

Abstract
Pure red cell aplasia (PRCA) is a syndrome defined by a normocytic normochromic anemia with severe reticulocytopenia and marked reduction or absence of erythroid precursors from the bone marrow. Diamond-Blackfan anemia is a congenital form of PRCA. Acquired PRCA may be either a primary disorder or secondary to some other disorder or agent. Primary acquired PRCA is an autoimmune disorder that is frequently antibody-mediated. Myelodysplastic syndromes may also present with the morphologic appearance of PRCA. Secondary acquired PRCA may be associated with collagen vascular/autoimmune disorders such as systemic lupus erythematosus; lymphoproliferative disorders such as chronic lymphocytic leukemia or large granular lymphocyte leukemia; infections, particularly B19 parvovirus; thymoma and other solid tumors; or a variety of other disorders, drugs, or toxic agents. The therapeutic approach to PRCA typically involves immunosuppression, but specific pathogenic subtypes are associated with specific therapeutic approaches. Cyclosporine A, with or without concurrent corticosteroids, appears to be the single most effective immunosuppressive agent.
Learning Objectives
Diagnose and classify pure red cell aplasia
Determine treatment plans for primary and secondary immune-mediated pure red cell aplasia and for parvovirus-associated pure red cell aplasia
Pure red cell aplasia (PRCA) is a syndrome defined by a normocytic normochromic anemia with severe reticulocytopenia and marked reduction or absence of erythroid precursors from the bone marrow.1 Abnormalities from PRCA are limited to the red cell lineage; abnormalities in other cell lines usually reflect another concurrent disorder.
Classification of PRCA
A classification of PRCA is outlined in Table 1. Congenital PRCA largely represents Diamond-Blackfan anemia (DBA), which is frequently associated with physical morphologic abnormalities and results from mutations in the genes for various ribosomal proteins.2 Pearson syndrome, a congenital mitochondrial disorder associated with marrow failure and exocrine pancreatic insufficiency, also exhibits erythroid precursor hypoplasia and is sometimes categorized with PRCA, but is probably better classified as a congenital sideroblastic anemia.3
Table 1.
Classification of pure red cell aplasia
| Congenital PRCA |
| Diamond-Blackfan anemia |
| Acquired PRCA |
| Primary |
| Primary autoimmune PRCA (includes transient erythroblastopenia of childhood) |
| Primary myelodysplastic PRCA |
| Secondary, associated with: |
| Autoimmune/collagen vascular disorders |
| Systemic lupus erythematosus |
| Rheumatoid arthritis |
| Inflammatory bowel disease |
| Other immunologic mechanisms |
| ABO-incompatible stem cell transplantation |
| Pyoderma gangrenosum |
| Lymphoproliferative disorders |
| Chronic lymphocytic leukemia |
| LGL leukemia |
| Hodgkin disease |
| Non-Hodgkin lymphomas |
| Angioimmunoblastic lymphadenopathy |
| Multiple myeloma |
| Waldenstrom macroglobulinemia |
| Castleman disease |
| Other hematologic malignancies |
| Chronic myelogenous leukemia |
| Chronic myelomonocytic leukemia |
| Myelofibrosis with myeloid metaplasia |
| Essential thrombocythemia |
| Acute lymphocytic leukemia |
| Solid tumors |
| Thymoma |
| Gastric cancer |
| Breast cancer |
| Biliary cancer |
| Lung cancer |
| Thyroid cancer |
| Renal cell carcinoma |
| Carcinoma of unknown primary site |
| Infections |
| B19 parvovirus |
| Human immunodeficiency virus |
| T-cell leukemia-lymphoma virus |
| Infectious mononucleosis |
| Viral hepatitis (hepatitis A, B, C, and E) |
| Cytomegalovirus |
| Bacterial infections |
| Group C Streptococcus |
| Tuberculosis |
| Bacterial sepsis |
| Drugs and toxins |
| rhEpo-induced Epo antibody-associated PRCA |
| Other drugs (Table 2) |
| Other disorders |
| Pregnancy |
| Riboflavin deficiency |
Modified from Lipton et al.44
This review will primarily focus on acquired PRCA. Primary acquired PRCA is an autoimmune disorder in which an immune mechanism interrupts erythroid differentiation. This may be mediated by an autoantibody or by another immunologic process. When an autoantibody is involved, the target of the autoantibody is variable and may be at the erythroid precursor level.4 Myelodysplastic primary acquired PRCA is an uncommon presentation of myelodysplasia morphologically characterized by erythroid hypoplasia, but is pathophysiologically distinct from the other types of PRCA described in this discussion.5 Transient erythroblastopenia of childhood is an uncommon self-limited variant of primary acquired PRCA occurring between the ages of 3 months and 4 years.6 Although familial cases have been reported, it appears to be an autoimmune disorder in most cases.6
Secondary acquired PRCA may be associated with autoimmune/collagen vascular disorders; lymphoproliferative disorders, particularly chronic lymphocytic leukemia; infections, especially B19 parvovirus; pregnancy; hematologic malignancies; nonhematologic neoplasms, of which the association with thymoma is the best known; and drugs and toxic agents. Many of the mechanisms underlying secondary acquired PRCA are immunologic, although not always antibody-mediated. This is particularly true for PRCA associated with autoimmune/collagen vascular disorders. In a number of cases of the less common associations of disorders with PRCA, particularly nonlymphoproliferative hematologic malignancies such as chronic myelogenous leukemia or nonthymoma solid tumors, the clinical course of PRCA may run independent of the course of the associated syndrome, suggesting the association is coincidental, rather than pathophysiologic.7
Specific syndromes of secondary PRCA
B19 parvovirus-associated PRCA.
