

Abstract
Direct quantitation of proteins in complex biological matrices by mass spectrometry remains a challenge. Here we describe a novel top-down parallel reaction monitoring-mass spectrometry (PRM-MS) assay, enabled by microflow LC-nanospray MS using a silicon microfluidic LC-MS chip. We demonstrated direct analysis of intact proteins such as somatropin in human plasma, achieving sensitivity (0.1–1.0 fmole) and speed (1–5 mins) on par with ELISA.
Keywords: Top-down MS, PRM-MS, intact proteins, plasma, Orbitrap, somatropin
TOC
Technological advances in liquid chromatography (LC) and mass spectrometry (MS) have opened up tremendous opportunities for practical applications of LC-MS 1–3. However, compared to the currently dominant protein assay, i.e., enzyme-linked immunosorbent assay (ELISA), MS-based assays have to further increase the speed, sensitivity, robustness, simplification of sample preparation, and ease of operation, in order to gain wider adoptions in biopharmaceutical R&D and clinical diagnostics 4. Bottom-up MS quantifies proteins at the peptide level after proteolytic digestion, which has been shown to have suboptimal reproducibility, because the labor-intensive process depends on the enzymatic reaction and target protein properties 5, 6. On the other hand, quantitation at the protein level using intact protein MS or top-down MS lacks sensitivity due to the much lower efficiency of ionization and detection of large proteins 7, 8. Moreover, to analyze proteins from complex matrices, e.g., human plasma, additional sample preparation such as immunoprecipitation is typically performed in order to enrich the target protein prior to MS analysis 9. PRM-MS at the peptide level significantly increased the sensitivity and efficiency of targeted analysis of proteins 10, 11. However, it still requires a proteolytic digestion step before LC-MS. Multiple reaction monitoring (MRM) for quantitation of intact proteins using a triple quadrupole mass spectrometer has been described 12, 13. But the assay achieved much lower sensitivity than that of peptide-level MRM.
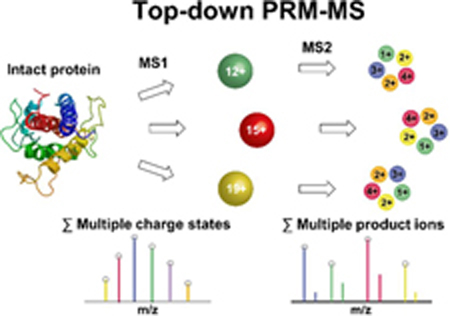
To overcome the above-mentioned challenges, we developed a novel, top-down PRM-MS assay for direct and rapid analysis of intact proteins in human plasma without sample fractionation and enzymatic digestion. It starts with rapid LC separation and fragmentation of precursor ions from multiple charge states of the intact protein, followed by simultaneous MS detection and quantitation of multiple fragment ions from each charge state of the protein. Our assay takes advantage of the high-sensitivity conferred by the peptide-level MS detection and augmented by PRM, the high-specificity conferred by targeted analysis of the intact proteins by high-resolution mass spectrometry (HRMS), and the high-speed and high-sensitivity of microflow LC-nanospray MS analysis enabled by our integrated silicon microfluidic LC-MS chips. We demonstrated its proof-of-principle application in directly quantifying exogenous somatropin in human plasma within 5 mins.
We first developed an integrated silicon microfluidic chip (i.e., MEA chip 14, 15) for rapid LC-MS analysis of intact proteins and peptides (Figure 1a). A LC column was integrated with a multinozzle emitter at the chip level with an internal swept volume of <15 nL. The length of the LC column was optimized to be 8 cm to ensure fast separation while maintaining high separation efficiency. The MEA chip was further interfaced to a Thermo Q Exactive Plus mass spectrometer for LC-MS using in-house designed adapters (Figure 1b). We next validated the performance of our MEA chip for both top-down and bottom-up MS (Figure 1c and 1d). MEA chip with a C4 column was used to perform LC-MS analysis of 1 μL of 100× diluted pooled human plasma, at a flow rate of 5 μL/min with a 5 min LC gradient. We observed good separation of HSA, HbA, and apoA-I (Figure 1c), similar to what we had published earlier for a 1-hour LC-MS run at 600 nL/min 3. MEA chip with a C18 column was used to perform LC-MS/MS of tryptic digests of HeLa cells, at a flow rate of 1 μL/min with a 60 min LC gradient. We were able to identify ~2,200 protein groups and ~12,000 peptides from 200 ng HeLa cell digest (Figure 1d). These results suggested our MEA chip could achieve high throughput with microflow LC while maintaining the MS detection sensitivity.
Figure 1.

MEA chip for LC-MS. (a) A MEA chip integrates (i) a multinozzle emitter, and (ii) a LC column. (b) The connections of a MEA chip and its interface to a mass spectrometer for LC-MS study. (c) Representative total ion chromatogram (TIC) for a 5-min LC-MS run of 1 μL of 100× diluted human plasma using a MEA chip with a C4 column. (d) Representative TIC for a 70 min LC-MS run of 200 ng HeLa cell digest using a MEA chip with a C18 column.
