

Abstract
Dysregulated alternative splicing (AS) event that contribute to diabetes pathogenesis have been identified, but little is known about the RNA binding proteins (RBPs) involved. We have previously found that the RBP CELF1 is upregulated in the diabetic hearts; however, it is unclear if CELF1 contributes to diabetes-induced AS changes. Utilizing genome wide approaches, we identified extensive changes in AS patterns in Type 1 diabetic (T1D) mouse hearts. We discovered that many aberrantly spliced genes in T1D hearts have CELF1 binding sites. CELF1-regulated AS affects key genes within signaling pathways relevant to diabetes pathogenesis. Disruption of CELF1 binding sites impairs AS regulation by CELF1. In sum, our results indicate that CELF1 target RNAs are aberrantly spliced in the T1D heart leading to abnormal gene expression. These discoveries pave the way for targeting RBPs and their RNA networks as novel therapies for cardiac complications of diabetes.
Keywords: Diabetic heart, CELF1, alternative splicing, RNA binding proteins
INTRODUCTION
Type 1 diabetes results from autoimmune-mediated obliteration of pancreatic beta islet cells that produce insulin. Uncontrolled diabetes can lead to complications that impair quality of life and even result in death [1–3]. Cardiovascular complications are the major causes of mortality among diabetic patients [4–6]. In addition to hypertension and atherosclerosis, diabetic conditions adversely affect the cardiac muscle cells leading to heart failure [7–9, 6, 9]. Diabetic conditions dramatically affect gene expression and AS contributing to disease pathogenesis [10, 11], but there is limited information on AS regulators responsible for such changes [11, 12].
AS allows selective inclusion or exclusion of alternative exons, introns, or parts of exons and introns into the mature mRNA; thereby controlling gene expression and generating different protein isoforms [13–15]. Precise regulation of AS is critical for heart function and development [16, 17]. AS is controlled by RBPs that bind RNA and interact with the splicing machinery. Although RBPs have emerging roles in diabetes, only a few RBPs are associated with cardiac complications of diabetes [12, 19, 20].
We have previously shown that the RBPs RBFOX2 and CELF1 are altered in T1D hearts. We have demonstrated the consequences of RBFOX2-mediated changes in diabetes cardiac pathogenesis [19]. However, the role of CELF1 in T1D-induced AS changes still remains unknown. CELF1 regulates AS, mRNA translation and mRNA decay [21–23]. Overexpression of CELF1 in mouse hearts leads to dilated cardiomyopathy and heart failure associated with defective AS patterns [24]. Therefore, in this study we comprehensively analyzed the cardiac transcriptome and tested whether CELF1 contributes to AS changes in T1D hearts. Our results indicate that many putative CELF1 targets with important functions in the heart, displayed altered AS patterns in T1D hearts. Importantly we show that CELF1 regulates AS of these transcripts that are differentially spliced in T1D hearts.
METHODS
Type 1 diabetes mouse models
The animal models used for this study are streptozotocin (STZ)-induced T1D and non-obese diabetic (NOD). All animal experiments were conducted in accordance with the NIH Guidelines and approved by the University of Texas Medical Branch Institutional Animal Care and Use Committee (Protocol # 1101001). T1D mouse models were described in [11, 25]. C57BL6J male mice (Jackson laboratories) were injected with citrate buffer (vehicle control) or 60 mg/kg of STZ daily for five consecutive days. Female NOD (NOD/ShiLtJ) and ICR control mice were obtained from Jackson laboratories [25]. Mice with hyperglycemia greater than 400mg/dL for at least 3 weeks were sacrificed and left ventricles were isolated for RNA extraction.
Quantitative RT-PCR and statistical analysis
For qRT-PCR, RNA was extracted from cells and mouse hearts using TRIzol (Invitrogen 15596–018) according to the manufacturer’s protocol. qRT-PCR was performed as described previously [19, 20]. Student t-test was used to determine statistical significance between two groups using Prism software.
Transfections
H9C2 cells were cultured and transfected as previously described [19, 20]. H9c2 cells were transfected with a pool of 10µM CELF1 specific (Invitrogen siRNA ID# s168616 and s168617), or scrambled siRNAs (Invitrogen AM4611) using Lipofectamine RNAiMAX (Invitrogen) and harvested 72hrs later. H9c2 cells were transfected with GFP or GFP-tagged CELF1 using neon nucleofection (Invitrogen) and harvested after 48hrs. H9c2 cells were transfected with 200ng WT, or CELF1 binding site mutant Tmem184b splicing reporter and harvested after 72hrs.
