

Supplemental Digital Content is available in the text.
Keywords: aneuploidy, apoptosis, aurora kinase, cell cycle, cell cycle checkpoints, cell death, cell proliferation, ENMD-2076, metastatic colorectal cancer, patient-derived xenograft model
Abstract
Cancer is a disease caused by several factors characterized by uncontrolled cell division, growth, and survival. ENMD-2076, is a novel orally active small molecule multikinase inhibitor targeting angiogenesis, proliferation, and the cell cycle. It is selectively active against the mitotic kinases aurora A and B, and kinases responsible for angiogenesis including VEGFR2/KDR and FGFR1 and 2. ENMD-2076 has been shown to inhibit tumor growth and prevent angiogenesis in vitro and in vivo in preclinical cancer models. Moreover, in a phase I trial, ENMD-2076 was well tolerated, exhibited a linear pharmacokinetic profile, and showed a promising antitumor activity in a number of solid tumors. In this study, we show that ENMD-2076 has antiproliferative effects, causes cell cycle arrest, and has activity in preclinical models of colorectal cancer (CRC), including patient-derived xenograft (PDX) models. Forty-seven human CRC cell lines were exposed in vitro to ENMD-2076 and analyzed for effects on cell cycle, apoptosis, and downstream effector proteins. The drug was then tested against 20 human CRC PDX models to further evaluate in-vivo antitumor activity. We show that ENMD-2076 exhibits a broad range of activity against a large panel of CRC cell lines with varying molecular characteristics. Mechanistically, ENMD-2076 exposure resulted in a G2/M cell cycle arrest, an increase in aneuploidy, and cell death in responsive cell lines. In addition, ENMD-2076 treatment resulted in a promising antitumor activity in CRC PDX models. These results support the continued development of ENMD-2076 in CRC including further exploration of rational combinations.
Introduction
In the last few decades, many targeted therapies have been approved for the treatment of metastatic colorectal cancer (CRC), enhancing the benefit of standard chemotherapy with a survival benefit shown in multiple randomized phase III clinical trials 1–4. Despite these recent advances, the 5-year survival rate for patients with metastatic CRC is still quite low 5. Improving the understanding of molecular pathways and mechanisms of acquired resistance to treatment may help us discover new strategies for metastatic CRC.
The aurora kinases are a family of highly conserved serine/threonine protein kinases involved in the regulation of critical events during the mitotic phase of the cell cycle 6. This family includes three different isoforms of aurora kinases (A, B, and C) that differ in their specific functions during mitosis 7.
Aurora A is involved in regulating multiple steps during mitosis including centromere formation, duplication, spindle assemble, microtubule–kinetochore interaction, chromosome condensation, alignment, and segregation 8–11. It is highly expressed in multiple human cancers including melanoma, breast, colorectal, ovarian, pancreatic, and bladder cancers. In breast cancer, aurora A overexpression is associated with reduced survival whereas in CRCs overexpression was significantly associated with chromosomal instability 12,13.
Aurora-B is thought to be a chromosomal passenger complex protein capable of regulating cell cycle through an interaction with other proteins 14. It is also involved in chromosome condensation, segregation, and proper microtubule attachment to the kinetochore, central spindle assembly, and cytokinesis 15. Aurora C functions are not yet well understood, but it is believed that its functions overlap and complement the function of aurora-B in mitosis with a possible role in spermatogenesis 16. Aurora kinases A and B are attractive targets for cancer therapy in many solid tumors and hematologic malignancies due to their key role in mitosis, overexpression in cancer, and an association with poor prognosis and chemoresistance 7.
ENMD-2076, developed by EntreMed Inc. (now CASI Pharmaceuticals), is an orally bioavailable molecule that is an active multitargeted tyrosine kinase and aurora kinase inhibitor. The main targets include vascular endothelial growth factor (VEGF) receptor 2 (VEGFR2/KDR), fibroblast growth factor receptor 1 (FGFR1), SRC, c-Kit, Flt-3, and aurora A and B. According to Fletcher et al. 17, ENMD-2076 is relatively selective for aurora A over B with IC50 values of 0.13 and 0.45 μmol/l, respectively. ENMD-2076 has exhibited in-vitro activity against a variety of hematologic and solid tumor cell lines, including CRC-derived cell lines 17. In-vivo studies showed both antiproliferative and antiangiogenic effects against human CRC models and triple-negative breast cancer xenografts 18,19. In a phase I trial, ENMD-2076 was well tolerated, exhibited a linear pharmacokinetic profile, and showed a promising antitumor activity in several solid tumors including CRC 20.
The present study was designed to specifically evaluate the antitumor activity of ENMD-2076 against CRC models in CRC cell lines in vitro and in patient-derived xenograft (PDX) models in vivo. In addition, the effects of ENMD-2076 treatment on cell cycle, ploidy, apoptosis, senescence, and downstream effectors were investigated.
Materials and methods
Drug
ENMD-2076 [2-(phenylvinyl-4-[4-methylpiperazin-1-yl])-6-(5-methy-2H-pyrazol-3-yl-amino)-pyrimidine] was provided by CASI Pharmaceuticals (Rockville, Maryland, USA) and prepared as a 10 mmol/l stock solution in dimethyl sulfoxide for in-vitro studies and at 25 mg/ml in sterile water for the in-vivo studies. The free base of ENMD-2076 was used for in-vitro experiments and the tartrate salt was used for oral gavage administration in vivo.
Cell lines, culture, and proliferation
Human CRC cell lines were obtained from American Type Culture Collection (Manassas, Virginia, USA), DSMZ Cell Line Bank (Braunschweig, Germany), ECACC (Sigma, St. Louis, Missouri, USA), and the Korean Cell Line Bank (Seoul, Korea). The GEO cell line was a generous gift from Prof. Fortunato Ciardiello (Seconda Università degli Studi di Napoli, Naples, Italy). All cell lines were cultured in RPMI media supplemented with 10% fetal bovine serum, 1% penicillin–streptomycin, and 1% MEM nonessential amino acids and routinely screened for the presence of mycoplasma (MycoAlert; Cambrex Bio Science, Baltimore, Maryland, USA). Cell lines were maintained at 37°C with 5% CO2. All CRC cell lines used in this study have been fully characterized and authenticated in the University of Colorado Cancer Center (UCCC) DNA Sequencing and Analysis Core (University of Colorado Cancer Center Support Grant P30-CA046934).
