

Abstract
The poor norepinephrine innervation and high density of Gi/o-coupled α2A and α2C adrenoceptors in the striatum and the dense striatal dopamine innervation have prompted the possibility that dopamine could be an effective adrenoceptor ligand. Nevertheless, the reported adrenoceptor agonistic properties of dopamine are still inconclusive. In this study, we analyzed the binding of norepinephrine, dopamine and several compounds reported as selective dopamine D2-like receptor ligands, such as the D3 receptor agonist 7-OH-PIPAT and the D4 receptor agonist RO-105824, to α2 adrenoceptors in cortical and striatal tissue, which express α2A adrenoceptors and both α2A and α2C adrenoceptors, respectively. The affinity of dopamine for α2 adrenoceptors was found to be similar to that for D1-like and D2-like receptors. Moreover, the exogenous dopamine receptor ligands also showed high affinity for α2A and α2C adrenoceptors. Their ability to activate Gi/o proteins through α2A and α2C adrenoceptors was also analyzed in transfected cells with bioluminescent resonance energy transfer techniques. The relative ligand potencies and efficacies were dependent on the Gi/o protein subtype. Furthermore, dopamine binding to α2 adrenoceptors was functional, inducing changes in dynamic mass redistribution, adenylyl cyclase activity and ERK1/2 phosphorylation. Binding events were further studied with computer modeling of ligand docking. Docking of dopamine at α2A and α2C adrenoceptors was nearly identical to its binding to the crystallized D3 receptor. Therefore, we provide conclusive evidence that α2A and α2C adrenoceptors are functional receptors for norepinephrine, dopamine and other previously assumed selective D2-like receptor ligands, which calls for revisiting previous studies with those ligands.
Keywords: α2-adrenoceptors, BRET, DMR, adenylyl cyclase, ERK1/2 phosphorylation, docking
Introduction
The neurotransmitter norepinephrine (NE) binds and activates three subfamilies of adrenoceptors: α1-adrenoceptors, subdivided into α1A, α1B and α1D; α2-adrenoceptors, subdivided into α2A, α2B and α2C; and β-adrenoceptors, subdivided into β1, β2 and β3 [1]. Classically, α1-, α2- and β-adrenoceptors couple to Gq/11, Gi/o and Gs, respectively [1, 2]. In mammalian species, α2A is the main subtype in most brain regions whereas α2B subtype has a limited distribution and is mostly expressed in the thalamus [3, 4]. The α2C subtype is found with particularly high density in the striatum [5, 6] with a moderately lower density than α2A [7, 8]. The high density of dorsal striatal α2A and α2C adrenoceptors prompted a fundamental question in view of the well-known paucity of striatal noradrenergic terminals [9-11] and the concomitant low extracellular levels of striatal NE [12]. Yet, a series of studies indicate that both types of receptors are fully functional in the striatum, where they seem to be localized mostly postsynaptically, preferentially in GABAergic striatal efferent neurons [13, 14]. There is also evidence for α2A adrenoceptors playing a role as autoreceptors localized in the sparse striatal noradrenergic terminals [15]. It was postulated by Zhang et al., [16] that dopamine (DA) could provide the endogenous neurotransmitter for striatal α2 adrenoceptors. In transfected mammalian cells, using radioligand binding experiments, they found only a small preferential affinity of NE versus DA at both α2A and α2C adrenoceptors. Similar results were more recently obtained from radioligand binding studies using transfected mammalian and insect cell lines [17] and with radioligand binding and autoradiographic experiments in bird and rat brain with a non-selective α2 adrenoceptor ligand [18]. However, Zhang et al., [16] reported a much lower potency of DA (in the micromolar range) than NE (in the nanomolar range) at the level of α2C adrenoceptor-mediated signaling (modulation of forskolin-induced adenylyl cyclase activation).