Differential alternative splicing (DAS) and differential gene expression (DEG) analysis and statistic analysis
An Illumina HiSeq 1000 system was used for paired end (2 × 15 cycles) RNA sequencing at the UTMB Next-Generation Sequencing Core facility and yielded ~200 million reads per sample [19, 20]. To identify AS changes, we performed DAS analysis as previously described in [26]. Briefly, raw RNA-Seq reads were first aligned to mouse (mm9) genome using STAR (version 2.5.1b) [27] with default settings and only uniquely mapped reads were retained for further analysis. The number of reads for each exon and each exon-exon junction in each RNA-Seq file was computed by using the Python package HTSeq [28] with the UCSC KnownGene (mm9) annotation [29]. Dirichlet-multinomial was used to model the counts of the reads aligned to each isoform of each event [30], and the likelihood ratio test was used to test the significance of splicing changes between STZ and control mice. We calculated the q-values from the p-values in the likelihood ratio test by the Benjamini-Hochberg procedure. PSI (Percent Spliced In; Ψ) was used to evaluate the percentage of the inclusion of variable exon relative to the total mature mRNA in the splicing events [31]. The DAS events were identified under |ΔΨ| > 0.05 and q < 0.05.
For DEG analysis, uniquely aligned reads were retained to calculate the read counts for each gene against the UCSC KnownGene annotation (mm9), and a count table was constructed by counting the number of the reads that were aligned uniquely to each of the genes for each sample. Normalization and DEG analysis were performed using DESeq [32]. FDR adjusted q-values were then calculated using the Benjamini-Hochberg procedure. The log2-fold changes of each comparison were also calculated for each gene. The differentially expressed genes were identified under two-fold changes and q < 0.05. Differential cassette exon (ES) events were overlapped with CELF1 binding sites that were extracted from the CELF1 cross-linking immunoprecipitation followed by RNA-seq (CELF1 CLIP-seq) datasets. CELF1 binding sites were examined within the 250bp intronic regions flanking the alternative exon and 50bp downstream of 5’ end of the alternative exons.
Plasmids
The Tmem184b splicing reporter was constructed as follows: pcDNA5-GFP-IL7R splicing reporter plasmid (from Mariano Garcia-Blanco) was mutated from TCTAGA to GATAGA to remove one of the two Xba1 sites. Mutated vector was then digested with Xba1 and Xho1 (Roche) and purified with the QIAquick PCR purification kit. Tmem184b genomic sequence that covers intron 9 (500nt)-exon9-intron 10 (316nt) or a mutant version of Tmem184b genomic sequence that lacks one of the CELF1 CLIP-seq peaks (62nt) in intron 10 (G-blocks from IDT) were ligated into the vector using T4 DNA ligase (Roche). All constructs were confirmed by Sanger DNA sequencing.
RESULTS
Widespread AS and gene expression changes in Type 1 diabetic mouse hearts
We have previously identified cassette exon splicing changes in T1D hearts [11]. In the current study, we extended our analyses and determined DEG as well as all forms of DAS in the T1D heart using our RNA-Seq data obtained from STZ:T1D mouse hearts (GSE80664) [20]. We identified seven major types of AS events including cassette exon splicing (ES), alternative 5’ splice site (A5SS), alternative 3’ splice site (A3SS), alternative first exon (AFE), alternative last exon (ALE), mutually exclusive exons (ME) and intron retention (IR) in diabetic hearts. We determined percent spliced in (PSI, Ψ) and found a total of 3768 statistically significant AS events altered in T1D hearts (Fig. 1A) with the criteria that |ΔΨ| > 0.05 and < 0.05 (Supp. Table S1). All major types of AS patterns were affected in T1D hearts, with the most common being the cassette exon splicing (1341/3768: 35.6% ES) (Fig. 1A). Of 1341 cassette exon splicing events detected in diabetic hearts, 910 were exon exclusion and 431 were exon inclusion events (Fig. 1B).