Cells were seeded in 96-well plates at 2000–8000 cells/well, depending on their logarithmic growth phase. Plates were incubated for 24 h to allow cells to attach. Cell lines were exposed to ENMD-2076 at increasing concentrations (0–5 µmol/l) for 72 h. After 72 h, cells were fixed with 1% trichloro-acetic acid and then stained with 0.4% sulforhodamine B. Dye was then solubilized with tris base and absorbance measured on a Synergy 2 microplate reader (Biotek Instruments Inc., Winooski, Vermont, USA). IC50 was calculated from at least three independent experiments for each cell line.
Apoptosis and cell cycle analysis
Cells were seeded in six-well plates (2×105/well) and allowed to adhere for 24 h. ENMD-2076 was added at the indicated concentrations for 24 and 48 h. Following exposure, cells were trypsinized, washed with PBS, and stained with annexin V and propidium iodide (BioTool, Huston, Texas, USA) and analyzed by flow cytometry. For cell cycle analysis, cells were plated and drugged as described above. Cells were trypsinized, washed in PBS, and resuspended in Krishan’s stain and incubated at 4°C for 24 h. Cells were analyzed for cell cycle and ploidy using flow cytometry performed by the UCCC Flow Cytometry Core.
Immunoblotting
CRC cell lines were seeded in six-well plates and allowed to attach for 24 h (density was determined for each cell line on the basis of growth rate). The following day, cell lines were treated with various concentrations of ENMD-2076 for 24, 48, and 72 h. Following exposure, cells were scraped into RIPA buffer containing protease and phosphatase inhibitors (Pierce, Santa Ana, California, USA). Cells were lysed with a Qsonica Q55 probe sonicator (Qsonica, Newtown, Connecticut, USA) for 20 s. Samples were centrifuged at 16 000g at 4°C for 10 min. Total protein was determined using the Pierce 660 nm Protein Assay (Pierce, Santa Ana, California, USA). Fifty micrograms of protein were electrophoresed on 4–12% Bis-Tris precast gels (Life Technologies, Carlsbad, California, USA) and transferred to nitrocellulose membrane using Pierce G2 Fast Blotter (Pierce, Santa Ana, California, USA). Membranes were blocked for 1 h in blocking buffer (0.1% Casein solution in 0.2× PBS) at room temperature. Membranes were incubated overnight at 4°C in blocking buffer plus 0.1% Tween-20 with the following primary antibodies: phosphor-aurora A/B/C, p53, BCL-XL, p21, BAX, pHH3, surviving, and actin. All primary antibodies were purchased from Cell Signaling Technology (Danvers, Massachusetts, USA) and diluted as per manufactures’ instructions. Blots were washed 3×10 min in 1× TBS containing 0.1% Tween-20 and incubated with the appropriate secondary goat anti-rabbit and goat anti-mouse immunoglobulin G (H+L) DyLight conjugated antibodies (Cell Signaling Technology) at a 1 : 15 000 dilution for 1 h at room temperature. Blots were washed 3×15 min and then developed using the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, Nebraska, USA). p53 signal was quantified using Image J software (National Institute of Mental Health, Bethesda, Maryland, USA) with identical area measured for each band and compared with their relative control.
Clonogenic colony formation assay
Overall, 2000–20 000 cells in logarithmic growth phase were seeded in a six-well plate. The cells were exposed to ENMD-2076 (0.1, 0.5, 1, and 2 µmol/l) for 72 h, then drug was removed, cells were washed and plain media was added for an additional 72 h. At the end of the incubation period, cells were fixed with methanol and stained with 1× crystal violet for 30 min. The plates were then washed three times with water and allowed to air-dry. Colonies formed after 72 h were compared with their relative control groups and quantified using Image J (Colony Area Plugin).
Senescence
Cells were seeded in 24-well plates, allowed to adhere overnight at 37°C, and treated with vehicle or ENMD-2076 (0.3, 0.6, 1.25, 2.5, and 5 µmol/l) once and then let sit 4–10 days, changing media every 2–3 days. Cells were fixed and stained for senescence-associated β-galactosidase (SA-β-gal) using the Senescence β-Galactosidase Staining Kit at pH of 5.9–6.1 (Cell Signaling Technology). Images were acquired using a Nikon inverted microscope (Nikon Instruments Inc. Melville, New York, USA) at ×20 magnification. Images from representative fields of view were taken at each time point and concentration. SA-β-gal stained positive cells were quantified using Image J. Each image was converted into RGB stock and a montage was created for the image. Upper and lower threshold levels were set to adjust and determine the intensity of staining. The fraction of image area stained with SA-β-gal was recorded and normalized to the corresponding cell count. Statistical analysis was performed by employing two-way analysis of variance followed by Dunnett’s post-hoc test.
Patient-derived xenograft models
Five to six-weeks-old female athymic nude mice were purchased from Harlan Laboratories (Indianapolis, Indiana, USA), caged in groups of five, kept on a 12 h light/dark cycle, and given sterile food and water ad libitum. The PDX models were generated as previously described 21. Briefly, a tumor specimen was collected at the time of surgery from a consenting patient at the University of Colorado Hospital. Tumor material remaining after histopathologic analysis was cut into ~2–3 mm3 pieces, submerged in Matrigel (Corning, Tewksbury, Massachusetts, USA), and injected subcutaneously into both flanks. After tumors were passage through at least the F3 generation, they were excised, cut, and injected into the left and right flanks of 5–6 mice (≥10 evaluable tumors) per group. When the average tumor size reached a volume of ∼150–300 mm3, the mice were randomized into either vehicle or ENMD-2076 treatment groups. Mice were monitored daily for signs of toxicity and weighted twice weekly. Mice were treated with ENMD-2076 (200 mg/kg) daily by oral gavage for at least 28 days. Tumor size was evaluated twice weekly by caliper measurements using the following equation: tumor volume=(length×width2)×0.52 and recorded in the Study Director Software package (Studylog Systems, South San Francisco, California, USA). The tumor growth inhibition index was calculated from average volume of the treated (Vt) and vehicle control (Vvc) groups, with the equation: TGII=100×(Vt final−Vt initial)/(Vvc final−Vvc intial). At the end of the treatment, mice were sacrificed by CO2 overdose followed by cervical dislocation before removal of the tumors for further analysis.