Due to the mismatch between dopaminergic and noradrenergic innervation and the density of their receptors in several brain areas, the controversy about the differential binding affinity of DA versus NE on adrenoceptors and the potential functional efficacy of this binding, we wanted to study in detail the ability of DA and several synthetic DA receptor ligands to bind to the orthosteric site of α2 adrenoceptors in transfected cells and in sheep brain. Moreover, we further analyzed the ability of these ligands to generate functional responses: activation of G proteins, inhibition of cAMP accumulation, and ERK1/2 phosphorylation. In the present study, we first analyzed the ability of DA and several DA receptor ligands to bind to α2 adrenoceptors in cortical tissue, which predominantly expresses α2A adrenoceptors, and striatal tissue, which expresses both α2A and α2C adrenoceptors. We also studied the potential dopaminergic function of α2A and α2C adrenoceptors using the same methodology that recently allowed us to demonstrate the potent activation of all Gi/o-coupled DA D2-like receptors by NE [19]. This methodology consists on sensitive Bioluminescent Resonance Energy Transfer (BRET)-based techniques that allow detection of ligand-dependent interactions between specific receptors and specific G proteins (G protein activation) or receptor-induced activation of effectors (adenylyl cyclase activity) in living cells [19]. Moreover, we compared the ability of NE, DA and DA receptor ligands to modulate dynamic mass redistribution (DMR) and to activate MAPK signaling. Lastly, we modeled the binding of DA at α2A and α2C adrenoceptors, as compared to its binding to the crystallized D3 receptor. Our results provide conclusive evidence for α2A and α2C adrenoceptors being not only NE but also DA receptors and common targets for other D2-like receptor ligands.
Materials and Methods
DNA Constructs and Transfection
For BRET experiments, human receptor constructs were used for α2A and α2C adrenoceptors (cDNA Resource Center). The following human G protein constructs were used: Gαi1-, Gαi2-, Gαi3-, Gαo1-, or Gαo2-Renilla luciferase 8 (RLuc8) with RLuc8 inserted at position 91, untagged Gβ1, and Gγ2 fused to full-length mVenus at its N terminus. The Gα-RLuc8 constructs were kindly provided by Céline Galés (INSERM, Toulouse, France). The cAMP sensor using YFP-Epac-Rluc (CAMYEL) biosensor was obtained from the American Type Culture Collection (no. MBA-277; ATCC, Manassas, VA) [20]. All the constructs were confirmed by sequencing analysis. A constant amount of plasmid cDNA (0.5 μg Gα-RLuc8, 4.5 μg Gβ1, 5 μg Gγ2-mVenus and 5 μg receptor) was transfected into HEK-293T cells using polyethylenimine (Sigma-Aldrich) in a 1:2 ratio in 10-cm dishes. Cells were maintained in culture with Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and kept in an incubator at 37°C and 5% CO2. The transfected amount and ratio among the receptor and heterotrimeric G proteins were tested for optimized dynamic range in drug-induced BRET. HEK-293T were also used in BRET experiments for determination of adenylyl cyclase inhibition (see below). For DMR and ERK1/2 phosphorylation assays, CHO cells were grown in Minimum Essential Medium (MEMα; Gibco) supplemented with 2 mM L-glutamine, 100 μg/mL sodium pyruvate, MEM nonessential amino acid solution (1/100), 100U/mL penicillin/streptomycin, and 5% (vol/vol) of heat-inactivated fetal bovine serum. These cells were transfected with human α2A-RLuc8 receptor using polyethylenimine in a 1:2 ratio in 25-cm2 cell culture flasks. All experiments were performed approximately 48-hours after transfection.
