vascular diseases [54]. What's more, Meng et al. demonstrated that the inhibition of ROS suppressed RIP-mediated human HK2 cell necroptosis, which may be the key mechanism of cisplatin-induced nephrotoxicity [55]. Although ROS-mediated necroptosis is harmful for the recovery of certain diseases, some researches also reported that it has the advantage on repressing the growth of tumor cells. Recently, Li et al. revealed that ROS-mediated necroptosis repressed human breast cancer growth possibly due to impaired angiogenesis [56]. Ma et al. reported mitochondrial ROS production induced by C/EBP homologous protein (CHOP) promoted A549 human lung cancer cell necroptosis and inhibited lung cancer progression [57]. These results showed that the function of ROS-induced necroptosis mainly depended on the cellular type and microenvironmental condition. In this study, we found that ROS accumulated significantly in activated HSCs under curcumol treatment. Pretreatment with ROS scavenger NAC, the curcumol-induced inhibition of cell viability was significantly impaired. Numerous studies have revealed that ROS is mainly produced in mitochondria, which in turn lead to the damage of organelles, or even cell death [58]. Besides, RIPK3 could upregulate ROS levels via activating GLUD1 localized at mitochondria, and enhance mitochondrial superoxide production [59]. Furthermore, Marshall et al. reported that mitochondrial respiratory chain inhibitors also decreased LDH release and RIPK1-RIPK3 interaction, suggesting that mitochondria ROS participated in necroptosis [60]. Consistently, we identified that inhibition of RIPK3 with siRNA markedly suppressed curcumol-induced generation of mitochondrial superoxide. It is well known that MDA can mark lipid peroxidation and the presence of excessive MDA represents that cell membranes are seriously damaged and defective. Additionally, SOD is a key antioxidant enzyme, which catalyzes the disproportionation of superoxide (O2-) radicals into H2O2 and O2. Furthermore, the GSH/GSSG ratio convincingly stands for intracellular redox status, which will affect cell survival. Our results demonstrated that curcumol-induced overproduction of ROS led to an increase of MDA levels and a decrease of SOD and GSH content, while RIPK3 siRNA significantly impaired the effect. In addition, RIPK3 siRNA attenuated curcumol-induced mitochondrial depolarization. These findings collectively indicate the mitochondrial superoxide and mitochondrial dysfunction are indispensable for curcumol-induced necroptosis in HSCs.
Prolonged JNK activation is implicated in a variety of hepatic pathologies. Our previous study revealed that DHA induced-JNK1/2 overexpression promoted HSC autophagy to control excessive inflammation [24]. Activated JNK also acted as a downstream mediator of RIPK3-driven necroptosis in hepatocyte [30]. A recent study reported that JNK activation increased intracellular ROS generation, which contributed to glioma cell parthanatos [29]. In the present study, curcumol promoted JNK activation in a time and concentration dependent manner, while RIPK3-deficiency reduced the phosphorylation of JNK in curcumol-treated HSCs, indicating that RIPK3 contributed to JNK activation. Importantly, the inhibition of JNK1/2 activity by selective JNK1/2 inhibitor (SP600125) led to the increase of cell viability and decrease of curcumol-induced mitochondrial ROS and MMP of HSCs. In accord with previous researches, our data shows that JNK1/2 activation serves as a downstream event of RIPK3, and triggers ROS overexpression in activated HSCs, which might be the key mechanism of curcumol-induced necroptosis to inhibit HSC activation. Importantly, we should also take into account the possibility that necroptosis induced by curcumol may have adverse effects on hepatocytes. Attractively, we found that the indicated dose of curcumol could not trigger ROS production and cell death in primary hepatocytes. By contrast, curcumol could significantly reduce the activities of supernatant ALT, AST and LDH in primary hepatocytes from CCl4-treated mice liver. Overall, these results show that curcumol the indicated dose of curcumol attenuates HSC activation by inducing HSC necroptosis specifically, and protects mice liver from CCl4-induced injury at the same time.