Figure 1: DAS and DEG analysis in T1D mouse hearts.
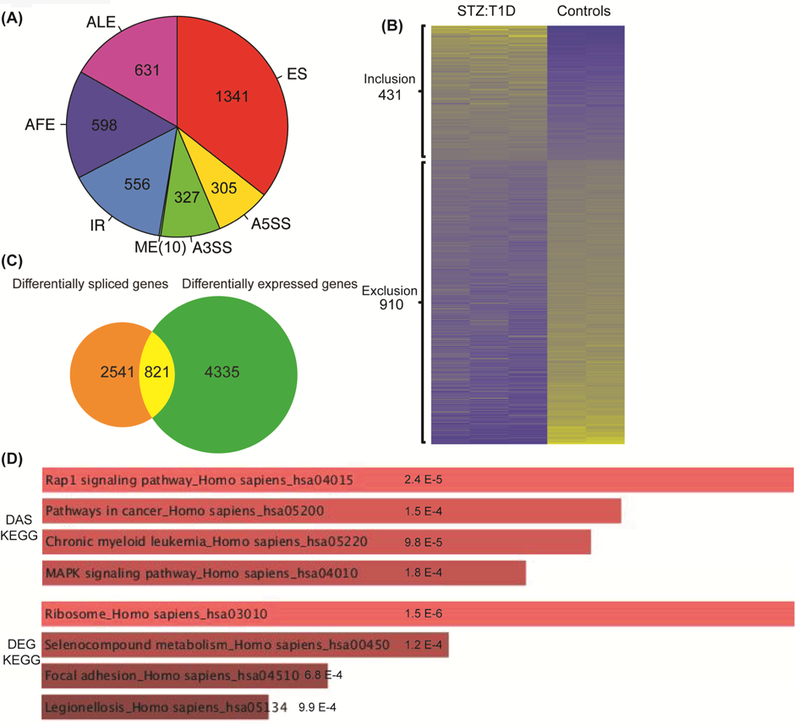
A) DAS analyses were performed using the RNA-seq data from STZ:T1D and mock treated, non-diabetic (Control) mouse hearts. AS changes in STZ:T1D mice were grouped into seven different categories based on the type of AS event: cassette Exon Splicing (ES), Alternative 5’ Splice Site (A5SS), Alternative 3’ Splice Site (A3SS), Mutually Exclusive exon (ME), Intron Retention (IR), Alternative First Exon (AFE) and Alternative Last Exon (ALE). The pie chart depicts the distribution of different splicing types identified in T1D hearts based on the criteria that |ΔΨ| > 0.05 and q < 0.05. The total DAS events was 3768. B) Heat map of alternative exons that are either included or excluded in T1D vs control mouse hearts based on percent spliced in (PSI) values. Yellow represents high PSI values and blue indicates low PSI values. C) Venn diagram comparison of DAS and DEG in STZ:T1D and control mouse hearts. 3362 DAS genes (some have multiple AS events) were compared to 5156 genes with a change in mRNA levels (>2 fold increase or decrease, and q < 0.05). D) The KEGG categories for DAS or DEG changes in T1D were identified using Enrichr (http://amp.pharm.mssm.edu/Enrichr). Significance is represented on the charts.
We identified 5156 differentially expressed genes in T1D hearts (>2 fold change and q < 0.05 ) (Supp. Table S2). Next, we checked whether mis-spliced genes display mRNA levels changes. Only 21.8% of mis-spliced transcripts (821 out of 3768) exhibited changes in mRNA levels (Fig. 1C and Supp. Table S3). KEGG pathway analysis of DAS genes identified Rap1 signaling, pathways in cancer, and MAPK signaling (DAS, Fig. 1D) while analysis of DEG in T1D hearts identified ribosomes, selenocompound metabolism, and focal adhesion processes to be affected in T1 hearts (DEG, Fig. 1D).
Since the majority of cassette exon splicing leads to removal of the protein coding regions affecting the protein output in T1D hearts, we focused on the top AS changes that favor exon exclusion. We validated three top changing exon exclusion events (Ablim3 exon 18, Per1 exon 16, and Gpr116 exon 12) that were not identified in our original AS analysis [20]. As expected, these pre-mRNAs displayed enhanced exon exclusion in STZ:T1D mice when compared to controls based on the number of reads mapped to these exons (Fig. 2A and Supp. Table S1) and by qRT-PCR (Fig. 2B). AS of these three genes were also affected in a different T1D mouse model known as NOD mice, favoring alternative exon exclusion (Fig. 2C). In sum, our DAS analysis successfully identified new AS events that were validated using two different T1D mouse models.
Figure 2: Validation of newly identified DAS events in two different T1D mouse models.
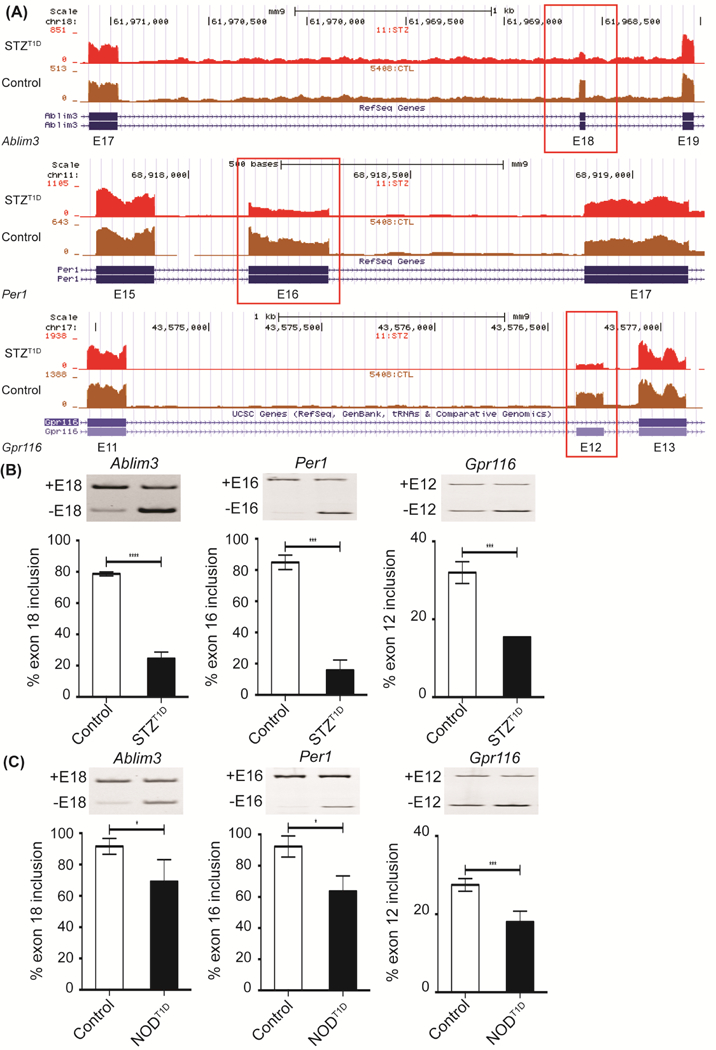
A) Representative genome browser images of AS of Ablim3 exon 18, Per1 exon 16, and Gpr116 exon 12 in mock treated, non-diabetic (Control) or STZ:T1D mice left ventricles based on RNA-Seq reads. % inclusion of Ablim3 exon 18, Per1 exon 16, and Gpr116 exon 12 was determined by qRT-PCR in B) Control or STZ:T1D (n≥3) and C) in non-diabetic (Control) or NOD:T1D mice left ventricles (n≥3). In AS gel figures +E# = Exon # inclusion and −E# = Exon # exclusion. Data represent means ± SD. P-values are represented as **** < 0.0001, *** < 0.001, * < 0.05.