Human B19 parvovirus is responsible for the aplastic crisis seen in patients with chronic hemolytic disorders such as sickle cell anemia, and can produce chronic PRCA in immunocompromised patients.8 B19 parvovirus directly infects human erythroid progenitors through the red cell surface P antigen (globoside). Individuals whose erythroid progenitors do not express P antigen are resistant to parvovirus infection.9
Recombinant human erythropoietin–induced antibody-mediated PRCA.
Although cases of primary autoimmune PRCA caused by de novo antibodies against endogenous erythropoietin (Epo) have been described, they are very rare. Beginning in the 1990s, reports of PRCA associated with anti-Epo antibodies in patients with renal failure treated with recombinant human Epo (rhEpo) for anemia began to appear.10 These cases were predominantly associated with subcutaneous rhEpo administration and primarily occurred outside the United States; more than 90% of cases involved a particular rhEpo product. Epidemiologic studies eventually implicated potential adjuvant effects of leachates from rubber stoppers in prefilled syringes and particular stabilizers in the specific rhEpo formulation. With resolution of these issues, new cases have become rare.11
Thymoma-associated PRCA.
Thymoma is the disorder with the strongest historical association with secondary PRCA. At one time, it was believed that thymoma was associated with up to 50% of cases of PRCA. The finding of PRCA may precede the finding of a thymoma or may occur after its resection. Recent series suggest PRCA is found in less than 5% of patients with thymoma.12 Among patients presenting with PRCA, the frequency of thymoma appears to be somewhere in the 7% to 10% range (the retrospective nature of many reports makes it difficult to definitively separate cases of PRCA that developed after thymectomy).13,14
Lymphoproliferative disorder–associated.
The lymphoproliferative disorders most frequently associated with PRCA are chronic lymphocytic leukemia and large granular lymphocyte (LGL) leukemia,15,16 although it has been reported with Hodgkin and non-Hodgkin lymphoma, as well as Waldenstrom macroglobulinemia, as outlined in Table 1. A number of small studies have suggested an increased frequency of otherwise unapparent clonal T-cell disorders in primary acquired PRCA.17 PRCA is typically immune mediated in these cases, although usually not by antibody-dependent mechanisms.18
Drugs and chemicals.
At least 50 drugs and chemicals have been associated with PRCA. The list includes a number of “usual suspects” for hematologic toxicity, such as diphenylhydantoin, azathioprine, and isoniazid.19 Table 2 lists several drugs implicated in PRCA, according to a PubMed review. Many of the reported associations derive from single case reports. Studies of underlying mechanisms are infrequent. PRCA associated with some drugs such as diphenylhydantoin and rifampicin has been linked to IgG-mediated inhibition of erythropoiesis.20,21 A number of agents implicated in PRCA do not appear in Table 2, such as benzene, halothane, methazolamide, phenobarbital, sulfathiazole, thiamphenicol, and tolbutamide, as these particular reports are not indexed in PubMed (frequently because they antedate the indexing range).
Table 2.
