

Abstract
The structure of the ribosomally synthesized and post-translationally modified peptide product, mycofactocin is unknown. Recently, the first step in mycofactocin biosynthesis was shown to be catalyzed by MftC in two S-adenosylmethionine dependent steps. In the first step, MftC catalyzes the oxidative decarboxylation of the MftA peptide to produce the styrene containing intermediate MftA**, followed by a subsequent C-C bond formation to yield the lactam containing MftA*. Here, we demonstrate the subsequent biosynthetic step catalyzed by MftE is specific for MftA*. The hydrolysis of MftA* leads to the formation of MftA(1–28) and 3-amino-5-[(p-hydroxyphenyl) methyl]-4,4-dimethyl-2-pyrrolidinone (AHDP). The hydrolysis reaction is Fe2+ dependent and addition of the metal to the reaction leads to a kobs ~ 0.2 min−1. Lastly, we validate the structure of AHDP by a 1H, 13C, and COSY NMR techniques as well as mass spectrometry.
Keywords: MftE, mycofactocin, peptidase, peptide, creatininase, MftA

Ribosomally synthesized and post-translationally modified peptides (RiPPs) make up a large class of secondary metabolites, encoding for natural products such as antibiotics, redox cofactors, and quorum sensing molecules1–6. Mycofactocin is an example of a RiPP natural product and is proposed to be a redox cofactor produced primarily in bacteria from the Mycobacterium genera7,8. Although the structure of mycofactocin is elusive, and its precise physiological function remains unknown, its biosynthetic pathway has been shown to be essential for M. tuberculosis growth on cholesterol9,10. To solve the structure of mycofactocin, and thereby gain insight into its physiological role, recent efforts have been made to elucidate its biosynthesis. An initial bioinformatics study indicated that mycofactocin is produced by the gene cluster mftABCDEF (Figure 1A)7. Furthermore, it was proposed that MftA, a small ~30 amino acid peptide with a conserved C-terminal sequence comprised of -CGVY, served as the backbone for mycofactocin. Consistent with this proposal, experiments have shown that MftC, a radical S-adenosylmethionine (SAM) protein, catalyzes the oxidative decarboxylation of the C-terminal tyrosine to form MftA**, in the presence of the peptide chaperone MftB (Figure 1B)11,12. A subsequent study suggested that MftE, a member of the creatinine amidohydrolase family, was the likely candidate for being the next processing enzyme in the pathway. It was proposed that MftE hydrolyzes the last two residues from MftA**, resulting in Val-p-(2-aminoethenyl)phenol (Figure 1B)13. However, upon a follow up study from our lab on the catalytic mechanism for MftC, it was shown that MftA** is an intermediate of a two-step reaction14. Indeed, MftC catalyzes the conversation of MftA to MftA**, followed by a subsequent SAM dependent C-C bond formation between the Cβ position of the penultimate valine and the C2 position of p-(2-aminoethenyl)phenol. It was proposed that this new bond results in the formation of MftA* which putatively contains a C-terminal 3-amino-5-[(p-hydroxyphenyl) methyl]-4,4-dimethyl-2-pyrrolidinone moiety (herein referred to as AHDP, Figure 1B)14.
Figure 1.