Transcripts that display CELF1 binding sites undergo AS changes in Type 1 diabetic hearts
Since CELF1 protein is elevated in T1D hearts [19], we tested if CELF1 contributes to AS changes in T1D hearts. In vivo targets of many RBPs have been identified by several methods including CLIP-seq [18]. Thus, we used the CELF1 CLIP-seq dataset obtained from mouse hearts [22] to determine CELF1 binding sites within 250nt of the upstream and downstream intronic sequences and 50nt of the 5’ and 3’ ends of the alternative exons, mis-spliced in T1D hearts. Out of 1318 differential cassette exon (ES) events (with one variable exon/event), 138 transcripts with aberrant AS patterns in T1D hearts displayed CELF1 binding sites (Fig. 3A and Supp. Table S4). Importantly, 72% of CELF1 targets displayed aberrant exon exclusion in T1D hearts (Fig. 3B), impacting the protein coding region of these genes. KEGG pathway analysis of CELF1 targets identified the cGMP-PKG signaling pathway, aldosterone synthesis and vascular smooth muscle contraction as top categories (Fig. 3C and Supp. Table S5). We first validated the AS pattern of putative CELF1 target histone deacetylase 7 (HDAC7) identified by our CELF1 CLIP-seq vs RNA-seq comparison. HDAC7 pre-mRNA has CELF1 binding sites indicated by the mapped CELF1 CLIP-seq reads (Fig. 3D, black rectangle). HDAC7 controls transcription by affecting chromatin remodeling and myocyte enhancer factor proteins [34] and is implicated in vascular integrity [35]. In T1D hearts, Hdac7 exon 8 was more excluded (Fig. 3E). This AS change is expected to remove an exon within the histone deacetylase domain affecting its function. To validate whether CELF1 regulates HDAC7 AS, we depleted CELF1 in heart-derived H9c2 cells and examined endogenous HDAC7 AS. CELF1 knockdown induced almost complete exclusion of HDAC7 exon 8, suggesting that CELF1 regulates this exon. These results validate our CLIP-seq and RNA-seq comparison and show that CELF1 target RNAs undergo AS changes in T1D hearts.
Figure 3: CELF1 binding sites are present within transcripts mis-spliced in T1D hearts.
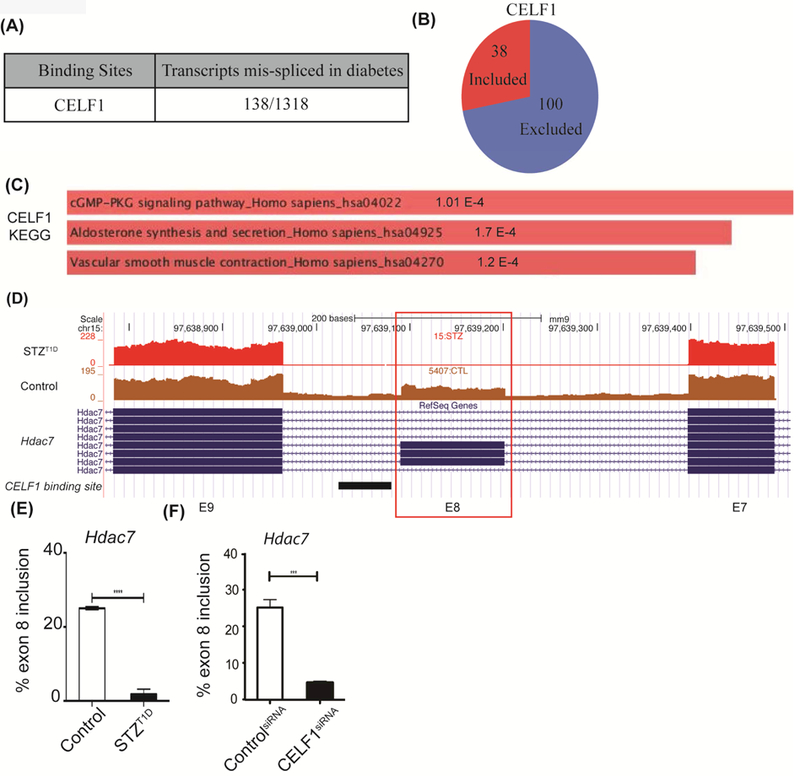
A) Number of transcripts differentially spliced in T1D mouse hearts that display CELF1 binding sites. CELF1 binding sites were identified by extracting data from CELF1 CLIP-Seq and overlapping these binding sites within transcripts (1318) mis-spliced in T1D hearts. To evaluate the significance of events that are positively or negatively regulated in both datasets, Fisher’s exact test was used to examine the enrichment. The Pearson correlation coefficient for regulated AS changes