Selected drugs associated with PRCA based on PubMed review
| Agent | Multiple reports | Mechanism investigated |
|---|---|---|
| Alemtuzumab | ||
| Allopurinol | ✓ | |
| Ampicillin | ||
| Azathioprine | ✓ | ✓ |
| Carbamazepine | ✓ | |
| Cephalothin | ||
| Cladribine | ||
| Chlorpropamide | ✓ | |
| Chloroquine | ||
| Clopidogrel | ||
| Dapsone/pyrimethamine | ✓ | |
| Diphenylhydantoin | ✓ | ✓ |
| Recombinant Epo | ✓ | ✓ |
| Estrogens | ||
| Fenoprofen | ✓ | |
| Fludarabine | ✓ | |
| Interferon-α | ✓ | |
| Isoniazid | ✓ | ✓ |
| Lamivudine | ✓ | |
| Leuprolide | ✓ | |
| Linezolid | ✓ | |
| Micafungin | ||
| Mycophenolate mofetil | ✓ | |
| d-Penicillamine | ✓ | |
| Phenylbutazone | ||
| Procainamide | ✓ | |
| Ribavirin | ✓ | |
| Rifampicin | ✓ | |
| Sulfasalazine | ✓ | |
| Sulindac | ||
| Tacrolimus | ✓ | |
| Trimethoprim/sulfamethoxazole | ✓ | |
| Valproic acid | ✓ | ✓ |
| Zidovudine | ✓ |
PubMed search performed May 15, 2016. Search terms used were “red cell aplasia” AND the name of the specific drug.
Pregnancy.
Pregnancy has been associated with PRCA and usually (although not always) resolves after delivery.22 Occurrence of PRCA with a pregnancy does not necessarily predict recurrence with a subsequent pregnancy. The possibility of B19 parvovirus infection should be considered.
ABO-incompatible stem cell transplantation.
PRCA after ABO-incompatible bone marrow or stem cell transplant is observed most commonly with the combination of a blood group A donor and a blood group O recipient. It was reported in 7.5% of cases of ABO-incompatible transplant in 1 recent series. Although there is a high frequency of spontaneous resolution after a period of transfusion support (sometimes lasting months), 30% to 40% of cases will result in chronic PRCA requiring additional measures.23
Clinical features and diagnosis
Clinical presentation
There is no specific clinical presentation of primary acquired PRCA; the signs and symptoms are only those associated with anemia. Because PRCA is a pure underproduction anemia, the gradual decline in hemoglobin concentration allows some degree of adaptation, and symptoms may be less than would be expected for the degree of anemia. Patients with secondary PRCA may of course manifest the symptomatology of the associated syndrome.
Laboratory findings/diagnostic evaluation
Peripheral blood counts.
Red cells in PRCA are normochromic and normocytic. The absolute reticulocyte count is always less than 10 000/µL (reticulocyte percentage, <1%), and in many cases is much lower. The diagnosis of PRCA should be questioned with higher reticulocyte values or if the reticulocyte percentage is only less than 1% when corrected for the degree of anemia. In general, the white blood count, white blood cell differential, and platelet count are normal. In the setting of concurrent inflammation, there may be some modest reduction in the total white blood count or a mild abnormality (either slightly high or slightly low) in the platelet count. There may also be a mild relative lymphocytosis.
Bone marrow morphology.
The diagnosis of PRCA requires a bone marrow examination. In primary acquired (autoimmune) PRCA, marrow cellularity and myeloid and megakaryocyte maturation are normal. The diagnosis of PRCA is based on the absence or near absence of erythroblasts from an otherwise normal marrow (<1% erythroblasts on the marrow differential count). In some cases, a few proerythroblasts and/or basophilic erythroblasts are seen, not exceeding 5% of the differential count. Large proerythroblasts with vacuolated cytoplasm and pseudopodia (“giant pronormoblasts”) are suggestive of B19 parvovirus infection, but are not diagnostic24 (Figure 1). There may be some slight increase in lymphocytes, lymphoid aggregates, and/or plasma cells, reflecting a degree of immune/inflammatory activation. Iron stains will typically be normal. Because of the paucity of erythroid precursors, ring sideroblasts are difficult to see. If present, ring sideroblasts favor a myelodysplastic syndrome, as would marked hypercellularity.
Figure 1.