BRET
BRET assays were performed to detect receptor ligand-induced events for Gi/o protein activation. Gi/o protein activation assay used RLuc-fused Gαi/o protein subunit and mVenus-fused Gγ2 protein for BRET pair. Receptor and untagged Gβ1 constructs were co-transfected. As reported previously [19, 20], cells were harvested, washed and resuspended in phosphate-buffered saline. Approximately 200,000 cells/well were distributed in 96-well plates, and 5 μM Coelenterazine H (substrate for luciferase) was added to each well. One minute after addition of Coelenterazine H, ligands (DA, NE, clonidine, quinpirole, 7-OH-PIPAT, and RO-105824) were added to each well. Antagonists were added 10 minutes before Coelenterazine. The fluorescence of the acceptor was quantified (excitation at 500 nm and emission at 540 nm for 1-second recordings) in Mithras LB940 (Berthold Technologies, Bad Wildbad, Germany) to confirm the constant expression levels across experiments. In parallel, the BRET signal from the same batch of cells was determined as the ratio of the light emitted by mVenus (510–540 nm) over that emitted by RLuc (485 nm). G protein-activation was calculated as the BRET change (BRET ratio for the corresponding drug minus BRET ratio in the absence of the drug) observed 10 minutes after the addition of the ligands. Emax values were expressed as the percentage of the effect of each ligand over the effect of NE. BRET curves were analyzed by non-linear regression using the commercial Prism 4 (GraphPad Software).
DMR
A global cell signaling profile or DMR was measured using an EnSpire Multimode Plate Reader (PerkinElmer, Waltham, Massachusetts, US). This label-free approach uses refractive waveguide grating optical biosensors, integrated into 384-well microplates. Changes in local optical density are measured in a detection zone up to 150 nm above the surface of the sensor. Cellular mass movements induced upon receptor activation are detected by illuminating the underside of the biosensor with polychromatic light and measured as changes in the wavelength of the reflected monochromatic light. These changes are a function of the refraction index. The magnitude of this wavelength shift (in picometers) is directly proportional to the amount of DMR. Briefly, after 24h of CHO cell transfection with α2A-RLuc8 receptor, cells were resuspended and seeded at a density of 7,000 cells per well in 384-well sensor microplates in 30 μl growing media and cultured for 24 h at 37°C and 5% CO2, to obtain monolayers at 70%–80% confluency. Before starting the assay, cells were washed twice with assay buffer (MEMα supplemented with 20 mM HEPES, pH 7.15, 0.1% DMSO and 0.1% BSA) and incubated 2 h in 40 μl per well in the reader at 24°C. Hereafter, the sensor plate was scanned, and a baseline optical signature was recorded for 10 min before adding 10 μl of the agonist dissolved in assay buffer and recorded for 90 min. Kinetic results were analyzed using EnSpire Workstation Software v 4.10.
Adenylyl cyclase activity
BRET assays were performed to detect receptor ligand-induced adenylyl cyclase activity. This assay used the CAMYEL biosensor construct which contains RLuc and YFP. The biosensor detects de conformational changes in Epac that are induced upon its binding to cAMP. The conformational change triggered by an increase in cAMP induced by forskolin results in a decrease in BRET due to the relative orientation change between donor and acceptor. A decrease in forskolin-induced cAMP levels is therefore observed as an increase in BRET [21]. To study Gαi dependent inhibition activity, cells were treated as above described but pre-stimulated for 10 minutes with 1 μM forskolin (Sigma-Aldrich), in the presence of 10 μM propranolol 10 minutes before sample reading to control for activation of endogenous β adrenergic receptors (see Results).
ERK1/2 phosphorylation
CHO cells were transfected with α2A-RLuc8 receptor, obtaining a transfection of about 0.3 pmols/mg protein. The day of the experiment, cells were starved by treating them with serum free media for 4h at 37°C. After that, cells were incubated with the indicated agonist for 5 minutes at 37°C. Then, cells were rinsed with ice-cold phosphate-buffered saline and lysed by adding 200 ml ice-cold lysis buffer (50 mM Tris-HCl [pH 7.4], 50 mM NaF, 150 mM NaCl, 45 mM β-glycerophosphate, 1% Triton X-100, 20 mM phenyl-arsine oxide, 0.4 mM NaVO4, and protease inhibitor cocktail). The cellular debris was removed by centrifugation at 13,000g for 5 minutes at 4°C, and the protein was quantified. To determine the level of ERK1/2 phosphorylation, equivalent amounts of protein were separated by electrophoresis on a denaturing 10% SDS polyacrylamide gel and transferred onto polyvinylidene fluoride membranes. Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE) was then added, and the membrane was rocked for 90 minutes. The membranes were then probed with a mixture of a mouse anti– phospho-ERK1/2 antibody (1:2500; Sigma-Aldrich) and rabbit anti-ERK1/2 antibody that recognizes both phosphorylated and nonphosphorylated ERK1/2 (1:40,000; Sigma-Aldrich) overnight at 4°C. The 42- and 44-kDa bands corresponding to ERK1 and ERK2 were visualized by the addition of a mixture of IRDye 800 (anti-mouse) antibody (1:10,000; Sigma-Aldrich) and IRDye 680 (antirabbit) antibody (1:10,000; Sigma-Aldrich) for 2 hours and scanned by the Odyssey infrared scanner (LICOR Biosciences). Band densities were quantified using the scanner software and exported to Excel (Microsoft, Redmond, WA). The level of phosphorylated ERK1/2 isoforms was normalized for differences in loading using the total ERK1/2 protein band intensities.