All the studies were carried out in accordance with the NIH guidelines for the care and use of laboratory animals, and in a facility accredited by the American Association for Accreditation of Laboratory Animal Care. Approval from University of Colorado Animal Care and Use Committee was obtained before the initiation of experiments. Obtaining tissue from CRC patients at the time of removal of a primary tumor or metastasectomy was carried out under a Colorado Multi-Institutional Review Board-approved protocol.
Immunohistochemistry
Formalin fixed, paraffin-embedded tumor in-vivo samples were processed and stained using Ki-67 Cline SP6 (Thermo Fisher Scientific, Waltham, Massachusetts, USA), Cleaved Caspase-3 (Cell Signaling Technology), p53 (Cell Marque, Rocklin, California, USA), CD34 (AbCam, Cambridge, Massachusetts, USA), and H&E by the UCCC Histology Core Facility. For the Ki-67, p53, and caspase-3 scoring, a manual count of the tumor cells stained/total number of tumor cells was performed. The area selected was chosen to be qualitatively average. The selected area was ~0.1 mm2. For the % necrosis, we measured the area of the entire tumor on the slide and the area of the necrotic region(s). The area measurement was performed by using a tool in Aperio ImageScope (Leica Biosystems Inc., Buffalo Grove, Illinois, USA). For the microvascular density, we measured the area of tumor as above and counted cross-sections of CD34 positive endothelial lined blood vessels within the tumor.
Data analysis
The one-way and two-way analysis of variance with Tukey and Dunnett post-tests were used to determine statistical significance between multiple groups. Analyses were performed with Prism, version 5.04 (GraphPad Software, La Jolla, California, USA). P values less than 0.05 was considered statistically significant.
Results
In-vitro characterization of ENMD-2076 activity in CRC cell lines
Forty-seven CRC cell lines were used to evaluate the activity of ENMD-2076. As shown in Fig. 1, a wide range of IC50 values were observed among the CRC cell lines tested and the results did not correlate with the KRAS, BRAF, PIK3CA, or p53 mutational status. Four cell lines (one sensitive/one moderate/two resistant) were chosen for further investigations: HCT116 (IC50=0.296380 µmol/l), GEO (IC50=1.03975 µmol/l), LS1034 (IC50=3.18 µmol/l), and LS123 (IC50=4.288 µmol/l) according to their spectrum of response to ENMD-2076.
Fig. 1.
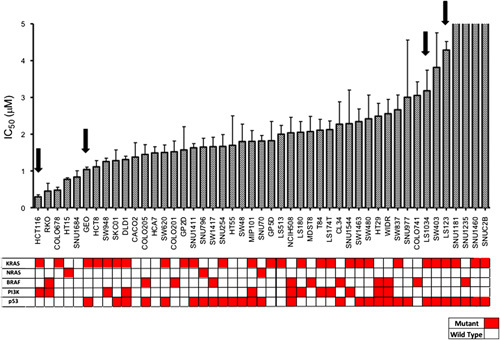
CRC cell lines exposed to ENMD-2076 to establish their IC50s. Cell lines were treated with increasing concentrations of ENMD-2076 and IC50 values were calculated. A broad range of sensitivity to the agent was observed. HCT116, GEO, LS1034, and LS123 (black arrows) were selected for further analysis.
Cell cycle regulation after ENMD-2076 treatment in CRC cell lines
To evaluate the effect of ENMD-2076 on cell cycle regulation, cell cycle analysis by flow cytometry was performed. It has previously been shown that inhibition of aurora A leads to a transient G2/M arrest causing aberrant cell division and aneuploidy 11. The flow cytometry analysis on the HCT116 (Fig. 2a) and GEO (Fig. 2c) CRC cell line treated with ENMD-2076 showed a dose-dependent increase in tetraploid cells, indicating a G2/M arrest (21–78%) after 24 h treatment exposure to ENMD-2076 at a concentration of 2.0 µmol/l, consistent with the selective activity of ENMD-2076 on the inhibition of aurora A as previously observed 17. By 48 h this increase was observed at all doses of ENMD-2076 (17–69% at 2.0 µmol/l) in the HCT116 (Fig. 2b) and at 1 and 2 µmol/l in GEO (Fig. 2d). By contrast, in the LS1034 and LS123 cell lines, no significant increase in ploidy was observed (Fig. 2e–h) with respect to the no drug control.
Fig. 2.
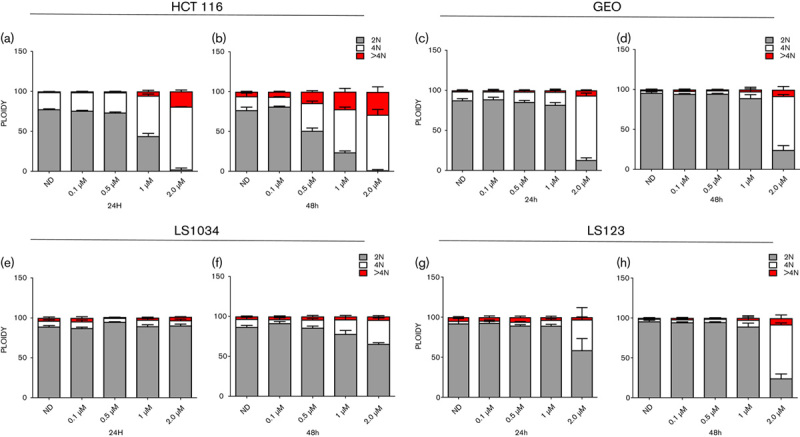
Cell cycle and ploidy analysis. Two sensitive (a–d) and two resistant (e–h) CRC cell lines were exposed for 24 and 48 h to ENMD-2076, stained with KRISHAN (propidium iodide) and flow analysis was performed. HCT116 and GEO cell lines showed a G2/M arrest following treatment with ENMD-2076 at 24 and 48 h.