was calculated as 0.74, higher than the correlation coefficient of non-regulated events. B) Distribution of cassette exon inclusion versus exclusion in CELF1 target transcripts differentially spliced in diabetes. C) The KEGG pathway categories for CELF1-regulated AS events in T1D hearts using Enrichr. D) Representative genome browser images of putative CELF1 target Hdac7 exon 8 in Control vs STZ:T1D mice left ventricles. CELF1 binding sites derived from CLIP-Seq data were represented below the genome browser image as a black rectangle. E) AS of Hdac7 exon 8 in Control (n=3) vs STZ:T1D mice left ventricles (n=2). F) AS analysis of endogenous Hdac7 exon 8 in H9c2 cell transfected with scrambled (Control) or CELF1-specific siRNA pools (n=3). Data represent means ± SD. P-values are represented as **** < 0.0001, *** < 0.001.
CELF1 binding is important for regulation of AS targets mis-spliced in T1D hearts
Since putative CELF1 targets affected in T1D mouse hearts are within the cGMP signaling pathway (Fig. 3C) relevant to cardiac complications of diabetes, we investigated AS of transmembrane protein 184b (Tmem184b). The family member of TMEM184B, TMEM184A, was identified as a regulator of cGMP-PKG and MAPK signaling pathways [36] (Fig. 1D). Deletion of TMEM184B in mice leads to cardiovascular defects that include abnormal heart morphology and enlarged hearts (http://www.mousephenotype.org/data/genes/MGI:2445179). Notably, Tmem184b pre-mRNA has CELF1 binding sites and has altered AS pattern in T1D hearts according to our DAS analysis. In two different T1D mouse models, Tmem184b exon 9 was more skipped (Fig. 4A, right and left panels). To determine the regulation by CELF1, we depleted or ectopically expressed CELF1 in H9c2 cells. CELF1 depletion but not overexpression of CELF1 resulted in increased inclusion of Tmem184b exon 9, suggesting that CELF1 is a repressor of this exon (Fig. 4B-C). Proper nuclear localization of GFP-CELF1 was confirmed by GFP-fluorescence (Supp. Fig. 1).
Figure 4: CELF1 contributes to diabetes-induced AS changes.
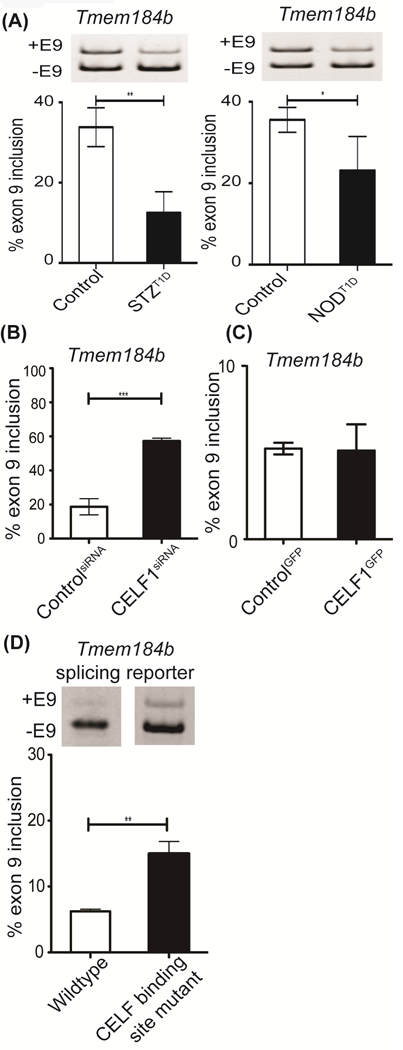
AS analysis of Tmem184b exon 9 A) in Control vs STZ:T1D (left graph) or Control vs NOD:T1D mice left ventricles (right graph) (n≥3), B) in H9c2 cell transfected with scrambled (Control) or CELF-specific siRNA pools (n=3), C) in H9c2 cells transfected with GFP (Control) or CELF1GFP (n=3), and D) in H9c2 cells transfected with wild-type or CELF1 binding site mutant Tmem184b splicing minigene reporter (n=4, two independent experiments). Data represent means ± SD. P-values are represented as *** < 0.001, ** < 0.01, * < 0.05.