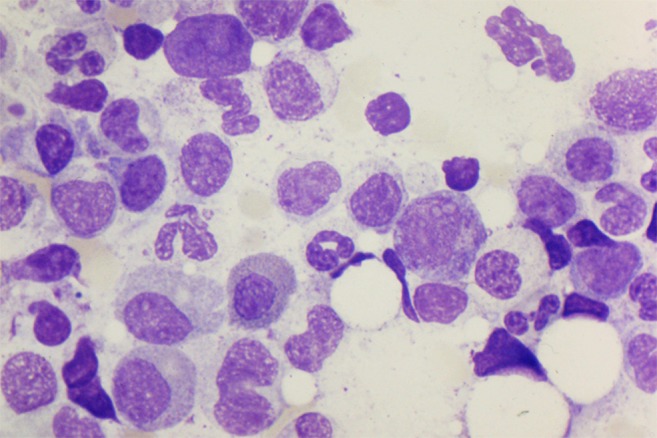
Bone marrow aspirate specimen from human immunodeficiency virus–infected patient with PRCA as a result of parvovirus. Giant pronormoblasts near the center of the field (100×).
Cellular immunology/cytogenetics.
As with all diagnostic examination of the bone marrow for cytopenias, material should be collected for cellular immunology, cytogenetics, and clonal analysis of T-cell receptors. Abnormal cytogenetics in the setting of a characteristic marrow for PRCA indicates the myelodysplastic variant of PRCA. If increased lymphocytes or plasma cells are present, they should be polyclonal in acquired immune PRCA. If clonal lymphocytes are present, it suggests PRCA secondary to an associated lymphoproliferative disorder. T-cell-receptor gene rearrangement studies should be performed routinely.17
Parvovirus studies.
In all patients with marrows diagnostic of PRCA, B19 parvovirus testing should be performed. The test of choice is polymerase chain reaction testing on peripheral blood.
Radiologic studies.
In adults with PRCA and no evidence of parvovirus infection or of a disorder associated with secondary PRCA, a computed tomography scan of the chest should be performed to rule out a thymoma, which would have potential therapy implications.
Treatment
General comments
The treatment of PRCA associated with thymoma, B19 parvovirus infection, ABO-incompatible stem cell transplantation, antibody-mediated inhibition induced by rhEpo, or pregnancy will be discussed later. DBA is typically treated with glucocorticoids, usually prednisone. Hematopoietic stem cell transplantation has also been used in patients unresponsive to glucocorticoids at sustainable doses.25 Myelodysplastic primary PRCA should be treated as appropriate for a myelodysplastic syndrome.5 If the patient is using a medication associated with PRCA, and no other PRCA-associated syndrome is present, a trial of drug discontinuation should be considered. If an infectious disorder associated with PRCA is present, specific treatment of that disorder should be initiated. Similarly, if chronic lymphocytic leukemia, Hodgkin or non-Hodgkin lymphoma, or other lymphoproliferative disorders are complicating PRCA, treatment of those disorders should be initiated.
Otherwise, for primary acquired autoimmune PRCA, PRCA with LGL leukemia, PRCA associated with solid tumors, or secondary PRCA refractory to other therapy, the treatment of choice is immunosuppression. In principle, PRCA secondary to autoimmune/collagen vascular disorders may respond to therapy specific to the management of those disorders. In practice, the treatment of these disorders is usually immunosuppression, and in cases where alternative disease-modulating therapy is available, the patient is usually referred to the hematologist because it was ineffective. The goal of treatment is to attain a normal hemoglobin concentration without any requirement for transfusion; a partial response is attainment of transfusion independence with a low but clinically acceptable hemoglobin concentration.
Immunosuppression/immunomodulation
Table 3 lists pooled data from 4 series evaluating immunosuppressive therapy in PRCA.1,13,26,27 Overall, repeated trials of immunosuppression using a succession of agents can produce a response in approximately 2/3 of patients with PRCA. The traditional immunosuppression approach began with oral corticosteroids, usually prednisone, in doses similar to those used for immune thrombocytopenic purpura (1 mg/kg). Overall, this approach appears to have a 40% response rate. Prednisone is tapered after a response is obtained, or decreased and used in association with another agent if remission is not obtained.
Table 3.