Effect of EMND-2076 treatment on the induction of apoptosis in CRC cell lines
Previous studies have shown that an aurora inhibitor selective for aurora A (MLN8054) induce an apoptotic response in HCT116 cell lines 22. To further characterize whether ENMD-2076 induced apoptosis in the HCT116 CRC cell line as well as others, annexin V/propidium iodide flow cytometry was performed following treatment with ENMD-2076 for 24 and 48 h. As depicted in Fig. 3a and c, a dose-dependent induction of apoptosis was observed in the HCT116 and GEO cell lines at 24 h (2 µmol/l; P<0.001), which was still detected at 48 h (Fig. 3b and d). No significant induction of apoptosis was detectable in the LS1034 and LS123 cell line at any of the tested doses (Fig. 3e–h).
Fig. 3.
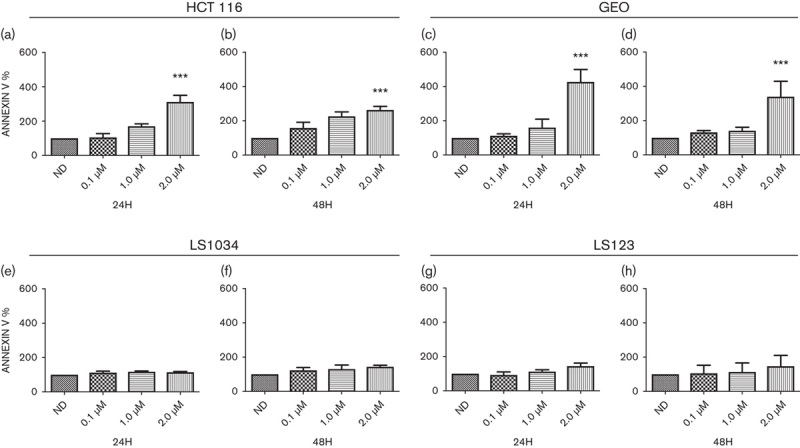
Assessment of apoptosis by annexin V staining. Two sensitive (a–d) and two resistant (e–h) CRC cell lines were treated for 24 and 48 h, stained with annexin V and propidium iodide and flow analysis was performed. HCT116 and GEO cell lines showed a greater increase of apoptosis at 2 µmol/l/treatment (P<0.001) compared with the LS1034 and LS123. ***P<0.001.
Evaluation of downstream effectors in CRC cell lines following treatment with ENMD-2076
To assess the effects of ENMD-2076 treatment on key intracellular downstream signaling pathways, immunoblotting was performed. A decrease in phosphorylated HH3 expression was observed after 24 and 48 h at 1 and 2 µmol/l, respectively, at 72 h (Fig. 4a and b) in HCT116 and GEO cell lines indicating that at higher doses ENMD-2076 also inhibits aurora-B. Moreover, a statistical significant increase in p53 was observed at 2 µmol/l in the HCT, 1 and 2 µmol/l in the GEO after 24, and at 1 and 2 µmol/l after 48 and 72 h exposure to ENMD-2076 in both cell lines (*≤0.05; **≤0.01; ***≤0.001; ***≤0.0001) (Supplementary Fig. 1, Supplemental digital content 1, http://links.lww.com/ACD/A265). The increase in p53 correlated with an increase in p21 at the same concentrations and time points indicating that ENMD-2076 upregulates and/or stabilizes p53 and p21. The decrease in the expression of survivin at 1 and 2 µmol/l after 24, 48, and 72 h exposure to the drug in the HCT116 and GEO cell lines correlates with ENMD-2076’s ability to induce apoptosis (Fig. 4a and b). None of these changes were observed in the LS1034 cell line (Fig. 4c). In the LS1034 a statistical significant decrease in p53 expression was observed after 48 h at 0.5 and 2 µmol/l and after 72 h at 1 and 2 µmol/l, with an increase at 0.5 µmol/l after 72 h (Supplementary Fig. 1, Supplemental digital content 1, http://links.lww.com/ACD/A265). In LS123 cells, we detected a statistical increase in p53 at 2 µmol/l after 24 and an increase of pHH3 at 1 and 2 µmol/l followed by a decrease at 48 and 72 h at all dose concentrations and a less pronounced reduction in survivin at 2 µmol/l after 48 and 72 h treatment (Fig. 4d) (Supplementary Fig. 1, Supplemental digital content 1, http://links.lww.com/ACD/A265). The reduction in the expression of pHH3 could be related to the inhibitory effect that ENMD-2067 has on aurora-B at higher doses, as observed in the HCT116 and GEO. ENMD-2076 (known to be relatively selective for aurora A) was able to illicit a decrease in the levels of phosphorylated HH3 expression in three out of the four cell lines analyzed, indicating that, at higher doses, ENMD-2076 also inhibits aurora-B 17. It is unclear whether the additional activity against aurora-B is important in tumorigenesis. However, there is emerging data to support the importance of aurora-B inhibition in cancer growth and progression 23–25. Regardless of sensitivity, a decrease in the aurora kinases expression was observed in the HCT116 and LS1034 at 72 h treatment and in the GEO and LS123 at all time points, confirming what we have observed with a different compound 26.
Fig. 4.
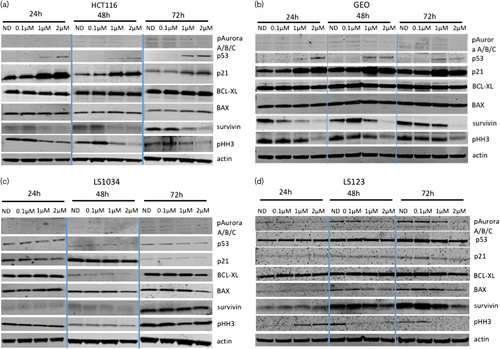
Effect of ENMD-2076 on downstream effectors. Two sensitive (a, b) and two resistant (c, d) CRC cell lines were exposed to ENMD-2067 for 12, 24, and 48 h. Western blot analysis of proteins following ENMD-2067 treatment.