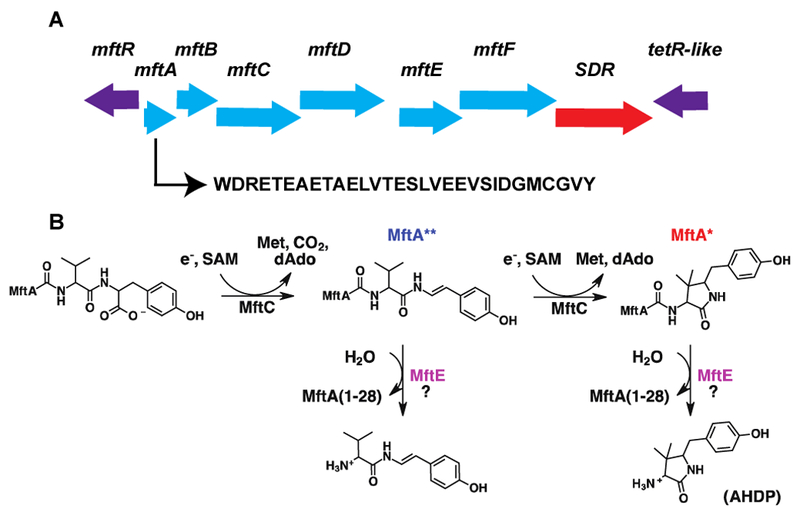
(A) A schematic representation of the mycofactocin biosynthetic pathway (blue) and the sequence of MftA used in this study. The gene encoding for a short-chain dehydrogenase (SDR) is shown in red. (B) MftC catalyzes the SAM dependent oxidative decarboxylation of the C-terminal tyrosine of MftA to form MftA** followed by a subsequent SAM dependent C-C bond formation between the penultimate valine and the β-carbon of tyrosine to form MftA*14. The subsequent MftE modification is unknown and is the focus of this study.
Since multiple products are produced by MftC and guiding experimental data regarding the structure of mycofactocin is lacking, a new question has now arisen: what is the identity of the precursor for mycofactocin, MftA* or MftA**? To address this question, we examined the substrate specificity and the metal dependency of MftE, and we characterized the products of the MftE reaction using HPLC-ESI-MS, 1H NMR, 13C NMR, and COSY. As a result of this study, we present a previously unknown natural product that clarifies the biosynthetic route of mycofactocin.
As previously described, MftE belongs to the creatinine amidohydrolase family13, a large protein family consisting of >11,000 protein sequences (IPR003785). Creatinine amidohydrolase is widely distributed binuclear Zn2+ enzyme responsible for the hydrolysis of creatinine to form creatine15,16. Interestingly, within this family are the two subfamilies FAPy deformylase and the mycofactocin peptidase, both of which are associated with cofactor or putative cofactor biosynthesis. FAPy deformylase is an Fe2+-dependent amidohydrolase responsible for the deformylation of 2-amino-5-formylamino-6-ribosylamino-4(3H) pyrimidinone 5´-monophosphate, a precursor to the redox cofactor F0 and F42017. To observe the relationship between the mycofactocin peptidase, MftE, and the remaining members of the amidohydrolase fold family, a sequence similarity network (SSN) was generated for the family IPR003785 using the Enzyme Function Initiative-Enzyme Similarity Tool (ESI-EST)18. The analysis, based on 6,279 Uniref 290 sequences, generated a network consisting of 2,433 of nodes where each node represents a set of protein sequences that are 50% identical (Figure 2). Notably, the mycofactocin peptidase cluster (purple, 309 sequences) and the FAPy deformylase cluster (green, 43 sequences) stand out from the central creatinine amidohydrolase clusters (1735 sequences). Indeed, the majority of sequences within the network belong to uncharacterized clusters that are independent of the creatinine amidohydrolase central clusters suggesting that this family of proteins may have evolved to hydrolyze a variety of substrates.
Figure 2.