Response of patients with PRCA to immunosuppression
| Agent used | Patients treated (responders/total) | % responding |
|---|---|---|
| Corticosteroids | 50/128 | 39% |
| Cytotoxic agents | 46/113 | 41% |
| Cyclosporine A | 37/48 | 77% |
| Antithymocyte globulin | 10/19 | 53% |
| Multiple agents | 91/133 | 68% |
The most effective immunosuppressive agent is cyclosporine A. Its overall response rate of more than 75% is particularly impressive, given that a number of the early series reporting its use consisted largely of patients who had failed a number of therapeutic modalities previously.4,13 Cyclosporine can be considered the agent of choice for immunosuppression in PRCA,1 or it can be considered the second-line agent after corticosteroids. A reasonable starting dose is 6 mg/kg daily. It is sometimes used in association with prednisone 30 mg/day.4 Cyclosporine trough levels should be monitored with target levels of 150 to 250 ng/mL.1 Renal and hepatic function should also be monitored. After normalization of the hemoglobin concentration, cyclosporine can be tapered slowly. Maintenance therapy may be required.14
Cytotoxic agents such as azathioprine or cyclophosphamide (usually in combination with oral corticosteroids) have been used in patients unresponsive to corticosteroids alone, or in patients who do not respond to cyclosporine. The response rate in this context is approximately 40%. Tacrolimus has also been reported to be effective in PRCA and may provide an alternative to cyclosporine,28 but has also been reported as a cause of PRCA (Table 2). Rituximab, a humanized anti-CD20 monoclonal antibody, appears to have some efficacy against primary autoimmune PRCA,29 but is mainly used in PRCA secondary to lymphoproliferative disorders.30 Antithymocyte globulin, in the doses used for treatment of aplastic anemia, has a 50% response rate in primary autoimmune PRCA (Table 3). Modalities used in refractory primary PRCA with relatively low response rates include intravenous immunoglobulin,31 plasma exchange,32 splenectomy,33 and bone marrow transplantation.34
The Japanese PRCA Consortium has provided long-term follow-up data on patients with PRCA after immunosuppressive therapy.14 The median survival for patients with primary autoimmune PRCA had not been reached at 250 months. For patients with LGL or thymoma-associated PRCA, median survivals were similar at 147.8 and 142.1 months, respectively. The principal causes of death were infection and organ failure. Factors predictive of death were refractoriness to therapy and relapse from response.14
Treatment of B19 parvovirus-associated PRCA
As noted earlier, every patient with diagnosis of PRCA should be tested for B19 parvovirus. A diagnosis of B19 parvovirus associated PRCA is an indication for intravenous immunoglobulin as specific and highly effective therapy. A course of intravenous immunoglobulin for this purpose uses the usual therapeutic dose employed for disorders such as immune thrombocytopenic purpura: 2 g/kg usually divided over 5 days (400 mg/kg/d).8 In a recent review of an institutional experience and the published literature,35 the vast majority were significantly immunocompromised. PRCA corrected after a first course of intravenous immunoglobulin in 93% of patients, but approximately one-third relapsed, at mean time to relapse of 4.3 months.35
Thymoma-associated PRCA
If a thymoma is present in a patient with PRCA, it generally should be resected. Although earlier studies reported more optimistic results, no more than a third of patients will experience remission, and responses may be less than normalization of hemoglobin concentration.36 Relapses are frequent, and PRCA may develop after resection of a thymoma in patients who did not have PRCA previously.37 Adjuvant immunosuppressive therapy is typically required.36,37 The question of the value of thymectomy in PRCA likely requires reexamination. This will be a challenge requiring a multiinstitutional effort, as no single institution is likely to have more than a few cases.
PRCA after ABO-incompatible stem cell transplantation
As discussed earlier, ABO-incompatible stem cell transplantation can result in the generation of isohemagglutinins directed against donor red cell antigens also expressed on red cell precursors, leading to PRCA.23,38 There is a high frequency of spontaneous resolution, but prolonged transfusion support may be required. If antidonor isohemagglutinins persist longer than 2 months after transplant, the likelihood of spontaneous remission is reported to be low.38 Approaches taken to mitigate this problem include adjustment in the immunosuppressive regimen, donor leukocyte infusion, plasma exchange, and rituximab treatment.38
rhEpo-induced antibody-mediated PRCA
As discussed earlier, recognition of the factors that contributed to the unexpected development of anti-Epo antibodies in patients with renal failure and consequent presentation with PRCA has largely eliminated this mechanism as a cause of new cases of PRCA.39 However, patients with this significant problem may still be under the care of hematologists. Immunosuppression should be initiated, with cyclosporine A (with or without corticosteroids) as the probable first choice. Kidney transplantation should also be considered, if that is a possibility.40 Successful rechallenge with rhEpo in patients who no longer have detectable antibodies has been described in various case reports, but should be regarded as high-risk and is generally not recommended. Rechallenge with intravenous rhEpo has been suggested as a potentially less immunogenic strategy, but even that approach has resulted in a recurrence of PRCA.41
Pregnancy-associated PRCA
Because of the rarity of this syndrome, there is very little experience to guide management recommendations. Most patients will have resolution of PRCA at the end of pregnancy and can be supported with transfusion as needed; prednisone has also been used for immunosuppression.22,42 The majority of reported pregnancies result in a live delivery at term if anemia is controlled.43 Cyclosporine and other immunosuppressive agents may have significant effects on fetal outcome and maternal morbidity and probably should be avoided. Testing for B19 parvovirus and treatment of patients with positive results is reasonable.