ENMD-2076 effect on colony formation
To assess the effects of ENMD-2076 exposure on colony formation, clonogenic assays were performed using the four selected cell lines. In the HCT116 and GEO cell lines (Fig. 5a and b), ENMD-2076 treatment for 72 h resulted in a dramatic decrease in colony formation as compared with the no drug control, whereas the LS1034 and LS123 cells maintained the ability to form colonies (Fig. 6c and d) despite an overall reduction compared with controls.
Fig. 5.
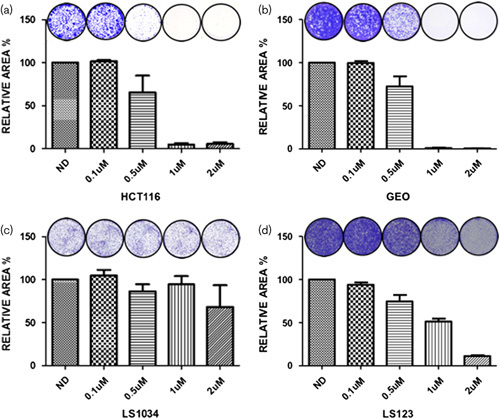
Clonogenic analysis of four CRC cell lines exposed to ENMD-2076. (a) HCT116, (b) GEO, (c) LS1034, (d) LS123 were plated in six-well plated and exposed to increasing concentrations of ENMD-2076 (0.1, 0.5, 1, and 2 µmol/l) for 72 h. Drug was removed and replaced with media to allow for regrowth of clones. Cells were stained with crystal violet, photographed, and quantitated using Image J software using the colony area plugin.
Fig. 6.
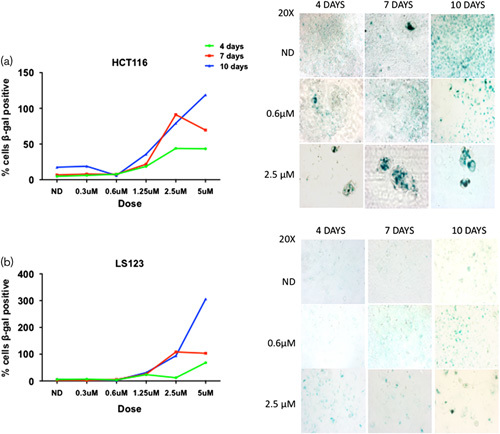
Effects of ENMD-2076 treatment on SA-ß-Gal. Senescence analysis of (a) HCT116 and (b) LS123 were chronically treated with vehicle control and ENMD-2076 for 4, 7, and 10 days at increasing concentrations (fresh media and drug was added every 2–3 days). Quantification of SA-β-gal staining was performed using Image J, showing percentage of positive cells. Representative images for SA-β-gal expression were acquired using a Zeiss microscope at ×20 magnification.
Senescence
Aurora kinase inhibitors have been shown to increase senescence and therefore we decided to investigate this in CRC cell lines 18,27. Senescence is classically described as an irreversible terminal cellular growth arrest that can be identified by changes in cellular morphology (large, flattened cells with increased vacuoles) and the presence of SA-β-galactosidase activity 28. To explore the induction of senescence, we exposed all the cell lines to ENMD-2076 up to 10 days of treatment and evaluated cells for morphologic changes and the presence of SA-β-gal activity. As depicted in Fig. 6a and b, treatment with ENMD-2076 resulted in increased SA-β-gal activity and cellular size at all time points tested in the HCT116 and LS123 cell lines as compared with vehicle control, regardless of sensitivity. The enlarged cellular size phenotype is commonly observed in senescent cells.
In-vivo antitumor activity of ENMD-2076
To confirm the antiproliferative effects observed in vitro, we decided to assess the activity of ENMD-2076 in a more clinically relevant model. In-vivo studies were carried out using 20 CRC PDX models. As observed previously in our in-vitro experiments, there was a wide range of response to ENMD-2076 treatment (Fig. 7a). None of our models showed regression (TGII≤0), but nine (45%) out of 20 models had a TGII of less than 20%, indicating a promising activity of the drug. KRAS, NRAS, BRAF, PIK3CA, and p53 mutational status did not predict response to ENMD-2076. Representative growth curves of sensitive and resistant models are shown in Fig. 7b and c.
Fig. 7.
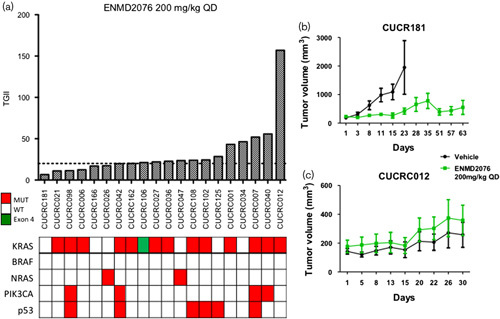
Tumor growth inhibition index (TGII) of all PDX models. (a) TGII, treated over control; thus, lower numbers indicate greater tumor reduction. Nine explants were found to be sensitive to ENMD-2076 (TGII<20%) regardless of mutational status (b) CRC181 and (c) CRC012: Growth curves of two of the 20 PDX models treated with vehicle and ENMD-2076 (200 mg/kg, QD) for more than 28 days. QD, daily.
Immunohistochemistry performed on the PDX models treated with ENMD-2076
As depicted in Fig. 7a, ENMD-2076 treatment resulted in a significant tumor growth inhibition in nine out of the 20 PDTX models tested. We selected three models that were deemed responsive to ENMD-2067 treatment and three models that were nonresponsive according to %TGII and performed Ki-67, p53, caspase-3, and CD34 (microvascular density marker) staining (Table 1). In the three nonresponsive models (CUCRC007, CUCRC012, CUCRC40), an increase in Ki-67 staining in the ENMD-2076 treated with respect to vehicle was observed (Supplementary Fig. 2, Supplemental digital content 1, http://links.lww.com/ACD/A265), whereas in the sensitive models (CUCRC 006, CUCRC 021, CUCRC181) we detected a reduction in the Ki-67 staining (%) in the treated tumors with respect to the vehicle control (Table 1). No major differences between the responsive and nonresponsive tumors were observed in p53 and CD34 stainings.
Table 1.