Radioligand binding
Brains of male and female sheep of 4-6 months old were freshly obtained from the local slaughterhouse. Brain tissues (cortex and dorsal striatum) and HEK-293T cell suspensions, were disrupted with a Polytron homogenizer (PTA 20 TS rotor, setting 3; Kinematica, Basel, Switzerland) for two 5 s-periods in 10 volumes of 50 mM Tris-HCl buffer, pH 7.4, containing a proteinase inhibitor cocktail (Sigma, St. Louis, MO, USA). Membranes were obtained by centrifugation twice at 105.000 g for 45 min at 4°C. The pellet was stored at −80°C, washed once more as described above and resuspended in 50 mM Tris-HCl buffer for immediate use. Membrane protein was quantified by the bicinchoninic acid method (Pierce Chemical Co., Rockford, IL, USA) using bovine serum albumin dilutions as standard. Binding experiments were performed with membrane suspensions at room temperature in 50 mM Tris-HCl buffer, pH 7.4, containing 10 mM MgCl2. For competition-binding assays, membrane suspensions (0.2 mg of protein/mL) were incubated for 2 h with a constant free concentration of 0.9 nM of the α2R antagonist [3H]RX821002, or 1.3 nM of the D1-like receptor antagonist [3H]SCH 23390 or 0.8 nM of the D2-like receptor antagonist [3H]YM-09151-2 and increasing concentrations of each tested ligand: NE, DA, clonidine, 7-OH-PIPAT, quinpirole and RO-105824. For α2R saturation-binding assays, membrane suspensions (0.2 mg of protein/mL) were incubated for 3 h at room temperature in 50 mM Tris-HCl buffer, pH 7.4, containing 10 mM MgCl2 with increasing concentrations of the α2R antagonist [3H]RX821002. Non-specific binding was determined in the presence of 10 μM of the non-radiolabeled antagonist RX821002 (for α2R) or 30 μM of DA (for D1R and D2R). In all cases, free and membrane-bound ligands were separated by rapid filtration of 500 μl aliquots in a cell harvester (Brandel, Gaithersburg, MD, USA) through Whatman GF/C filters embedded in 0.3% polyethylenimine that were subsequently washed for 5 s with 5 ml of ice-cold 50 mM Tris-HCl buffer. The filters were incubated with 10 mL of Ecoscint H scintillation cocktail (National Diagnostics, Atlanta, GA, USA) overnight at room temperature and radioactivity counts were determined using a Tri-Carb 2800 TR scintillation counter (PerkinElmer) with an efficiency of 62%.