We next developed a top-down PRM-MS assay for analyzing intact proteins in human plasma using a Thermo Q Exactive Plus mass spectrometer. We used the growth hormone somatropin (SOMA) as an example because there are strong interests in studying its drug metabolism and pharmacokinetics (DMPK) 16, 17. Our workflow includes five major steps (Figure 2a): (i) LC separation of intact proteins spiked into human plasma on a MEA chip; (ii) selection of precursor ions of target proteins at various charge states by the quadrupole; (iii) fragmentation of target proteins at different charge states in the collision cell; (iv) selection and detection of all fragment ions for PRM-MS by Orbitrap; and (v) identification and quantitation of target proteins from the selected fragment ions using Skyline.
Figure 2.

Top-down PRM-MS assay using a MEA chip. (a) Schematic diagram showing the process flow of top-down PRM-MS of intact proteins: (i) Intact proteins separated from human plasma using a C4 column are selected as precursor ions. (ii) After mass selection, the precursor ions are fragmented into product ions. (iii) With ion selection, product ions go into the Orbitrap. (iv) All product ions are scanned by the Orbitrap to generate a MS/MS spectrum. (v) Protein identification and quantitation are based on MS/MS spectra. (b) Comparison of the total ion chromatograms for a 5-min LC-MS run of SOMA spiked in human plasma using (i) full MS scan; and (ii) top-down PRM-MS. (iii) SOMA sequence showing the fragment ions used for top-down PRM-MS. Blue arrows indicate the site of the b-ions selected for analysis. (c) Quantitation of SOMA by top-down PRM-MS: (i) The extracted ion chromatogram of SOMA with different charge states analyzed with Skyline. (ii) Sensitivity comparison among different numbers of precursor ions with various charge states (1 or 8) and product ions (1 or 14), using 0.78 fmole SOMA. (iii) Limit of detection (LOD) for the top-down PRM-MS assay. For SOMA spiked in human plasma, we obtained a LOD of 0.57 fmole. (iv) Accuracy and precision assessment of the top-down PRM-MS assay. The SOMA USP reference standard was used to generate a calibration curve. Another SOMA sample was measured with an accuracy of 94.5% at 5 fmole (CV = 1.6%), and 99.7% at 60 fmole (CV = 0.8%), compared to the theoretical values (both normalized to 100%).
To setup PRM-MS, we ran a full-scan LC-MS of plasma samples spiked with SOMA (Figure 2b-i). SOMA was separated from HSA, HbA, and apoA-I, and its retention time was determined to be ~3.01 min. We then scheduled the MS detection time window of 2.9–3.2 min for PRM-MS and selected 8 charge states ranging from +12 to +19 of SOMA as the precursor ions (Figure 2b-ii). As expected, we detected a single peak in the total ion chromatogram (TIC) representing SOMA using the top-down PRM mode (Figure 2b-ii). We performed data-dependent MS/MS (dd-MS2) analysis and optimized the fragmentation conditions for producing SOMA product ions. The fragmentation pattern was consistent between pure SOMA and SOMA spiked in plasma, and further confirmed by the ProSightPC 3.0 software. Finally, we selected the common 14 b-ions (b4, b11, b25-b36) as product ions for each charge state for PRM-MS quantitation of SOMA (Figure 2b-iii, and Table S1).
We summed up the intensities of all 14 product ions for each charge state using Skyline software (Figure 2c-i). We further summed up the intensities of all charge states for protein quantitation, assuming the same fragmentation efficiency of each charge state. As a direct demonstration of the dramatic increase in sensitivity (peak area intensity) using our top-down PRM-MS assay, we observed a 10.8-fold increase in sensitivity using 8 charge-states/14 product b-ions, a 4.3-fold increase using 1 charge-state (+15)/14 product b-ions, compared to 1 charge-state (+15)/ one highest-intensity product ion (b26), for detection of SOMA at 0.78 fmole (Figure 2c-ii). We showed a linear correlation between the amount of SOMA spiked in plasma and the total peak area intensity for PRM-MS (Figure 2c-iii). The limit of detection (LOD) of SOMA was determined as
| (1) |
In which σ is the standard deviation of the response of the calibration curve and S is the slope of the calibration curve. We spiked SOMA into 1 μL of 100× diluted pooled human plasma with descending amounts ranged from 60 to 0.45 fmole. We performed PRM-MS with ascending amounts and determined the limit of detection (LOD) to be 0.57 fmole based on the equation (1). To validate the accuracy and precision of our assay, we generated a calibration curve using the SOMA USP reference standard and measured the concentration of a separate SOMA sample. We obtained an accuracy of 94.5% at 5 fmole (CV = 1.6%), and 99.7% at 60 fmole (CV = 0.8%), compared to the theoretical values (both normalized to 100%) (Figure 2c-iv).