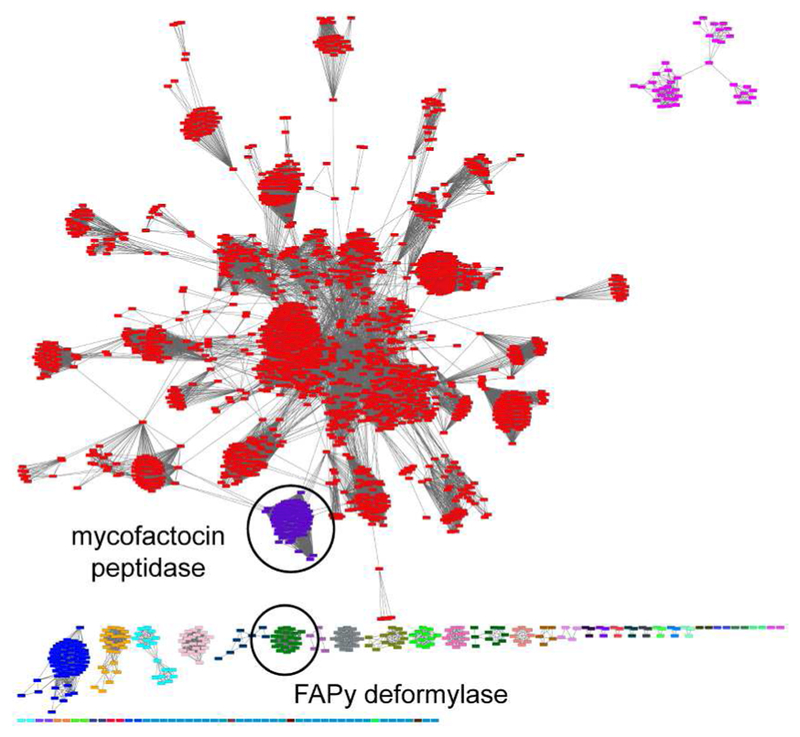
A sequence similarity network for the creatinine amidohydrolase family (IPR003785) shows the divergence of the mycofactocin peptidase (purple) and the FAPy deformylase (green) from the main body of creatinine amidohydrolases.
Analysis of MftE began with the gene from M. ulcerans being cloned into, overexpressed in, and isolated from E. coli. Recombinant MftE was purified to >95% purity, as determined by SDS-PAGE (Figure S1A). The purified protein showed high homogeneity as determined by size exclusion chromatography, with >95% of the total protein appearing at a retention time consistent with a mass of an octamer (Figure S1B). MftE was previously shown to bind iron and zinc however, efforts to fully reconstitute the protein with iron and zinc and their influence on catalysis were not reported. To address this, metal quantification assays of as-purified and reconstituted MftE were performed. Consequently, analysis of the as-purified MftE by ICP-MS revealed that it binds 0.51 ± 0.01 eq of Zn and 0.10 ± 0.01 eq of Fe (Table S1), consistent with the previously reported values13. Following reconstitution with Zn2+, MftE was found to bind 0.56 eq of Zn while maintaining ~0.10 eq of Fe. Likewise, when Fe2+ was added, MftE was found to bind 0.70 ± 0.02 eq of Fe and maintained 0.15 ± 0.01eq of Zn. Upon reconstitution with both Fe2+ and Zn2+, MftE was found to bind 0.71 ± 0.01 eq of Fe and 0.55 ± 0.01 eq of Zn per polypeptide chain (Table S1). These results are consistent with MftE binding one Fe2+ and Zn2+ atom, similar to the FAPy deformylase protein ArpB17. The influence of metal binding on catalysis will be discussed later.
Next, we set out to address if MftE is specific for MftA* or MftA**. Previous studies have shown that MftE from M. smegmatis hydrolyzes MftA** to produce Val-p-(2-aminoethenyl)phenol and the remaining MftA peptide13. However, these results were largely dependent upon difficult HPLC-ESI-MS analysis of a complex mixture and with the inability to distinguish the substrate (e.g. MftA* v. MftA**) or the structure of the product. Additionally, the mass estimated for the small molecule is consistent with the hydrolyzed products from both MftA* and MftA**. To circumvent these obstacles, MftA* and MftA** were separately purified from reactions containing MftA, MftB, MftC, DTT, SAM, and dithionite (DTH), as previously described (see Supporting Information for details)14, and individually used in reactions with MftE. Reconstituted MftE and purified MftA, MftA* or MftA** were incubated for 3 hr and analyzed by HPLC monitoring the absorbance of tryptophan and tyrosine at 280 nm (see Supporting Information for details). In the control experiment, MftA eluted at ~13 min (Figure 3, black) and the addition of MftE to MftA did not result in a significant change in the chromatogram (Figure 3, grey). Likewise, when MftE was added to MftA**, no significant change in the chromatogram is observed (Figure 3, blue and light blue). However, the addition of MftE to MftA* resulted in the formation of two new product peaks (12.5 and 13.9 min) with the concomitant disappearance of the MftA* peak (Figure 3, red and orange). Moreover, even in the presence of MftA**, MftE retains its activity towards MftA* (Figure S2). Taken together, this suggests that MftE is highly specific for MftA* and does not recognize the unmodified peptide MftA nor the intermediate MftA**. Therefore, we propose that the MftA* is the correct precursor in mycofactocin biosynthesis.