To further investigate CELF-mediated regulation, we generated a splicing minigene reporter by inserting intron9-exon9-intron10 of Tmem184b in between the open reading frame of GFP [37]. We also constructed a mutant Tmem184b splicing minigene that removed one of the CELF1 CLIP-seq peaks that represents a CELF1 binding cluster. The removal of only one CELF1 binding peak led to more inclusion of exon 9 in comparison to WT Tmem184b (Fig. 4D). Knockdown of CELF1 (Fig. 4B) or removal of CELF1 binding site within Tmem184b pre-mRNA (Fig. 4D) led to increased exon inclusion indicating that CELF1 is a repressor of this exon. These results show that CELF1 binding site is important for repression of Tmem184b exon 9 inclusion and that CELF1 contributes to the aberrant AS of Tmem184b in T1D hearts.
DISCUSSION
In this study, we comprehensively analyzed transcriptome changes in STZ:T1D mouse hearts and found extensive DAS and DEG changes. We identified different types of AS events that are altered in diabetic hearts: 36% cassette exon splicing, ~15% intron retention, ~16% first exon splicing, and ~17% alternative last exon splicing (Fig. 1A). Although the majority of AS changes in diabetic hearts were independent of changes in mRNA levels (Fig. 1C), 821 transcripts exhibited changes in both AS and mRNA levels (Fig. 1). Of the cassette exon changes, exon exclusion events that remove protein-coding regions are favored in T1D hearts. Importantly, we validated these AS changes in two different T1D mouse models.
Previously, we have shown that RBFOX2 is a major contributor to AS dysregulation and cardiomyocyte dysfunction in the heart [20]. In this study, our goal was to determine whether other RBPs play a role in AS changes in T1D hearts. Our previous data indicate that CELF1 protein is increased in diabetic hearts [19]. In this study, we identified 138 CELF1 targets mis-spliced in T1D hearts and showed that CELF1 regulates AS of targets with binding sites (Figs. 3 and 4). Importantly, the majority of CELF1 target genes with roles in muscle contraction and PKG signaling display exon exclusion, which likely impacts protein function (Fig. 3B). Importantly, transgenic mice conditionally overexpressing CELF1 in cardiomyocytes develop dilated cardiomyopathy and heart failure associated with aberrant AS patterns. Therefore, we hypothesize that upregulation of CELF1 in the diabetic hearts could contribute to cardiomyopathy by affecting AS of critical genes.
Although CELF1 is upregulated in T1D hearts, our AS analysis of CELF1 targets indicate that diabetes-induced AS changes are consistent with CELF1 depletion or low CELF1 splicing activity. RBFOX2 splicing activity is reduced in diabetic hearts despite an increase in its protein levels [19, 20]. It is possible that an inactive isoform of CELF1 is also increased in T1D hearts or CELF1-regulated alternative exons are also controlled by a complex network of RBPs affected in T1D hearts. CELF1 and RBFOX2 oppositely regulate AS [22, 38, 39] and CELF1 and RBFOX2 have overlapping targets mis-spliced in diabetes [39]. Thus, we propose a model that changes in RBFOX2 and CELF1 jointly contribute to genome wide AS changes in the T1D hearts.
Supplementary Material
Table 1:
Primer information used for qRT-PCR
| Gene Name |
Species | Gene ID | Analysis Target |
Forward Primer (5’ to 3’) |
Reverse Primer (5’ to 3’) |
|---|---|---|---|---|---|
| Ablim3 | Mouse | ENSMU SG0000 0032735 |
Exon 18 | AAGCTCCTATG CGGATCCTT |
GCAGCCCATTT CTCCTGTAG |
| Per1 | Mouse | ENSMU SG0000 0020893 |
Exon 16 | GCCTCTGATGA TGACAAGCA |
GTTGGGTCAGG GGCTACTGT |
| Gpr116 | Mouse | ENSMU SG0000 0056492 |
Exon 12 | GTGTCCCAGTG GGTCTTCTG |
GGTCCCGGGTT ATTGTTAGG |
|
Tmem184 b |
Mouse | ENSMU SG0000 0009035 |
Exon 9 | GGCATGCCTTC ACCTACAAG |
GTGGACTGCTG CGTGTACTG |
|
Tmem184 b |
Rat | ENSRN OG0000 0022802 |
Exon 9 | GGCATGCCTTC ACCTACAAG |
GTGGACTGCTG CGTGTACTG |
| GFP | CCACAAGTTCA GCGTGTCCG |
CGTCCTTGAAG AAGATGGTG |
|||
| Hdac7 | Mouse | ENSMU SG0000 0022475 |
Exon 8 |
TTCCTCCCCCA GTAGTAGCA |
CGAGGGCCTAA AGTTGAATG |
| Hdac7 | Rat | Exon 8 |
TCCTCCCCCAG TAGTAGCAG |
CCAAAGTTGAAT GGGTCCTG |
Acknowledgements