Summary/conclusion
PRCA should be suspected in patients with an isolated anemia associated with marked reticulocytopenia. The diagnosis requires bone marrow examination demonstrating the absence or near absence of erythroblasts from an otherwise normal marrow. Evaluation of acquired PRCA should focus on identifying patients with myelodysplastic syndrome presenting with erythroid hypoplasia or PRCA associated with drugs, with B19 parvovirus infection, with thymoma, or with lymphoproliferative disorders, for whom syndrome-specific management is required. For other patients with primary or secondary acquired PRCA, the treatment of choice is immunosuppression. Cyclosporine A, with or without concurrent corticosteroids, appears to be the single most effective immunosuppressive agent.
Acknowledgments
Many years ago, the author was introduced to PRCA by Sanford Krantz and Emmanuel Dessypris and learned the techniques of hematopoietic cell culture from Ken-ichi Sawada, who, with his colleagues from the Japanese PRCA Consortium, continues the rich tradition of translational research in PRCA. The mentorship and intellectual generosity of these friends and colleagues are gratefully acknowledged.
References
- 1.Sawada K, Fujishima N, Hirokawa M. Acquired pure red cell aplasia: updated review of treatment. Br J Haematol. 2008;142(4):505-514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruggero D, Shimamura A. Marrow failure: a window into ribosome biology. Blood. 2014;124(18):2784-2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fleming MD. The genetics of inherited sideroblastic anemias. Semin Hematol. 2002;39(4):270-281. [DOI] [PubMed] [Google Scholar]
- 4.Means RT, Dessypris EN, Krantz SB. Treatment of refractory pure red cell aplasia with cyclosporine A: disappearance of IgG inhibitor associated with clinical response. Br J Haematol. 1991;78:114-119. [DOI] [PubMed] [Google Scholar]
- 5.Cerchione C, Catalano L, Cerciello G, et al. . Role of lenalidomide in the management of myelodysplastic syndromes with del(5q) associated with pure red cell aplasia (PRCA). Ann Hematol. 2015;94(3):531-534. [DOI] [PubMed] [Google Scholar]
- 6.van den Akker M, Dror Y, Odame I. Transient erythroblastopenia of childhood is an underdiagnosed and self-limiting disease. Acta Paediatr. 2014;103(7):e288-e294. [DOI] [PubMed] [Google Scholar]
- 7.Dessypris EN, McKee CL Jr, Metzantonakis C, Teliacos M, Krantz SB. Red cell aplasia and chronic granulocytic leukaemia. Br J Haematol. 1981;48(2):217-225. [DOI] [PubMed] [Google Scholar]
- 8.Frickhofen N, Chen ZJ, Young NS, Cohen BJ, Heimpel H, Abkowitz JL. Parvovirus B19 as a cause of acquired chronic pure red cell aplasia. Br J Haematol. 1994;87(4):818-824. [DOI] [PubMed] [Google Scholar]
- 9.Brown KE, Hibbs JR, Gallinella G, et al. . Resistance to parvovirus B19 infection due to lack of virus receptor (erythrocyte P antigen). N Engl J Med. 1994;330(17):1192-1196. [DOI] [PubMed] [Google Scholar]
- 10.Carson KR, Evens AM, Bennett CL, Luminari S. Clinical characteristics of erythropoietin-associated pure red cell aplasia. Best Pract Res Clin Haematol. 2005;18(3):467-472. [DOI] [PubMed] [Google Scholar]
- 11.Bennett CL, Starko KM, Thomsen HS, et al. . Linking drugs to obscure illnesses: lessons from pure red cell aplasia, nephrogenic systemic fibrosis, and Reye’s syndrome. a report from the Southern Network on Adverse Reactions (SONAR). J Gen Intern Med. 2012;27(12):1697-1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernard C, Frih H, Pasquet F, et al. . Thymoma associated with autoimmune diseases: 85 cases and literature review. Autoimmun Rev. 2016;15(1):82-92. [DOI] [PubMed] [Google Scholar]