Ki67, p53, caspase-3, and CD34 (microvascular density marker) staining

Discussion
ENMD-2076 is a multitargeted orally bioavailable small molecule with a mechanism of action involving different targets responsible for tumor growth and survival: VEGFR2/KDR, FGFR1, SRC, c-Kit, Flt-3, and aurora A and B. As angiogenesis is a validated target in CRC and regorafenib, a small molecule with similar angiogenic targets is approved as third-line therapy, we hypothesized that ENMD-2076, with additional cell cycle targets of aurora A and B kinases, would exhibit antitumor activity in preclinical CRC models.
An improved understanding of CRC cancer biology has led to the development of novel molecularly targeted agents and new options in treatment algorithms for advanced disease. For example, the introduction of monoclonal antibodies targeting the epidermal growth factor receptor (EGFR), has improved the overall survival of metastatic CRC patients. However, studies demonstrating that patients whose tumors harbor KRAS exon 2 mutant CRC did not respond to the EGFR-targeting monoclonal antibodies cetuximab and panitumumab have provided evidence that these agents are only clinically active against a specific molecular subtype of CRC 29–32. This negative selection of CRC patients for EGFR-directed therapy has further evolved to include extension of KRAS mutant to the RAS family (KRAS exons 3 and 4 and NRAS exons 2, 3, and 4), identification of BRAF mutant CRC as a poorly responsive subgroup, and more recent data indicating differential efficacy according to tumor location 33,34. In fact, the presence of microsatellite instability is one of the few positive selection tools in CRC representing just about 5% of metastatic disease, enabling the use of immunotherapy 35. Otherwise, there remains a large unmet need in effective strategies for patients with advanced CRC, which continues to increase as we gain more insight into patient selection.
In the present study, in-vitro and in-vivo experiments confirmed that ENMD-2076 possesses anticancer activity by inhibiting cell proliferation and inducing apoptosis in a subset of CRC cell lines. KRAS/BRAF/PI3KCA mutations did not predict response to ENMD-2076, indicating potential activity against a broad array of CRC subtypes. Our observations were consistent with previously reported data demonstrating induction of cell cycle arrest and apoptosis following exposure to aurora kinase inhibitors in the HCT116 p53 wild-type (WT) colon cancer cell line 26,36.
Aurora A is known to be a key regulatory component of p53. When overexpressed, aurora A leads to increased degradation of p53 by S315 phosphorylation, thus affecting cell cycle regulation and associated apoptotic activity. Moreover, p53 destabilization may impact downstream effectors, such as p21 37. The role of p53 and aurora kinase inhibition among cancer types is controversial in the literature. For example, in a study utilizing CRC cell lines, one group showed differential behavior among clones containing functional p53 with respect to clones that were p53 deficient. The p53-deficient clones appeared to be more sensitive to the aurora inhibitors with an enhanced mitotic arrest leading to the conclusion that loss of p53 function was predictive for increased anticancer activity 38. Meanwhile, in a triple-negative breast cancer cell model, knockdown of p53 WT led to resistance to aurora kinase inhibitors 39. Interestingly in our study, there was a significant induction of p53 following exposure to ENMD-2076 in both the p53 WT HCT116 and p53 mutant GEO cell lines at all time points, regardless of p53 status. The increase in p53 expression was also associated with the induction of p21 at the same concentrations and time points, indicative of p53 and p21 stabilization and cell cycle arrest, similar to previous data obtained by our group investigating the aurora A inhibitor alisertib alone and in combination with the MEK inhibitor TAK-733 26,36. Hence, p53 status may be a relevant factor in terms of response to ENMD-2076 in CRC, wherein patients harboring wild-type p53 or specific functional mutants of this tumor suppressor may be more responsive; however, further studies are needed to fully elucidate this mechanism.
The induction of apoptosis in ENMD-2076-sensitive cell lines was observed by flow cytometry analysis and correlated with a significant reduction in survivin expression by immunoblotting in the HCT116 and GEO cell lines. Survivin, a member of the apoptosis inhibitor family, exhibits increased expression in human cancers of various origins, and has been shown to inhibit apoptosis by caspase inhibition as well as promote mitosis by aurora-B kinase activation 40. Interestingly, the observed increase in p53 expression followed by a decrease in survivin expression in responding cell lines indicates the capacity of ENMD-2076 to induce apoptosis by inhibiting aurora A and stabilizing p53.
Previous reports have assessed the effects of MLN8054 (aurora A inhibitor) in the HCT116 xenograft model in vivo, suggesting that this agent results in senescence as a terminal phenotype; however, no comparison was performed in resistant cell line xenografts or PDX models 27. Therefore, we investigated whether ENMD-2076 would induce senescence in our CRC cell lines having observed an increase in p53/p21 expression. Using β-galactosidase activity as a read out we observed induction of senescence in both the sensitive and resistant cell lines. Data on the role of senescence as a mechanism of cell death or resistance in CRC are lacking in existing in the literature; thus, further studies using preclinical in-vivo modeling will be necessary to better define the role of senescence in sensitive or resistant CRC and to better characterize the role of aurora A inhibitors with regard to p53 mutational status. In fact, the tumor suppressors p53 and p21 are particularly important in regulating cellular senescence, and if p53 is phosphorylated from aurora A kinase it abrogates p53 DNA binding and transactivation activity with a consequent p53 and p21 instabilities 41. Further studies using correlative tissue samples obtained from patients enrolled on clinical trials of aurora kinase inhibitors may lead to a better understanding of the relationship between senescence and clinical antitumor activity.
The Food and Drug Administration granted marketing approval for the use of the multityrosine kinase inhibitor regorafenib, in metastatic CRC on the basis of the modest clinical benefits as compared with placebo. When comparing the in-vivo activity of ENMD-2076 to data obtained in a previous preclinical study with regorafenib against our CRC PDX models, we observed a trend toward better antiproliferative effects with ENMD-2076 versus regorafenib. When comparing the TGII% (tumor growth inhibitions index) in five out of the six PDX models used in these studies, ENMD-2076 showed superiority in suppressing tumor grown with respect to regorafenib, with a mean TGII of 24.5 and 60% for ENMD-2076 and regorafenib, respectively 42.