Binding data analysis
Data were analyzed according to the ‘two-state dimer model’ of Casadó et al. [22]. The model assumes GPCR dimers as a main functional unit and provides a more robust analysis of parameters obtained from saturation and competition experiments with orthosteric ligands, as compared with the commonly used ‘two-independent-site model’ [22, 23]. In saturation experiments with the radioligand, the model analyzes the total number of radioligand binding sites (Bmax; more specifically it calculates RT, the total number of dimers, where Bmax = 2RT), the affinity of the radioligand for the first protomer in the unoccupied dimer (KDA1), the affinity of the radioligand for the second protomer when the first protomer is already occupied by the radioligand (KDA2) and an index of cooperativity of the radioligand (DCA). A positive or negative value of DCA implies either an increase or a decrease in affinity of KDA2 versus KDA1 and its absolute value provides a measure of the degree of increase or decrease in affinity. In competition experiments the model analyzes the interactions of the radioligand with a competing ligand and it provides the affinity of the competing ligand for the first protomer in the unoccupied dimer (KDB1), the affinity of the competing ligand for the second protomer when the first protomer is already occupied by the competing ligand (KDB2) or the radioligand (KDAB) and an index of cooperativity of the competing ligand (DCB). A positive or negative value of DCB implies either an increase or a decrease in affinity of KDB2 versus KDB1 and its absolute value provides a measure of the degree of increase or decrease in affinity.
Radioligand competition and saturation curves were analyzed by nonlinear regression using the commercial Grafit curve-fitting software (Erithacus Software, Surrey, UK), by fitting the binding data to the mechanistic two-state dimer receptor model, as described in detail elsewhere [24]. The equation describing the saturation experiment with the radioligand A in non-cooperative conditions (KDA2/KDA1 = 4) is: Abound = 2 A RT / (2 KDA1 + A), where A represents the radioligand concentration. To calculate the macroscopic equilibrium dissociation constants from competition experiments, the following general equation must be applied: Abound = (KDA2 A + 2A2 + KDA2 A B / KDAB) RT / (KDA1 KDA2 + KDA2 A + A2 + KDA2 A B/ KDAB + KDA1 KDA2 B / KDB1 + KD A1 KDA2 B2 / (KDB1 KDB2)), where B represents the assayed competing compound concentration. F. For A non-cooperative and non-allosteric modulation between A and B, the equation is simplified due to the fact that KDA2 = 4KDA1 and KDAB = 2KDB1; Abound = (4KDA1 A + 2A2 + 2KDA1 A B / KDB1) RT / (4KDA12 + 4KDA1 A + A2 + 2KDA1 A B / KDB1 + 4KDA12 B / KDB1 + 4KDA12 B2 / (KDB1 KDB2)). For A and B non-cooperative and non-allosteric modulation between A and B, the equation can be simplified due to the fact that: KDA2 = 4KDA1, KDB2 = 4KDB1 and KDAB = 2KDB1; Abound = (4KDA1 A + 2A2 + 2KDA1 A B / KDB1) RT / (4KDA12 + 4KDA1 A + A2 + 2KDA1 A B / KDB1 + 4KDA12 B / KDB1 + KDA12 B2 / KDB12).
Statistical analysis
In binding assays, goodness of fit was tested according to reduced chi-squared value given by the regression program. The test of significance for two different model population variances was based upon the F-distribution. Using this F-test, a probability greater than 95% (p < 0.05) was considered the criterion to select a more complex model (cooperativity) over the simplest one (non-cooperativity). In all cases, a probability of less than 70% (p > 0.30) resulted when one model was not significantly better than the other. In all cases, results are given as parameter values ± S.E.M. and statistical differences were analyzed with Prism 4.
Drugs
Dopamine hydrochloride and L-(-)-norepinephrine (+)-bitartrate salt monohydrate were purchased from Sigma. (-) quinpirole hydrochloride, clonidine hydrochloride, 7-OH-PIPAT maleate, RO-105824 dihydrochloride, RX821002 and yohimbine hydrochloride were purchased from Tocris. [3H]RX821002 (63.9 Ci/mmol), [3H]SCH 23390 (81.9 Ci/mmol) and [3H]YM-09151-2 (84.4 Ci/mmol) were from Perkin-Elmer. Pertussis toxin was purchased from Sigma.