In conclusion, our results demonstrate that curcumol could have a significant protective effect against liver injury and fibrosis in CCl4-treated mice. Mechanistically, curcumol inhibits HSC activation through promoting RIPK1/RIPK3 complex formation, followed by JNK1/2 activation and subsequently mitochondrial dysfunction, which ultimately leads to HSC cell death. The necroptosis of activated HSCs induced by curcumol may prove to be a valuable therapeutic strategy for the treatment of liver fibrosis.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81600483, 31571455, 31401210), University Science Research Project of Jiangsu Province (16KJB310010), the Open Project Program of Jiangsu Key Laboratory for Pharmacology and Safety Evaluation of Chinese Materia Medica (No. JKLPSE 201502), Project of the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX-181577).
Acknowledgments
Conflict of interest
The authors declare no conflict of interest.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2018.09.007.
Contributor Information
Jiangjuan Shao, Email: jjshao1976@163.com.
Shizhong Zheng, Email: nytws@163.com.
Appendix A. Supplementary material
Fig. S1.

Curcumol reduces inflammation in mice liver. (A) ELISA measurement of IL-6, IL-8, IL-10 and TNF-α levels in serum. (B) Immunochemical staining for CD45 and F4/80 of liver sections. Scale bar, 100 µm. Data are expressed as mean ± SD (n = 6); *P < 0.05 versus CCl4, **P < 0.01 versus CCl4, and ***P < 0.001 versus CCl4, ###P < 0.001 versus vehicle control.
Fig. S2.

The deficiency of RIPK1 protein might switch the cell death from necroptosis to apoptosis in curcumol-treated HSCs. Human HSC-LX2 cells were pretreated with 50 μM Nec-1 for 1 h, and then were treated with the indicated concentration of curcumol for 24 h. Immunoblot analysis of including cleaved caspase-3, pro-caspase-3, cleaved caspase-9, pro-caspase-9 were performed. Data are expressed as mean ± SD (n = 3); **P < 0.01 versus DMSO, and ***P < 0.001 versus DMSO.
Fig. S3.

Curcumol induces HSC death and suppresses CCl4-induced cell damage in hepatocytes. Primary HSCs and hepatocytes were isolated from male ICR mice under CCl4 and the indicated dose of curcumol treatment. (A) Trypan blue staining was used to detect cell death. Scale bar, 100 µm. (B) ROS production was assessed by DCFH-DA staining, Scale bar, 50 µm. (C) Determination of supernatant ALT, AST, LDH levels in Primary hepatocytes. Data are expressed as mean ± SD (n = 6); **P < 0.01 versus CCl4, and ***P < 0.001 versus CCl4.
References
- 1.Chen Y. Differential effects of sorafenib on liver versus tumor fibrosis mediated by stromal-derived factor 1 alpha/c-x-c receptor type 4 axis and myeloid differentiation antigen-positive myeloid cell infiltration in mice. Hepatology. 2014;59(4):1435–1447. doi: 10.1002/hep.26790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shackel N.A. Beyond liver fibrosis: hepatic stellate cell senescence links obesity to liver cancer by way of the microbiome. Hepatology. 2014;59(6):2413–2415. doi: 10.1002/hep.26932. [DOI] [PubMed] [Google Scholar]
- 3.Kisseleva T., Brenner D.A. Role of hepatic stellate cells in fibrogenesis and the reversal of fibrosis. J. Gastroenterol. Hepatol. 2007;22(Suppl. 1):S73–S78. doi: 10.1111/j.1440-1746.2006.04658.x. [DOI] [PubMed] [Google Scholar]
- 4.Roderfeld M. Matrix metalloproteinase functions in hepatic injury and fibrosis. Matrix Biol. 2017 doi: 10.1016/j.matbio.2017.11.011. [DOI] [PubMed] [Google Scholar]
- 5.Rockey D.C. Translating an understanding of the pathogenesis of hepatic fibrosis to novel therapies. Clin. Gastroenterol. Hepatol. 2013;11(3):224–231. doi: 10.1016/j.cgh.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kisseleva T., Brenner D.A. Hepatic stellate cells and the reversal of fibrosis. J. Gastroenterol. Hepatol. 2006;21(Suppl. 3):S84–S87. doi: 10.1111/j.1440-1746.2006.04584.x. [DOI] [PubMed] [Google Scholar]