Figure 3.
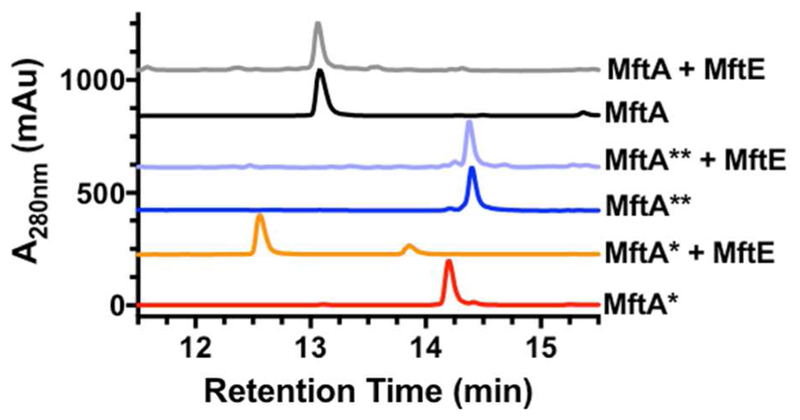
The HPLC chromatograms of reactions containing 50 μM of MftA variant with and without 50 μM MftE, indicate that MftE preferentially hydrolyses MftA* to form two new products at retention times 12.5 min and 13.9 min (orange). All reactions were carried out in the presence of 50 μM Zn2+ and 50 μM Fe2+.
Considering that MftE binds both Zn2+ and Fe2+, we set out to determine their effects on MftE catalysis. Initially, reactions were set up under steady state ([MftA*]>>>[MftE]) conditions in the presence of 1 eq Zn2+ and/or Fe2+ however, we observed that MftE catalyzes only a single turnover, under these conditions. Limited attempts to modify reaction conditions to promote multiple turnovers (e.g. addition of ascorbate, DTT, Fe2+, or Fe3+) were unsuccessful (Figure S3). Therefore, reactions were set up under single turnover conditions ([MftE]>[MftA*]) and analyzed by HPLC. The relative integrated peak area of MftA* was plotted versus time (Figure S4) and reaction rates were extracted from single exponential decay fits and are reported as kobs (Table 1). Under these conditions, the rate of hydrolysis for as-purified MftE is a modest 0.001 min+1. The addition of Zn2+ had a limited impact on the rate (kobs = 0.018 min−1) whereas the addition of Fe2+ increased the rate >200-fold to 0.232 min−1. The addition of both Zn2+ and Fe2+ had roughly the same effect on the reaction rate as Fe2+ alone. These data suggest that Fe2+ is catalytically active in MftE and that Zn2+ could be playing an auxiliary role. The observations made here are consistent with those found for AprB where the addition of Fe2+ increased the catalytic rate tenfold17. In ArpB, it was proposed that the addition of Fe2+ replaced a catalytically inactive Fe3+, resulting in the increased activity. However, unlike MftE, ArpB is capable of multiple turnovers which suggests that we are missing an important cofactor in our reactions.
Table 1.
Reaction rates for MftE with the addition of the indicated metals. Errors are reported as the sum of squared errors (SSE).