Homology modeling of α2A and α2C adrenoceptors
Homology models of α2A and α2C adrenoceptors were constructed from multiple templates using RosettaCM [25] with a protocol previously described [26]. Sequences of each adrenoceptor were aligned with sequences of the following receptors: D3 (PDB ID: 3PBL [27]), β1 (PDB ID: 4BVN [28]), β2 (PDB ID: 2RH1 [29]), 5HT1B (PDB ID: 4IAR [30]), and 5HT2B (PDB ID: 4IB4 [31]) using BLAST and modified to ensure alignment of secondary structure elements and conserved residues (Fig. S1). The N-terminus was truncated through residue 28 and 46 and the C-terminus was deleted after residue 442 and 456, respectively. Additionally, the long intracellular loop 3 was deleted at residues 229-372 in α2A and 243-381 in α2C and replaced with an 8-residue poly-Gly linker. These sequences were threaded onto each template and hybridized to generate full length, energy minimized structures. Models were clustered using automatic radius detection in Rosetta and the low energy cluster centers from the top 5 clusters were selected for additional modeling.
Protein-Ligand Docking
The tridimensional structure of DA was obtained from PubChem (ID 3713609). Conformers of DA were generated using the BCL [32]. To identify the initial starting coordinates for ligand docking, homology models were aligned with the crystal structure of β2 adrenoceptor (PDB ID: 4LDO (33)) and DA was aligned with the crystallized ligand. Ligand docking was performed in RosettaLigand using the small perturbation of ligand position protocol and swapping of ligand conformers (34, 35). 1000 models for each protein-ligand complex were generated. Models were sorted initially by total energy and then culled to the top 5% of models by interface energies for analysis. Per-residue ΔΔG analysis was performed to identify residues involved in ligand binding.
Results
Binding of DA and DA receptor ligands to α2 adrenoceptors in cortical and striatal tissue
First, we analyzed the ability of NE, DA, the non-selective α adrenoceptor agonist clonidine, the non-selective D2-D3-D4 receptor agonist quinpirole, the selective D3 receptor agonist 7-OH-PIPAT and the selective D4 receptor agonist RO-105824 to displace the binding of the non-selective α2 adrenoceptor antagonist radioligand [3H]RX821002 in membrane preparations from sheep cortex and striatum with competitive inhibition experiments. See Materials and Methods and refs. [22-24] for description of the variables. Saturation experiments with [3H]RX821002 for cortical and striatal tissue provided Bmax values for α2 adrenoceptors of 0.33 ± 0.02 and 0.13 ± 0.02 pmol/mg protein and affinity values (KDA1) of 0.06 ± 0.01 and 0.07 ± 0.01 nM (n=4-8), respectively. This implies that the density of α2 adrenoceptors in the cortex, which is mostly represented by α2A adrenoceptors [3,4], is three times higher than in the striatum, which expresses similar densities for both α2A and α2C adrenoceptors [8]. To test the binding selectivity of [3H]RX821002 for α2 adrenoceptors and not for D2-like receptors, we developed competition experiments of the D2-like receptor antagonist [3H]YM-09151-2 with increasing concentrations of unlabeled RX821002 in sheep striatal membranes. RX821002 did not displace the radioligand binding at any concentration up to 10 μM (Fig. 1A). The same experiments were also performed in membranes from HEK-293T cells stably transfected with human D2, D3 or D4 receptors, with identical results (data not shown). These results demonstrate that the radioligand [3H]RX821002 does not bind to D2-like receptors.
Figure 1. Radioligand binding to D2-like and D1-like receptors in brain striatum.