- 7.Zoubek M.E. Reversal of liver fibrosis: from fiction to reality. Best. Pract. Res. Clin. Gastroenterol. 2017;31(2):129–141. doi: 10.1016/j.bpg.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Bangen J.M. Targeting CCl4 -induced liver fibrosis by rna interference-mediated inhibition of cyclin e1 in mice. Hepatology. 2017;66(4):1242–1257. doi: 10.1002/hep.29275. [DOI] [PubMed] [Google Scholar]
- 9.Tsuchida T., Friedman S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017;14(7):397–411. doi: 10.1038/nrgastro.2017.38. [DOI] [PubMed] [Google Scholar]
- 10.Qu Y. Exosomes derived from mir-181-5p-modified adipose-derived mesenchymal stem cells prevent liver fibrosis via autophagy activation. J. Cell. Mol. Med. 2017;21(10):2491–2502. doi: 10.1111/jcmm.13170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin H. Activation of ppargamma/p53 signaling is required for curcumin to induce hepatic stellate cell senescence. Cell Death Dis. 2016;7:e2189. doi: 10.1038/cddis.2016.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Z. Autophagy regulates turnover of lipid droplets via ros-dependent rab25 activation in hepatic stellate cell. Redox Biol. 2017;11:322–334. doi: 10.1016/j.redox.2016.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Tran D.D.H. Myc target gene, long intergenic noncoding rna, linc00176 in hepatocellular carcinoma regulates cell cycle and cell survival by titrating tumor suppressor micrornas. Oncogene. 2018;37(1):75–85. doi: 10.1038/onc.2017.312. [DOI] [PubMed] [Google Scholar]
- 14.Roychowdhury S. Receptor interacting protein 3 protects mice from high-fat diet-induced liver injury. Hepatology. 2016;64(5):1518–1533. doi: 10.1002/hep.28676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Afonso M.B. Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin. Sci. 2015;129(8):721–739. doi: 10.1042/CS20140732. [DOI] [PubMed] [Google Scholar]
- 16.Chang Y.J. Gallic acid induces necroptosis via tnf-alpha signaling pathway in activated hepatic stellate cells. PLoS One. 2015;10(3):e0120713. doi: 10.1371/journal.pone.0120713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galluzzi L., Kroemer G. Necroptosis: a specialized pathway of programmed necrosis. Cell. 2008;135(7):1161–1163. doi: 10.1016/j.cell.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 18.Hitomi J. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135(7):1311–1323. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou W., Yuan J. Snapshot: necroptosis. Cell. 2014;158(2) doi: 10.1016/j.cell.2014.06.041. (464-464 e461) [DOI] [PubMed] [Google Scholar]
- 20.Rickard J.A. Ripk1 regulates ripk3-mlkl-driven systemic inflammation and emergency hematopoiesis. Cell. 2014;157(5):1175–1188. doi: 10.1016/j.cell.2014.04.019. [DOI] [PubMed] [Google Scholar]
- 21.Mulay S.R. Cytotoxicity of crystals involves ripk3-mlkl-mediated necroptosis. Nat. Commun. 2016;7:10274. doi: 10.1038/ncomms10274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fulda S. Regulation of necroptosis signaling and cell death by reactive oxygen species. Biol. Chem. 2016;397(7):657–660. doi: 10.1515/hsz-2016-0102. [DOI] [PubMed] [Google Scholar]
- 23.Marshall K.D., Baines C.P. Necroptosis: is there a role for mitochondria? Front. Physiol. 2014;5:323. doi: 10.3389/fphys.2014.00323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Z. Ros-jnk1/2-dependent activation of autophagy is required for the induction of anti-inflammatory effect of dihydroartemisin in liver fibrosis. Free Radic. Biol. Med. 2016;101:272–283. doi: 10.1016/j.freeradbiomed.2016.10.498. [DOI] [PubMed] [Google Scholar]