Finally, we performed bottom-up PRM-MS assay of SOMA spiked in plasma (Figure S1). Different from the top-down PRM, the bottom-up PRM requires an additional step of proteolytic digestion before precursor ions selection, which could take more than 12 hours prior to LC-MS analysis. We selected the peptide 1FPTIPLSR8 as the precursor ion with 6 of its product ions for quantitation in a 5 min LC-MS/MS run. Using Skyline, we generated a linear calibration curve and determined the LOD to be 0.41 fmole based on equation (1). Therefore, our top-down PRM-MS could achieve high detection sensitivity for quantitation of intact proteins similar to that of the bottom-up PRM-MS, while dramatically simplifying the overall LC-MS workflow by removing the lengthy and suboptimal proteolytic digestion step, as well as the additional enrichment step such as immunoprecipitation.
In summary, we have developed a top-down PRM-MS assay for rapid, direct, and sensitive quantitation of intact proteins in human plasma. With this assay, we demonstrated identification and quantitation of SOMA spiked in human plasma within 5 mins (could go down to 2 mins) with a limit of detection of 0.57 fmole. The assay takes advantage of the high-resolution provided by Orbitrap MS, and the high-sensitivity and high-throughput of microflow LC-nanospray MS enabled by our MEA chip. By further increasing the LC separation efficiency of our MEA chip and implementing the multiplex of endogenous and exogenous proteins in blood samples, our top-down PRM-MS assay will find wide applications to complement ELISA, and in some cases even replace ELISA, when specific antibodies are not available or multiple posttranslational modifications of the same proteins are analyzed simultaneously.
Supplementary Material
Acknowledgements
The work was supported by the National Institutes of Health award GM109682 (to Newomics Inc.). The authors also acknowledged supports from NIH awards AG046025, AI106100, AT008297, ES022360, ES023529, and HHSN261201300033C. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors thank current and former colleagues at Newomics Inc. including Dr. Geuncheol Gil for helpful discussions and technical assistance.
Footnotes
Conflict of Interest
Y. C., P. M., and D. W. are employees of Newomics Inc., which commercializes some of the technologies described in this work.
References
- (1). Anderson L; Hunter CL Mol Cell Proteomics. 2006, 5, 573–588. [DOI] [PubMed] [Google Scholar]
- (2). Chen R; Mias GI; Li-Pook-Than J; Jiang L; Lam HY; Miriami E; Karczewski KJ; Hariharan M; Dewey FE; Cheng Y; Clark MJ; Im H; Habegger L; Balasubramanian S; O’Huallachain M; Dudley JT; Hillenmeyer S; Haraksingh R; Sharon D; Euskirchen G; Lacroute P; Bettinger K; Boyle AP; Kasowski M; Grubert F; Seki S; Garcia M; Whirl-Carrillo M; Gallardo M; Blasco MA; Greenberg PL; Snyder P; Klein TE; Altman RB; Butte AJ; Ashley EA; Gerstein M; Nadeau KC; Tang H; Snyder M Cell. 2012, 148, 1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3). Mao P; Wang D J Proteome Res. 2014, 13, 1560–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4). Lequin RM Clin Chem. 2005, 51, 2415–2418. [DOI] [PubMed] [Google Scholar]
- (5). Burkhart JM; Schumbrutzki C; Wortelkamp S; Sickmann A; Zahedi RP J Proteomics. 2012, 75, 1454–1462. [DOI] [PubMed] [Google Scholar]
- (6). Bittremieux W; Tabb DL; Impens F; Staes A; Timmerman E; Martens L; Laukens K Mass Spectrom Rev. 2018, 37, 697–711. [DOI] [PubMed] [Google Scholar]
- (7). Toby TK; Fornelli L; Kelleher NL Annu Rev Anal Chem (Palo Alto Calif). 2016, 9, 499–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8). Chen B; Brown KA; Lin Z; Ge Y Anal Chem. 2018, 90, 110–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9). Whiteaker JR; Zhao L; Anderson L; Paulovich AG Mol Cell Proteomics. 2010, 9, 184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10). Peterson AC; Russell JD; Bailey DJ; Westphall MS; Coon JJ Mol Cell Proteomics. 2012, 11, 1475–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11). Lawrence RT; Searle BC; Llovet A; Villen J Nat Methods. 2016, 13, 431–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12). Wang EH; Combe PC; Schug KA J Am Soc Mass Spectrom. 2016, 27, 886–896. [DOI] [PubMed] [Google Scholar]
- (13). Wang EH; Appulage DK; McAllister EA; Schug KA J Am Soc Mass Spectrom. 2017, 28, 1977–1986. [DOI] [PubMed] [Google Scholar]
- (14). Mao P; Gomez-Sjoberg R; Wang D Anal Chem. 2013, 85, 816–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15). Mao P; Wang HT; Yang P; Wang D Anal Chem. 2011, 83, 6082–6089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16). Yang Z; Hayes M; Fang X; Daley MP; Ettenberg S; Tse FL Anal Chem. 2007, 79, 9294–9301. [DOI] [PubMed] [Google Scholar]
- (17). Gucinski AC; Boyne MT 2nd Anal Chem. 2012, 84, 8045–8051. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.