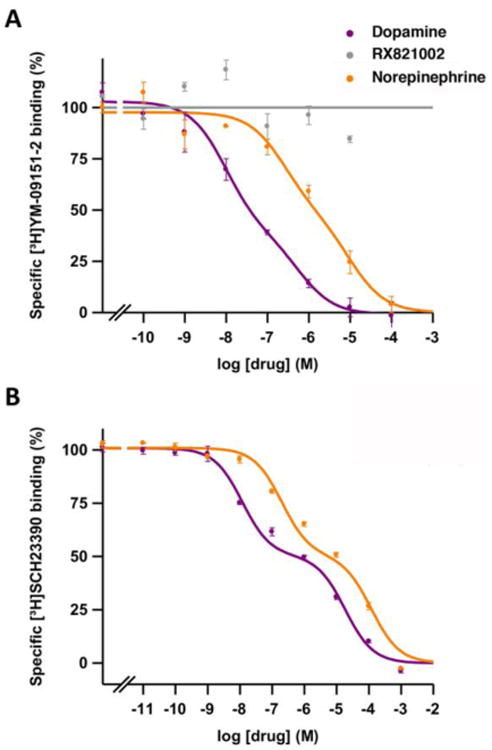
Representative competition curves of D2-like receptor antagonist [3H]-YM-09151-2 vs. increasing concentrations of free competitors NE, DA and RX821002 (A) and of D1-like receptor antagonist [3H] SCH23390 vs. increasing concentrations of DA and NE (B) in sheep brain striatum. Values are expressed as a percentage of the specific binding (100% is 0.13 ± 0.01 pmol/mg protein in A and 0.43 ± 0.04 pmol/mg protein in B). Experimental data were fitted to the two-state dimer receptor model equations, as described in the Materials and Methods section. Values are mean ± S.E.M. from a representative experiment (n = 3-5) performed in triplicate.
Competition experiments of [3H]RX821002 with NE, DA, clonidine and the D2-like receptor ligands in cortical and striatal sheep membranes are shown in Fig. 2A and 2B, respectively, and the KDB1, KDB2 and DCB values obtained are presented in Table 1. In both tissues, NE, DA and clonidine showed high affinity for [3H]RX821002 binding sites with an order of potency of clonidine>NE>DA (Fig. 2). The three ligands showed negative cooperativity (negative DCB values). The affinity of NE was higher in the cortex than in the striatum, with higher striatal KDB1, KDB2 and DCB values (stronger negative cooperativity) (Table 1). The affinity of DA was very similar in both tissues, with similar KDB1 values and a moderately but significantly higher KDB2 value in the striatum, resulting in similar DCB values (Table 1). The affinity of clonidine was also higher in the cortex, with a significantly higher striatal KDB1 value and similar DCB values (Table 1). 7-OH-PIPAT and quinpirole also displaced [3H]RX821002 binding with nanomolar and submicromolar affinities, respectively (Table 1 and Fig. 2). Interestingly, 7-OH-PIPAT showed negative cooperativity in the cortex, but not in striatum. The only measurable affinity parameter of 7-OH-PIPAT in the striatum, KDB1, was significantly higher than in the cortex and it was almost ten times lower than the cortical KDB2 value (Table 1). Quinpirole also showed differences in the binding parameters between the cortex and striatum, such as a lower KDB1 value but negative cooperativity in the striatum. Finally, RO-105824 also displaced [3H]RX821002 binding from the cortex and striatum with high affinity (subnanomolar). No cooperativity (DCB = 0) was obtained, except for RO-105824 in the cortex (DCB = -4.3) (Table 1).
Figure 2. Radioligand binding of dopaminergic and adrenergic ligands to α2 adrenoceptors in brain tissue.
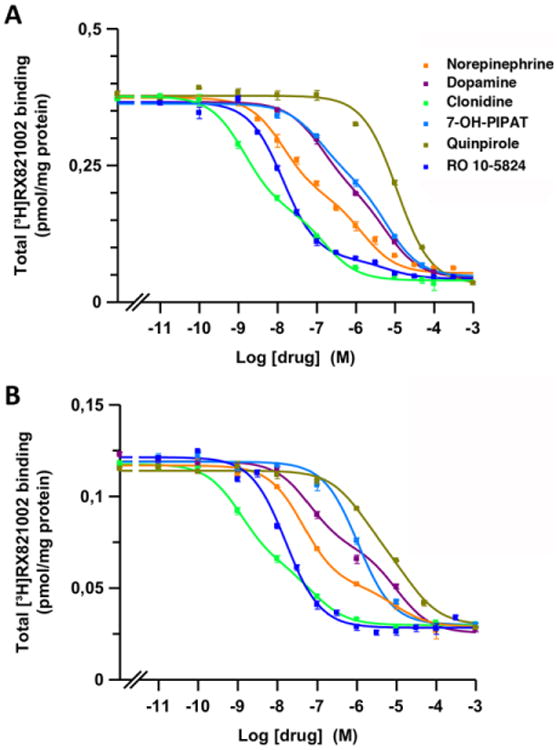
Representative competition curves of α2 adrenoceptor antagonist [3H]RX821002 vs. increasing concentrations of free competitors (NE, DA, clonidine, quinpirole, 7-OH-PIPAT and RO-105824) in sheep brain cortical (A) and striatal (B) membranes. Experimental data were fitted to the two-state dimer receptor model equations, as described in the Materials and Methods section. Values are mean ± S.E.M. from a representative experiment (n = 3) performed in triplicate.