- 25.Park H. Myricetin treatment induces apoptosis in canine osteosarcoma cells by inducing DNA fragmentation, disrupting redox homeostasis, and mediating loss of mitochondrial membrane potential. J. Cell. Physiol. 2018;233(9):7457–7466. doi: 10.1002/jcp.26598. [DOI] [PubMed] [Google Scholar]
- 26.Zheng W. Zearalenone altered the cytoskeletal structure via er stress- autophagy- oxidative stress pathway in mouse tm4 sertoli cells. Sci. Rep. 2018;8(1):3320. doi: 10.1038/s41598-018-21567-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chocry M. Reversion of resistance to oxaliplatin by inhibition of p38 mapk in colorectal cancer cell lines: involvement of the calpain/nox1 pathway. Oncotarget. 2017;8(61):103710–103730. doi: 10.18632/oncotarget.21780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Teixeira J. Extracellular acidification induces ros- and mptp-mediated death in hek293 cells. Redox Biol. 2018;15:394–404. doi: 10.1016/j.redox.2017.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng L. Jnk activation contributes to oxidative stress-induced parthanatos in glioma cells via increase of intracellular ros production. Mol. Neurobiol. 2017;54(5):3492–3505. doi: 10.1007/s12035-016-9926-y. [DOI] [PubMed] [Google Scholar]
- 30.Roychowdhury S. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology. 2013;57(5):1773–1783. doi: 10.1002/hep.26200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen X. Curcumol exhibits anti-inflammatory properties by interfering with the jnk-mediated ap-1 pathway in lipopolysaccharide-activated raw264.7 cells. Eur. J. Pharmacol. 2014;723:339–345. doi: 10.1016/j.ejphar.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 32.Wang J. Identification and validation nucleolin as a target of curcumol in nasopharyngeal carcinoma cells. J. Proteom. 2018;182:1–11. doi: 10.1016/j.jprot.2018.04.025. [DOI] [PubMed] [Google Scholar]
- 33.Chen G. Curcumol induces hsc-t6 cell death through suppression of bcl-2: involvement of pi3k and nf-kappab pathways. Eur. J. Pharm. Sci. 2014;65:21–28. doi: 10.1016/j.ejps.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 34.Pan R.L. Low-molecular-weight fibroblast growth factor 2 attenuates hepatic fibrosis by epigenetic down-regulation of delta-like1. Hepatology. 2015;61(5):1708–1720. doi: 10.1002/hep.27649. [DOI] [PubMed] [Google Scholar]
- 35.Mridha A.R. Nlrp3 inflammasome blockade reduces liver inflammation and fibrosis in experimental nash in mice. J. Hepatol. 2017;66(5):1037–1046. doi: 10.1016/j.jhep.2017.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pawlak M. Molecular mechanism of pparalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015;62(3):720–733. doi: 10.1016/j.jhep.2014.10.039. [DOI] [PubMed] [Google Scholar]
- 37.Friedman S.L. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008;88(1):125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang J. Ndrg2 ameliorates hepatic fibrosis by inhibiting the tgf-beta1/smad pathway and altering the mmp2/timp2 ratio in rats. PLoS One. 2011;6(11):e27710. doi: 10.1371/journal.pone.0027710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luedde T. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology. 2014;147(4):765–783. doi: 10.1053/j.gastro.2014.07.018. (e764) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang X. The end of ripk1-ripk3-mlkl-mediated necroptosis in acetaminophen-induced hepatotoxicity? Hepatology. 2016;64(1):311–312. doi: 10.1002/hep.28263. [DOI] [PubMed] [Google Scholar]
- 41.Han W. Necrostatin-1 reverts shikonin-induced necroptosis to apoptosis. Apoptosis. 2009;14(5):674–686. doi: 10.1007/s10495-009-0334-x. [DOI] [PubMed] [Google Scholar]