We thank Kuyumcu-Martinez lab members and Dr. Eric Wagner for critically reading the manuscript and Dr. Mariano Garcia-Blanco for providing the Ad-GFP splicing reporter. This work was supported, in part, by an American Heart Association Grant [15GRNT22830010]; UTMB Department of Biochemistry and Molecular Biology Bridging funds; and a grant from the National Institutes of Health/ National Heart Lung Blood Institute [1R01HL135031] to M.N.K-M. The contents of the manuscript are solely the responsibility of the authors and do not necessarily represent the official views of NIH. This work was also supported by startup funding to P.Y. from the ECE department and Engineering Experiment Station/Dwight Look College of Engineering; by funding from TEES-AgriLife Center for Bioinformatics and Genomic Systems Engineering (CBGSE), by TEES seed grant; and by CAPES Research Grant Program at Texas A&M University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].El-Beblawy NM, Andrawes NG, Ismail EA, Enany BE, El-Seoud HS, Erfan MA, Serum and Urinary Orosomucoid in Young Patients With Type 1 Diabetes: A Link Between Inflammation, Microvascular Complications, and Subclinical Atherosclerosis, Clin Appl Thromb Hemost, 22 (2016) 718–726. [DOI] [PubMed] [Google Scholar]
- [2].Schneider AL, Barzilay JI, Diabetes, the brain, and cognition: More clues to the puzzle, Neurology, 87 (2016) 1640–1641. [DOI] [PubMed] [Google Scholar]
- [3].Coppini DV, Enigma of painful diabetic neuropathy: can we use the basic science, research outcomes and real-world data to help improve patient care and outcomes?, Diabet Med, 33 (2016) 1477–1482. [DOI] [PubMed] [Google Scholar]
- [4].Caprio S, Wong S, Alberti KG, King G, Cardiovascular complications of diabetes, Diabetologia, 40 Suppl 3 (1997) B78–82. [DOI] [PubMed] [Google Scholar]
- [5].Harcourt BE, Penfold SA, Forbes JM, Coming full circle in diabetes mellitus: from complications to initiation, Nature reviews. Endocrinology, 9 (2013) 113–123. [DOI] [PubMed] [Google Scholar]
- [6].Boudina S, Abel ED, Diabetic cardiomyopathy revisited, Circulation, 115 (2007) 3213–3223. [DOI] [PubMed] [Google Scholar]
- [7].Bugger H, Abel ED, Molecular mechanisms of diabetic cardiomyopathy, Diabetologia, 57 (2014) 660–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Miki T, Yuda S, Kouzu H, Miura T, Diabetic cardiomyopathy: pathophysiology and clinical features, Heart Fail Rev, 18 (2013) 149–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Mizamtsidi M, Paschou SA, Grapsa J, Vryonidou A, Diabetic cardiomyopathy: a clinical entity or a cluster of molecular heart changes?, Eur J Clin Invest, 46 (2016) 947–953. [DOI] [PubMed] [Google Scholar]
- [10].Arikawa E, Ma RC, Isshiki K, Luptak I, He Z, Yasuda Y, Maeno Y, Patti ME, Weir GC, Harris RA, Zammit VA, Tian R, King GL, Effects of insulin replacements, inhibitors of angiotensin, and PKCbeta’s actions to normalize cardiac gene expression and fuel metabolism in diabetic rats, Diabetes, 56 (2007) 1410–1420. [DOI] [PubMed] [Google Scholar]
- [11].Nutter CA, Jaworski EA, Verma SK, Deshmukh V, Wang Q, Botvinnik OB, Lozano MJ, Abass IJ, Ijaz T, Brasier AR, Garg NJ, Wehrens XHT, Yeo GW, Kuyumcu-Martinez MN, Dysregulation of RBFOX2 Is an Early Event in Cardiac Pathogenesis of Diabetes, Cell Rep, 15 (2016) 2200–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nutter CA, Kuyumcu-Martinez MN, Emerging roles of RNA-binding proteins in diabetes and their therapeutic potential in diabetic complications, Wiley Interdiscip Rev RNA, 9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Black DL, Mechanisms of alternative pre-messenger RNA splicing, Annual review of biochemistry, 72 (2003) 291–336. [DOI] [PubMed] [Google Scholar]
- [14].Chabot B, Directing alternative splicing: cast and scenarios, Trends in genetics: TIG, 12 (1996) 472–478. [DOI] [PubMed] [Google Scholar]
- [15].Breitbart RE, Andreadis A, Nadal-Ginard B, Alternative splicing: a ubiquitous mechanism for the generation of multiple protein isoforms from single genes, Annual review of biochemistry, 56 (1987) 467–495. [DOI] [PubMed] [Google Scholar]
- [16].Cooper TA, Alternative splicing regulation impacts heart development, Cell, 120 (2005) 1–2. [DOI] [PubMed] [Google Scholar]
- [17].Kalsotra A, Xiao X, Ward AJ, Castle JC, Johnson JM, Burge CB, Cooper TA, A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart, Proceedings of the National Academy of Sciences of the United States of America, 105 (2008) 20333–20338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Konig J, Zarnack K, Luscombe NM, Ule J, Protein-RNA interactions: new genomic technologies and perspectives, Nat Rev Genet, 13 (2012) 77–83. [DOI] [PubMed] [Google Scholar]