| Protien | kobs(min−1) |
|---|---|
| As-purifed MftE | 0.001 ± 0.001 |
| MftE + Zn2+ | 0.018 ± 0.006 |
| MftE + Fe2+ | 0.23 ± 0.04 |
| MftE + Zn2+ + Fe2+ | 0.12 ± 0.01 |
Having found that MftE is specific for MftA*, we next focused on characterizing the products of the reaction. To begin with, the new peaks were isolated and analyzed by HPLC-ESI-MS. First, the mass spectrum of MftA* shows an abundant species at m/z = 1111.85 ([M+H]3+ + H2O, theoretical 1111.85) consistent with a 46 Da loss in the mass from the unmodified peptide MftA (Figure S5A and S5B). The difference of 46 Da is indicative of the loss of a 1C, 2O, and 2H, consistent with the decarboxylation of C-terminal tyrosine and subsequent C-C bond formation between Val29 and Tyr3014. The mass spectrum of the 12.5 min peak from the MftE reaction shows a prominent species at m/z = 1550.21 ([M+H]2+). This mass is 217 Da less than the mass of MftA* (Figure S5C) and the difference is consistent with the loss of the last two residues on MftA* plus the addition of OH from the hydrolysis of the Gly28-Val29 amide bond (theoretical [M+H]2+ m/z = 1550.21). The mass spectrum of the 13.9 min peak from the MftE reaction shows an abundant mass at m/z = 235.1436 ([M+H]1+, Figure S5D). This new mass is consistent with the mass of AHDP (theoretical ([M+H]1+ m/z = 235.1441). These results demonstrate that MftE acts as selective peptidase by hydrolyzing the amide bond between Gly28 and Val29 of MftA* to form two new products: MftA (residues 1–28) and putatively, AHDP. To corroborate this finding, we turned to 1H NMR. Here, we rationalized that if MftE cleaves the last two residues of MftA*, we would expect to see the disappearance of the two aromatic doublet peaks from tyrosine in the 1H NMR spectrum of the cleaved peptide MftA (1–28). Indeed, the 1H NMR spectrum of MftA (1–28) shows the absence of the two-tyrosine aromatic doublets at δ = 7.12 ppm and 6.77 ppm when compared to the 1H NMR spectrum of MftA* (Figure S6A and B).
Currently, the only evidence for the existence of the AHDP precursor comes from deuterium labelling in reactions with MftC and limited 13C NMR experiments with MftA*. Therefore, we set out to solve the structure of the putative AHDP molecule by using 1H NMR, COSY, and 13C NMR techniques. To do so, sub-milligram quantities of the molecule was purified from one pot reactions containing MftA, MftB, MftC, MftE, SAM, DTT, and DTH (Figure S7). To obtain the 13C enriched AHPD for 13C NMR experiments, MftA labelled with both 13C9, 15N enriched tyrosine and 13C5, 15N enriched valine was used in the aforementioned one pot reaction. Heavy MftA was produced via in vivo expression of the peptide in E. coli grown in M9 media supplemented with the heavy amino acids (see Supporting Information for details). These procedures resulted in the heterogenous incorporation of 13C in AHDP (Figure S8).
One dimensional 1H NMR analysis (Figure S9A) of the putative AHDP is consistent with the structure containing a para substituted aromatic (δ = 6.79 and 7.13 ppm), two non-equivalent methyl groups (δ = 0.81 and 1.04 ppm), an isolated hydrogen near an amino group (δ = 3.30 ppm), two non-equivalent benzylic hydrogens (δ = 2.48 and 2.78 ppm), and a hydrogen within a substituted lactam (δ = 3.56 ppm). The chemical shifts of the benzylic hydrogens and the lactam hydrogen are similar to those found in the authentic model compound 5-benzylpyrrolidin-2-one and suggest that the molecule consists of a 5-membered lactam (Figure S9B). COSY spectral analysis (Figure 4, S9C) indicates several cross-peak couplings between proton peaks. These include cross-peak couplings between aromatic hydrogens on C9-C10 (Figure 3A) and between aliphatic hydrogens on C7-C1 and C5-C1 (Figure 4B). Assignments of these couplings are highlighted in Figure 4C and are consistent with the proposed structure for AHDP. Lastly, 13C NMR analysis (Figure S10) provided the chemical shifts of the carbon signals that are consistent with the proposed structure for AHDP. Taken together with the MS data, the NMR data presented here is consistent with the product of the MftE reaction with MftA* being AHDP.
Figure 4.