- 42.Han W. Nec-1 enhances shikonin-induced apoptosis in leukemia cells by inhibition of rip-1 and erk1/2. Int. J. Mol. Sci. 2012;13(6):7212–7225. doi: 10.3390/ijms13067212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roca F.J., Ramakrishnan L. Tnf dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell. 2013;153(3):521–534. doi: 10.1016/j.cell.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhong C.Q. Quantitative phosphoproteomic analysis of rip3-dependent protein phosphorylation in the course of tnf-induced necroptosis. Proteomics. 2014;14(6):713–724. doi: 10.1002/pmic.201300326. [DOI] [PubMed] [Google Scholar]
- 45.Chen W. Diverse sequence determinants control human and mouse receptor interacting protein 3 (rip3) and mixed lineage kinase domain-like (mlkl) interaction in necroptotic signaling. J. Biol. Chem. 2013;288(23):16247–16261. doi: 10.1074/jbc.M112.435545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qu Y. Ripk3 interactions with mlkl and camkii mediate oligodendrocytes death in the developing brain. Cell Death Dis. 2017;8(2):e2629. doi: 10.1038/cddis.2017.54. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Friedman S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134(6):1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rizza S. S-nitrosylation of the mitochondrial chaperone trap1 sensitizes hepatocellular carcinoma cells to inhibitors of succinate dehydrogenase. Cancer Res. 2016;76(14):4170–4182. doi: 10.1158/0008-5472.CAN-15-2637. [DOI] [PubMed] [Google Scholar]
- 49.Jing L. Mlkl-pitpalpha signaling-mediated necroptosis contributes to cisplatin-triggered cell death in lung cancer a549 cells. Cancer Lett. 2018;414:136–146. doi: 10.1016/j.canlet.2017.10.047. [DOI] [PubMed] [Google Scholar]
- 50.Yang C. Regulation of rip3 by the transcription factor sp1 and the epigenetic regulator uhrf1 modulates cancer cell necroptosis. Cell Death Dis. 2017;8(10):e3084. doi: 10.1038/cddis.2017.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dai M.C. Curcumin protects against iron induced neurotoxicity in primary cortical neurons by attenuating necroptosis. Neurosci. Lett. 2013;536:41–46. doi: 10.1016/j.neulet.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 52.Newton K. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–1360. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- 53.Mandal P. RIP3 induces apoptosis independent of pro-necrotic kinase activity. Mol. Cell. 2014;56(4):481–495. doi: 10.1016/j.molcel.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Y.S. Amp-activated protein kinase protects against necroptosis via regulation of keap1-pgam5 complex. Int. J. Cardiol. 2018;259:153–162. doi: 10.1016/j.ijcard.2018.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meng X.M. Nadph oxidase 4 promotes cisplatin-induced acute kidney injury via ros-mediated programmed cell death and inflammation. Lab. Invest. 2018;98(1):63–78. doi: 10.1038/labinvest.2017.120. [DOI] [PubMed] [Google Scholar]
- 56.Li Y. Bufalin inhibits human breast cancer tumorigenesis by inducing cell death through the ros-mediated rip1/rip3/parp-1 pathways. Carcinogenesis. 2018;39(5):700–707. doi: 10.1093/carcin/bgy039. [DOI] [PubMed] [Google Scholar]
- 57.Ma Y.M. Novel chop activator lgh00168 induces necroptosis in a549 human lung cancer cells via ros-mediated er stress and nf-kappab inhibition. Acta Pharmacol. Sin. 2016;37(10):1381–1390. doi: 10.1038/aps.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Summers D.W. Mitochondrial dysfunction induces sarm1-dependent cell death in sensory neurons. J. Neurosci. 2014;34(28):9338–9350. doi: 10.1523/JNEUROSCI.0877-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang D.W. Rip3, an energy metabolism regulator that switches tnf-induced cell death from apoptosis to necrosis. Science. 2009;325(5938):332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 60.Basit F. Mitochondrial complex i inhibition triggers a mitophagy-dependent ros increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017;8(3):e2716. doi: 10.1038/cddis.2017.133. [DOI] [PMC free article] [PubMed] [Google Scholar]