ENMD-2076 has also showed antitumor activity in a phase I clinical trial and is currently being tested in numerous ongoing phase 2 clinical trials including fibrolamellar carcinoma, triple-negative breast cancer, advanced/metastatic soft tissue sarcoma, and advanced ovarian clear cell carcinoma 43,44. The mechanism of action appears to be a combination of its inhibition of aurora kinases and angiogenesis, through inhibition of VEGFR2 (KDR) and other angiogenic kinases, which leads to tumor apoptosis and senescence 17. However, neither a predictive biomarker, nor a subset of CRC patients likely to benefit for EMND-2076 has been identified. Fletcher et al. 45 recently presented data showing that an increase in antitumor response was observed when combining ENMD-2076 with an anti-PD1 antibody in syngeneic CRC mouse models (MC38 or CT26). In addition, recent data indicates that some of the targets of ENMD-2076, such as Aurora A, FAK c-KIT and KDR, when inhibited, may increase the activity of immune checkpoint inhibitors 46. In CRC, immunotherapy has emerged as an active strategy in microsatellite instability CRC patients, whereas little activity has been observed in MSS CRC 35. The data observed in the current study and in other studies in combination with anti-PD1, indicate a potential rational combination that should be further assessed as a potential strategy for patients with advanced CRC.
Supplementary Material
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.anti-cancerdrugs.com.
Acknowledgements
Conflicts of interest
There are no conflicts of interest.
References
- 1.Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013; 381:303–312. [DOI] [PubMed] [Google Scholar]
- 2.Mayer RJ, Van Cutsem E, Falcone A, Yoshino T, Garcia-Carbonero R, Mizunuma N, et al. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N Engl J Med 2015; 372:1909–1919. [DOI] [PubMed] [Google Scholar]
- 3.Tabernero J, Yoshino T, Cohn AL, Obermannova R, Bodoky G, Garcia-Carbonero R, et al. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): a randomised, double-blind, multicentre, phase 3 study. Lancet Oncol 2015; 16:499–508. [DOI] [PubMed] [Google Scholar]
- 4.Van Cutsem E, Tabernero J, Lakomy R, Prenen H, Prausova J, Macarulla T, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol 2012; 30:3499–3506. [DOI] [PubMed] [Google Scholar]
- 5.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016; 66:7–30. [DOI] [PubMed] [Google Scholar]
- 6.Crane R, Gadea B, Littlepage L, Wu H, Ruderman JV. Aurora A, meiosis and mitosis. Biol Cell 2004; 96:215–229. [DOI] [PubMed] [Google Scholar]
- 7.Keen N, Taylor S. Aurora-kinase inhibitors as anticancer agents. Nat Rev Cancer 2004; 4:927–936. [DOI] [PubMed] [Google Scholar]
- 8.Dutertre S, Cazales M, Quaranta M, Froment C, Trabut V, Dozier C, et al. Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to the G2-M transition. J Cell Sci 2004; 117:2523–2531. [DOI] [PubMed] [Google Scholar]
- 9.Kunitoku N, Sasayama T, Marumoto T, Zhang D, Honda S, Kobayashi O, et al. CENP-A phosphorylation by Aurora-A in prophase is required for enrichment of Aurora-B at inner centromeres and for kinetochore function. Dev Cell 2003; 5:853–864. [DOI] [PubMed] [Google Scholar]
- 10.Marumoto T, Honda S, Hara T, Nitta M, Hirota T, Kohmura E, et al. Aurora-A kinase maintains the fidelity of early and late mitotic events in HeLa cells. J Biol Chem 2003; 278:51786–51795. [DOI] [PubMed] [Google Scholar]
- 11.Marumoto T, Zhang D, Saya H. Aurora-A: a guardian of poles. Nat Rev Cancer 2005; 5:42–50. [DOI] [PubMed] [Google Scholar]
- 12.Baba Y, Nosho K, Shima K, Irahara N, Kure S, Toyoda S, et al. Aurora-A expression is independently associated with chromosomal instability in colorectal cancer. Neoplasia 2009; 11:418–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nadler Y, Camp RL, Schwartz C, Rimm DL, Kluger HM, Kluger Y. Expression of Aurora A (but not Aurora B) is predictive of survival in breast cancer. Clin Cancer Res 2008; 14:4455–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adams RR, Carmena M, Earnshaw WC. Chromosomal passengers and the (aurora) ABCs of mitosis. Trends Cell Biol 2001; 11:49–54. [DOI] [PubMed] [Google Scholar]
- 15.Kollareddy M, Dzubak P, Zheleva D, Hajduch M. Aurora kinases: structure, functions and their association with cancer. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 2008; 152:27–33. [DOI] [PubMed] [Google Scholar]
- 16.Hu HM, Chuang CK, Lee MJ, Tseng TC, Tang TK. Genomic organization, expression, and chromosome localization of a third aurora-related kinase gene, Aie1. DNA Cell Biol 2000; 19:679–688. [DOI] [PubMed] [Google Scholar]
- 17.Fletcher GC, Brokx RD, Denny TA, Hembrough TA, Plum SM, Fogler WE, et al. ENMD-2076 is an orally active kinase inhibitor with antiangiogenic and antiproliferative mechanisms of action. Mol Cancer Ther 2011; 10:126–137. [DOI] [PubMed] [Google Scholar]
- 18.Diamond JR, Eckhardt SG, Tan AC, Newton TP, Selby HM, Brunkow KL, et al. Predictive biomarkers of sensitivity to the aurora and angiogenic kinase inhibitor ENMD-2076 in preclinical breast cancer models. Clin Cancer Res 2013; 19:291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tentler JJ, Bradshaw-Pierce EL, Serkova NJ, Hasebroock KM, Pitts TM, Diamond JR, et al. Assessment of the in vivo antitumor effects of ENMD-2076, a novel multitargeted kinase inhibitor, against primary and cell line-derived human colorectal cancer xenograft models. Clin Cancer Res 2010; 16:2989–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diamond JR, Bastos BR, Hansen RJ, Gustafson DL, Eckhardt SG, Kwak EL, et al. Phase I safety, pharmacokinetic, and pharmacodynamic study of ENMD-2076, a novel angiogenic and Aurora kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res 2011; 17:849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pitts TM, Tan AC, Kulikowski GN, Tentler JJ, Brown AM, Flanigan SA, et al. Development of an integrated genomic classifier for a novel agent in colorectal cancer: approach to individualized therapy in early development. Clin Cancer Res 2010; 16:3193–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manfredi MG, Ecsedy JA, Meetze KA, Balani SK, Burenkova O, Chen W, et al. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc Natl Acad Sci USA 2007; 104:4106–4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alferez DG, Goodlad RA, Odedra R, Sini P, Crafter C, Ryan AJ, et al. Inhibition of Aurora-B kinase activity confers antitumor efficacy in preclinical mouse models of early and advanced gastrointestinal neoplasia. Int J Oncol 2012; 41:1475–1485. [DOI] [PubMed] [Google Scholar]