- 13.Dessypris EN. Pure red cell aplasia. Baltimore, MD: Johns Hopkins University Press; 1988. [Google Scholar]
- 14.Hirokawa M, Sawada K, Fujishima N, et al. ; PRCA Collaborative Study Group. Long-term outcome of patients with acquired chronic pure red cell aplasia (PRCA) following immunosuppressive therapy: a final report of the nationwide cohort study in 2004/2006 by the Japan PRCA collaborative study group. Br J Haematol. 2015;169(6):879-886. [DOI] [PubMed] [Google Scholar]
- 15.Fujishima N, Sawada K, Hirokawa M, et al. ; PRCA Collaborative Study Group. Long-term responses and outcomes following immunosuppressive therapy in large granular lymphocyte leukemia-associated pure red cell aplasia: a Nationwide Cohort Study in Japan for the PRCA Collaborative Study Group. Haematologica. 2008;93(10):1555-1559. [DOI] [PubMed] [Google Scholar]
- 16.Visco C, Barcellini W, Maura F, Neri A, Cortelezzi A, Rodeghiero F. Autoimmune cytopenias in chronic lymphocytic leukemia. Am J Hematol. 2014;89(11):1055-1062. [DOI] [PubMed] [Google Scholar]
- 17.Masuda M, Teramura M, Matsuda A, et al. . Clonal T cells of pure red-cell aplasia. Am J Hematol. 2005;79(4):332-333. [DOI] [PubMed] [Google Scholar]
- 18.Hirokawa M, Sawada K, Fujishima N, et al. . Acquired pure red cell aplasia associated with malignant lymphomas: a nationwide cohort study in Japan for the PRCA Collaborative Study Group. Am J Hematol. 2009;84(3):144-148. [DOI] [PubMed] [Google Scholar]
- 19.Thompson DF, Gales MA. Drug-induced pure red cell aplasia. Pharmacotherapy. 1996;16(6):1002-1008. [PubMed] [Google Scholar]
- 20.Dessypris EN, Redline S, Harris JW, Krantz SB. Diphenylhydantoin-induced pure red cell aplasia. Blood. 1985;65(4):789-794. [PubMed] [Google Scholar]
- 21.Mariette X, Mitjavila MT, Moulinie JP, et al. . Rifampicin-induced pure red cell aplasia. Am J Med. 1989;87(4):459-460. [DOI] [PubMed] [Google Scholar]
- 22.Choudry MA, Moffett BK, Laber DA. Pure red-cell aplasia secondary to pregnancy, characterization of a syndrome. Ann Hematol. 2007;86(4):233-237. [DOI] [PubMed] [Google Scholar]
- 23.Aung FM, Lichtiger B, Bassett R, et al. . Incidence and natural history of pure red cell aplasia in major ABO-mismatched haematopoietic cell transplantation. Br J Haematol. 2013;160(6):798-805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Au WY, Cheng VC, Wan TS, Ma SK. Myelodysplasia masquerading as parvovirus-related red cell aplasia with giant pronormoblasts. Ann Hematol. 2004;83(10):670-671. [DOI] [PubMed] [Google Scholar]
- 25.Narla A, Vlachos A, Nathan DG. Diamond Blackfan anemia treatment: past, present, and future. Semin Hematol. 2011;48(2):117-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lacy MQ, Kurtin PJ, Tefferi A. Pure red cell aplasia: association with large granular lymphocyte leukemia and the prognostic value of cytogenetic abnormalities. Blood. 1996;87(7):3000-3006. [PubMed] [Google Scholar]
- 27.Charles RJ, Sabo KM, Kidd PG, Abkowitz JL. The pathophysiology of pure red cell aplasia: implications for therapy. Blood. 1996;87(11):4831-4838. [PubMed] [Google Scholar]
- 28.Yoshida S, Konishi T, Nishizawa T, Yoshida Y. Effect of tacrolimus in a patient with pure red-cell aplasia. Clin Lab Haematol. 2005;27(1):67-69. [DOI] [PubMed] [Google Scholar]
- 29.Auner HW, Wölfler A, Beham-Schmid C, Strunk D, Linkesch W, Sill H. Restoration of erythropoiesis by rituximab in an adult patient with primary acquired pure red cell aplasia refractory to conventional treatment. Br J Haematol. 2002;116(3):727-728. [DOI] [PubMed] [Google Scholar]