- [19].Verma SK, Deshmukh V, Liu P, Nutter CA, Espejo R, Hung ML, Wang GS, Yeo GW, Kuyumcu-Martinez MN, Reactivation of fetal splicing programs in diabetic hearts is mediated by protein kinase C signaling, J Biol Chem, 288 (2013) 35372–35386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nutter CA, Jaworski EA, Verma SK, Deshmukh V, Wang Q, Botvinnik OB, Lozano MJ, Abass IJ, Ijaz T, Brasier AR, Garg NJ, Wehrens XH, Yeo GW, Kuyumcu-Martinez MN, Dysregulation of RBFOX2 Is an Early Event in Cardiac Pathogenesis of Diabetes, Cell Rep, 15 (2016) 2200–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ladd AN, Charlet N, Cooper TA, The CELF family of RNA binding proteins is implicated in cell-specific and developmentally regulated alternative splicing, Molecular and cellular biology, 21 (2001) 1285–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang ET, Ward AJ, Cherone JM, Giudice J, Wang TT, Treacy DJ, Lambert NJ, Freese P, Saxena T, Cooper TA, Burge CB, Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins, Genome Res, 25 (2015) 858–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chaudhury A, Cheema S, Fachini JM, Kongchan N, Lu G, Simon LM, Wang T, Mao S, Rosen DG, Ittmann MM, Hilsenbeck SG, Shaw CA, Neilson JR, CELF1 is a central node in post-transcriptional regulatory programmes underlying EMT, Nat Commun, 7 (2016) 13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Koshelev M, Sarma S, Price RE, Wehrens XH, Cooper TA, Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1, Human molecular genetics, 19 (2010) 1066–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nutter CA, Jaworski E, Verma SK, Perez-Carrasco Y, Kuyumcu-Martinez MN, Developmentally regulated alternative splicing is perturbed in type 1 diabetic skeletal muscle, Muscle Nerve, 56 (2017) 744–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Li J, Yu P, Genome-wide transcriptome analysis identifies alternative splicing regulatory network and key splicing factors in mouse and human psoriasis, Sci Rep, 8 (2018) 4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: ultrafast universal RNA-seq aligner, Bioinformatics, 29 (2013) 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Anders S, Pyl PT, Huber W, HTSeq--a Python framework to work with high-throughput sequencing data, Bioinformatics, 31 (2015) 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hsu F, Kent WJ, Clawson H, Kuhn RM, Diekhans M, Haussler D, The UCSC Known Genes, Bioinformatics, 22 (2006) 1036–1046. [DOI] [PubMed] [Google Scholar]
- [30].Yu P, Shaw CA, An efficient algorithm for accurate computation of the Dirichlet-multinomial log-likelihood function, Bioinformatics, 30 (2014) 1547–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Katz Y, Wang ET, Airoldi EM, Burge CB, Analysis and design of RNA sequencing experiments for identifying isoform regulation, Nat Methods, 7 (2010) 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Anders S, Huber W, Differential expression analysis for sequence count data, Genome Biol, 11 (2010) R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bugger H, Abel ED, Rodent models of diabetic cardiomyopathy, Dis Model Mech, 2 (2009) 454–466. [DOI] [PubMed] [Google Scholar]
- [34].van Rooij E, Fielitz J, Sutherland LB, Thijssen VL, Crijns HJ, Dimaio MJ, Shelton J, De Windt LJ, Hill JA, Olson EN, Myocyte enhancer factor 2 and class II histone deacetylases control a gender-specific pathway of cardioprotection mediated by the estrogen receptor, Circulation research, 106 (2010) 155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chang S, Young BD, Li S, Qi X, Richardson JA, Olson EN, Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10, Cell, 126 (2006) 321–334. [DOI] [PubMed] [Google Scholar]
- [36].Pugh RJ, Slee JB, Farwell SL, Li Y, Barthol T, Patton WA, Lowe-Krentz LJ, Transmembrane Protein 184A Is a Receptor Required for Vascular Smooth Muscle Cell Responses to Heparin, J Biol Chem, 291 (2016) 5326–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Evsyukova I, Bradrick SS, Gregory SG, Garcia-Blanco MA, Cleavage and polyadenylation specificity factor 1 (CPSF1) regulates alternative splicing of interleukin 7 receptor (IL7R) exon 6, RNA, 19 (2013) 103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ohsawa N, Koebis M, Mitsuhashi H, Nishino I, Ishiura S, ABLIM1 splicing is abnormal in skeletal muscle of patients with DM1 and regulated by MBNL, CELF and PTBP1, Genes Cells, 20 (2015) 121–134. [DOI] [PubMed] [Google Scholar]
- [39].Gazzara MR, Mallory MJ, Roytenberg R, Lindberg JP, Jha A, Lynch KW, Barash Y, Ancient antagonism between CELF and RBFOX families tunes mRNA splicing outcomes, Genome Res, 27 (2017) 1360–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.