- 24.Azzariti A, Bocci G, Porcelli L, Fioravanti A, Sini P, Simone GM, et al. Aurora B kinase inhibitor AZD1152: determinants of action and ability to enhance chemotherapeutics effectiveness in pancreatic and colon cancer. Br J Cancer 2011; 104:769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie F, Zhu H, Zhang H, Lang Q, Tang L, Huang Q, et al. In vitro and in vivo characterization of a benzofuran derivative, a potential anticancer agent, as a novel Aurora B kinase inhibitor. Eur J Med Chem 2015; 89:310–319. [DOI] [PubMed] [Google Scholar]
- 26.Pitts TM, Bradshaw-Pierce EL, Bagby SM, Hyatt SL, Selby HM, Spreafico A, et al. Antitumor activity of the aurora a selective kinase inhibitor, alisertib, against preclinical models of colorectal cancer. Oncotarget 2016; 7:50290–50301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huck JJ, Zhang M, McDonald A, Bowman D, Hoar KM, Stringer B, et al. MLN8054, an inhibitor of Aurora A kinase, induces senescence in human tumor cells both in vitro and in vivo. Mol Cancer Res 2010; 8:373–384. [DOI] [PubMed] [Google Scholar]
- 28.Lawless C, Wang C, Jurk D, Merz A, Zglinicki T, Passos JF. Quantitative assessment of markers for cell senescence. Exp Gerontol 2010; 45:772–778. [DOI] [PubMed] [Google Scholar]
- 29.Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008; 359:1757–1765. [DOI] [PubMed] [Google Scholar]
- 30.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 2008; 26:1626–1634. [DOI] [PubMed] [Google Scholar]
- 31.Van Cutsem E, Lenz HJ, Kohne CH, Heinemann V, Tejpar S, Melezinek I, et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J Clin Oncol 2015; 33:692–700. [DOI] [PubMed] [Google Scholar]
- 32.Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 2013; 369:1023–1034. [DOI] [PubMed] [Google Scholar]
- 33.Loree JM, Pereira AAL, Lam M, Willauer AN, Raghav K, Dasari A, et al. Classifying colorectal cancer by tumor location rather than sidedness highlights a continuum in mutation profiles and consensus molecular subtypes. Clin Cancer Res 2018; 24:1062–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hutchins G, Southward K, Handley K, Magill L, Beaumont C, Stahlschmidt J, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol 2011; 29:1261–1270. [DOI] [PubMed] [Google Scholar]
- 35.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015; 372:2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davis SL, Robertson KM, Pitts TM, Tentler JJ, Bradshaw-Pierce EL, Klauck PJ, et al. Combined inhibition of MEK and Aurora A kinase in KRAS/PIK3CA double-mutant colorectal cancer models. Front Pharmacol 2015; 6:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F, et al. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet 2004; 36:55–62. [DOI] [PubMed] [Google Scholar]
- 38.Marxer M, Ma HT, Man WY, Poon RY. p53 deficiency enhances mitotic arrest and slippage induced by pharmacological inhibition of Aurora kinases. Oncogene 2014; 33:3550–3560. [DOI] [PubMed] [Google Scholar]
- 39.Tentler JJ, Ionkina AA, Tan AC, Newton TP, Pitts TM, Glogowska MJ, et al. p53 family members regulate phenotypic response to aurora kinase A inhibition in triple-negative breast cancer. Mol Cancer Ther 2015; 14:1117–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Furuya M, Tsuji N, Kobayashi D, Watanabe N. Interaction between survivin and aurora-B kinase plays an important role in survivin-mediated up-regulation of human telomerase reverse transcriptase expression. Int J Oncol 2009; 34:1061–1068. [DOI] [PubMed] [Google Scholar]
- 41.Ferbeyre G, de Stanchina E, Lin AW, Querido E, McCurrach ME, Hannon GJ, et al. Oncogenic ras and p53 cooperate to induce cellular senescence. Mol Cell Biol 2002; 22:3497–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scott AJ, Arcaroli JJ, Bagby SM, Yahn R, Huber KM, Serkova NJ, et al. Cabozantinib exhibits potent antitumor activity in colorectal cancer patient-derived tumor xenograft models via autophagy and signaling mechanisms. Mol Cancer Ther 2018. DOI: 10.1158/1535-7163.MCT-17-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matulonis UA, Lee J, Lasonde B, Tew WP, Yehwalashet A, Matei D, et al. ENMD-2076, an oral inhibitor of angiogenic and proliferation kinases, has activity in recurrent, platinum resistant ovarian cancer. Eur J Cancer 2013; 49:121–131. [DOI] [PubMed] [Google Scholar]
- 44.Loong HHF, Blackstein ME, Gupta AA, Hogg D, Culala L, Nyquist-Schultz K, et al. Phase II study of oral ENMD-2076 administered to patients (pts) with advanced soft tissue sarcoma (STS). J Clin Oncol 2014; 32:10528–10528. [Google Scholar]
- 45.Fletcher GC, Kiarash R, Bray MR, Hu AS, Ren KK. Abstract 1642: evaluation of ENMD-2076 in combination with anti-PD1 in syngeneic cancer models. Cancer Res 2017; 77:1642–1642. [Google Scholar]
- 46.Symeonides SN, Anderton SM, Serrels A. FAK-inhibition opens the door to checkpoint immunotherapy in pancreatic cancer. J Immunother Cancer 2017; 5:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.anti-cancerdrugs.com.