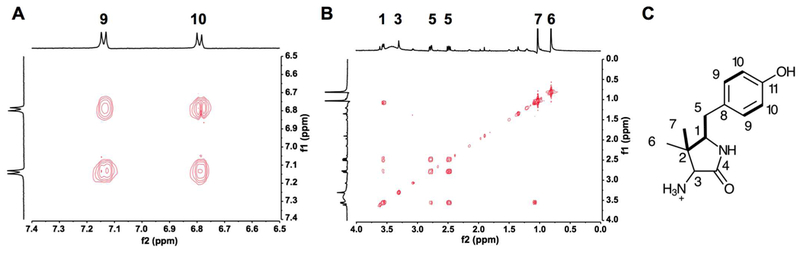
(A) COSY NMR spectra of AHPD in the aromatic region showing the cross-peak coupling between hydrogens on C9-C10. (B) COSY NMR spectra of AHPD in the saturated region showing the cross-peak coupling between hydrogens on C7-C1 and C5-C1. (C) Structural representation of the results from COSY NMR. Cross-peak couplings are represented by thick bonds.
Lastly, to reconcile the different activities reported for M. smegmatis MftE versus those reported here, we cloned Ms mftE, purified the protein from E. coli, and tested its substrate specificity. Since the last 30 amino acids of MftA from M. ulcerans and M. smegmatis are nearly identical (Mu v. Ms MftA: ΔAla10, Ser16Thr) and since the last eight residues are strictly conserved, we used MuMftA* and MuMftA** in the assay. Reactions with purified MsMftE were carried out as described for MuMftE. Similar to MuMftE, we observed that MsMftE is highly specific towards MftA* and does not react with MftA** (Figure S11). These observations are contrary to those previously reported for MsMftE. However, it should be noted that the initial report for MsMftE came prior to the discovery that MftC catalyzed a two-step reaction leading to the formation of MftA*. Bruender et al were operating under the best information at the time which suggested that MftA** was the only product from the MftC reaction. Further complications arise from the fact that Val-p-(2-aminoethenyl)phenol and AHDP are isomers and HPLC-ESI-MS and MS/MS were the sole techniques used to analyze the MsMftE reaction, making it difficult to distinguish the two molecules. Nevertheless, we provide evidence that MftE from two species is specific for MftA*.
CONCLUSIONS
We set out to elucidate the structure of mycofactocin by examining the chemistry of each of its biosynthetic components. Earlier work demonstrated that multiple products can be formed from the first biosynthetic step with MftC thereby doubling the possibilities of subsequent modifying steps. The work presented here conclusively demonstrates that one MftC product, MftA*, is the likely precursor for mycofactocin biosynthesis. By showing that MftE specifically hydrolyzes MftA*, resulting in the formation of the now structurally-characterized AHDP, we have clarified how mycofactocin is made. As a result, our future efforts will focus on modifications of AHDP by subsequent biosynthetic enzymes.
Supplementary Material
ACKNOWLEDGMENT
The authors of this paper thank Dr. Monika Dzieciatkowka (University of Colorado, Anschutz Medical Campus) for running the HPLC-ESI-MS experiments.
Funding Sources
This work was supported by National Institutes of Health Grant GM 124002 to J.A.L.