- 30.D’Arena G, Vigliotti ML, Dell’Olio M, et al. . Rituximab to treat chronic lymphoproliferative disorder-associated pure red cell aplasia. Eur J Haematol. 2009;82(3):235-239. [DOI] [PubMed] [Google Scholar]
- 31.Mouthon L, Guillevin L, Tellier Z. Intravenous immunoglobulins in autoimmune- or parvovirus B19-mediated pure red-cell aplasia. Autoimmun Rev. 2005;4(5):264-269. [DOI] [PubMed] [Google Scholar]
- 32.Freund LG, Hippe E, Strandgaard S, Pelus LM, Erslev AJ. Complete remission in pure red cell aplasia after plasmapheresis. Scand J Haematol. 1985;35(3):315-318. [DOI] [PubMed] [Google Scholar]
- 33.Zaentz DS, Krantz SB, Sears DA. Studies on pure red cell aplasia. VII. Presence of proerythroblasts and response to splenectomy: a case report. Blood. 1975;46(2):261-270. [PubMed] [Google Scholar]
- 34.Kochethu G, Baden HS, Jaworska E, Chang J, Chopra R. Reduced intensity conditioning bone marrow transplantation for pure red cell aplasia: successful outcome but difficult post transplant course. Bone Marrow Transplant. 2005;36(1):81-82. [DOI] [PubMed] [Google Scholar]
- 35.Crabol Y, Terrier B, Rozenberg F, et al. ; Groupe d’experts de l’Assistance Publique-Hôpitaux de Paris. Intravenous immunoglobulin therapy for pure red cell aplasia related to human parvovirus b19 infection: a retrospective study of 10 patients and review of the literature. Clin Infect Dis. 2013;56(7):968-977. [DOI] [PubMed] [Google Scholar]
- 36.Thompson CA, Steensma DP. Pure red cell aplasia associated with thymoma: clinical insights from a 50-year single-institution experience. Br J Haematol. 2006;135(3):405-407. [DOI] [PubMed] [Google Scholar]
- 37.Hirokawa M, Sawada K, Fujishima N, et al. ; PRCA Collaborative Study Group. Long-term response and outcome following immunosuppressive therapy in thymoma-associated pure red cell aplasia: a nationwide cohort study in Japan by the PRCA collaborative study group. Haematologica. 2008;93(1):27-33. [DOI] [PubMed] [Google Scholar]
- 38.Worel N. ABO-mismatched allogeneic hematopoietic stem cell transplantation. Transfus Med Hemother. 2016;43(1):3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Macdougall IC, Casadevall N, Locatelli F, et al. ; PRIMS study group. Incidence of erythropoietin antibody-mediated pure red cell aplasia: the Prospective Immunogenicity Surveillance Registry (PRIMS). Nephrol Dial Transplant. 2015;30(3):451-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Macdougall IC, Roger SD, de Francisco A, et al. . Antibody-mediated pure red cell aplasia in chronic kidney disease patients receiving erythropoiesis-stimulating agents: new insights. Kidney Int. 2012;81(8):727-732. [DOI] [PubMed] [Google Scholar]
- 41.Shimizu H, Saitoh T, Ota F, et al. . Pure red cell aplasia induced only by intravenous administration of recombinant human erythropoietin. Acta Haematol. 2011;126(2):114-118. [DOI] [PubMed] [Google Scholar]
- 42.Moussa M, Hassan MF. Newly diagnosed adult-onset Still’s disease with pure red cell aplasia in pregnancy. Arch Gynecol Obstet. 2014;290(1):195-198. [DOI] [PubMed] [Google Scholar]
- 43.Kashyap R, Pradhan M. Maternal and fetal outcome in pregnancy-associated pure red cell aplasia. J Obstet Gynaecol. 2010;30(7):733-734. [DOI] [PubMed] [Google Scholar]
- 44.Lipton J, Glader B, Means RT. Red cell aplasia: acquired and congenital disorders. In: Greer JP, Arber DA, Glader BE, List A, Means RT, Paraskevas F, Rodgers GM, eds. Wintrobe’s Clinical Hematology. 13th ed Philadelphia, PA: Lippincott Williams & Wilkins; 2013:975-89. [Google Scholar]