ABBREVIATIONS
- AHDP
3-amino-5-[(p-hydroxyphenyl) methyl]-4,4-dimethyl-2-pyrrolidinone
- DTT
dithiothreitol
- DTH
dithionite
- SAM
S-adenosylmethionine
Footnotes
ASSOCIATED CONTENT
Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- (1).Arnison PG et al. (2013) Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature. Nat. Prod. Rep 30, 108–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Babasaki K, Takao T, Shimonishi Y, and Kurahashi K (1985) Subtilosin A, a new antibiotic peptide produced by Bacillus subtilis 168: isolation, structural analysis, and biogenesis. J. Biochem 98, 585–603. [DOI] [PubMed] [Google Scholar]
- (3).Kawulka KE, Sprules T, Diaper CM, Whittal RM, Mckay RT, Mercier P, Zuber P, and Vederas JC (2004) Structure of subtilosin A, a cyclic antimicrobial peptide from Bacillus subtilis with unusual sulfur to carbon cross-links. Biochemistry 43, 3385–3395. [DOI] [PubMed] [Google Scholar]
- (4).Rea MC, Sit CS, Clayton E, O’Connor PM, Whittal RM, Zheng J, Vederas JC, Ross RP, and Hill C (2010) Thuricin CD, a posttranslationally modified bacteriocin with a narrow spectrum of activity against Clostridium difficile. Proc. Natl. Acad. Sci. U. S. A 107, 9352–9357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ibrahim M, Guillot A, Wessner F, Algaron F, Besset C, Courtin P, Gardan R, and Monnet V (2007) Control of the transcription of a short gene encoding a cyclic peptide in Streptococcus thermophilus: a new quorum-sensing system? J. Bacteriol 189, 8844–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Goodwin PM, and Anthony C (1998) The biochemistry, physiology and genetics of PQQ and PQQ-containing enzymes. Adv. Microb. Physiol 40, 1–80. [DOI] [PubMed] [Google Scholar]
- (7).Haft DH (2011) Bioinformatic evidence for a widely distributed, ribosomally produced electron carrier precursor, its maturation proteins, and its nicotinoprotein redox partners. BMC Genomics 12, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Haft DH et al. (2017) Mycofactocin-associated mycobacterial dehydrogenases with non-exchangeable NAD cofactors. Sci. Rep 7, 41074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Griffin JE, Gawronski JD, DeJesus MA, Ioerger TR, Akerley BJ, and Sassetti CM (2011) High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog 7, e1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Pandey AK, and Sassetti CM (2008) Mycobacterial persistence requires the utilization of host cholesterol. Proc. Natl. Acad. Sci. U. S. A 105, 4376–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Khaliullin B, Aggarwal P, Bubas M, Eaton GR, Eaton SS, and Latham JA (2016) Mycofactocin biosynthesis: Modification of the peptide MftA by the radical S-adenosylmethionine protein MftC. FEBS Lett 590, 2538–2548. [DOI] [PubMed] [Google Scholar]
- (12).Bruender NA, and Bandarian V (2016) The radical S-adenosyl-L-methionine Enzyme MftC Catalyzes an Oxidative Decarboxylation of the C-Terminus of the MftA Peptide. Biochemistry 55, 2813–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bruender NA, and Bandarian V (2017) The creatininase homolog MftE from Mycobacterium smegmatis catalyzes a peptide cleavage reaction in the biosynthesis of a novel ribosomally synthesized posttranslationally modified peptide (RiPP). J. Biol. Chem 292, 4371–4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Khaliullin B, Ayikpoe R, Tuttle M, and Latham JA (2017) Mechanistic elucidation of the mycofactocin+biosynthetic radical S-adenosylmethionine protein, MftC. J. Biol. Chem 292, 13022–13033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tsuru D, Oka I, and Yoshimoto T (1976) Creatinine Decomposing Enzymes in Pseudomonas putida. Agric. Biol. Chem 40, 1011–1018. [Google Scholar]
- (16).Yoshimoto T, Oka I, and Tsuru D (1976) Creatine amidinohydrolase of Pseudomonas putida: Crystallization and some properties. Arch. Biochem. Biophys 177, 508–515. [DOI] [PubMed] [Google Scholar]
- (17).Grochowski LL, Xu H, and White RH (2009) An iron(II) dependent formamide hydrolase catalyzes the second step in the archaeal biosynthetic pathway to riboflavin and 7,8-didemethyl-8-hydroxy-5-deazariboflavin. Biochemistry 48, 4181–4188. [DOI] [PubMed] [Google Scholar]
- (18).Gerlt JA, Bouvier JT, Davidson DB, Imker HJ, Sadkhin B, Slater DR, and Whalen KL (2015) Enzyme function initiativeenzyme similarity tool (EFI-EST): A web tool for generating protein sequence similarity networks. Biochim. Biophys. Acta - Proteins Proteomics 1854, 1019–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.