Table 1. Competitive inhibition experiments of [3H]RX821002 versus NE, DA, clonidine and D2-like receptor ligands in sheep brain cortex and striatum.
| LIGAND | BINDING PARAMETERS | |
|---|---|---|
|
| ||
| CORTEX | STRIATUM | |
| KDB1: 0.3 ± 0.2 * | KDB1: 0.8 ± 0,1 | |
| NE | KDB2: 250 ± 100 | KDB2: 5000 ± 3000 |
| DCB: -2.3 | DCB: -3.2 | |
|
| ||
| KDB1: 6.9 ± 0.2 | KDB1: 6 ± 1 | |
| DA | KDB2: 350 ± 10 * | KDB2: 1000 ± 200 |
| DCB: -1.1 | DCB: -1.6 | |
|
| ||
| KDB1: 0.014 ± 0.003 * | KDB1: 0.036 ± 0.005 | |
| Clonidine | KDB2: 40 ± 20 | KDB2: 20 ± 10 |
| DCB: -2.8 | DCB: -2.1 | |
|
| ||
| KDB1: 9 ± 2 ** | KDB1: 51 ± 6 | |
| 7-OH-PIPAT | KDB2: 430 ± 80 | |
| DCB: -1.1 | DCB: 0 | |
|
| ||
| KDB1: 530 ± 50 ** | KDB1: 110 ± 10 | |
| Quinpirole | KDB2: 2700 ± 400 | |
| DCB: 0 | DCB: -0.8 | |
|
| ||
| KDB1: 0.055 ± 0.003 *** | KDB1: 0.42 ± 0.03 | |
| RO-105824 | KDB2: 4000 ± 2000 | |
| DCB: -4.3 | DCB: 0 | |
Binding parameters from competitive-inhibition experiments of [3H]RX821002 versus NE, DA, clonidine and D2-like receptor ligands in membrane preparations from sheep brain cortex and striatum (Fig. 2). KDB1, KDB2 and DCB values were obtained according to the two-state dimer model (see Materials and Methods and ref. 22). KDB1 and KDB2 (in nM) are expressed as means ± S.E.M. of 3 experiments performed in triplicate. Statistical differences between affinity parameters of cortical versus striatal adrenoceptors were calculated by non-paired, two-tailed Student's t test;
p<0.05,
p<0.01 and
p<0.001.
Binding of DA and NE to D1-like and D2-like receptors in striatal tissue
Next, we compared the affinity of endogenous DA and NE binding to DA D1-like and D2-like receptors with the affinity, above determined, for α2 adrenoceptors. In addition to competition experiments with the D2-like radioligand antagonist [3H]YM-09151-2 (Fig. 1A), we performed competition experiments with the D1-like radioligand antagonist [3H]SCH 23390 in sheep striatal preparations (Fig. 1B). The equilibrium binding parameters are shown in Table 2. Both NE and DA showed negative cooperativity (Fig. 1) with negative Dc values (Table 2). The affinity of DA for the first protomer of the D2-like receptor dimer (KDB1), mostly represented by the D2R subtype in dorsal striatum (36), was similar to the affinity for α2 adrenoceptors and D1-like receptors (Fig. 1, Table 2). The affinity of DA for the second protomer (KDB2) of the D1-like receptor was even about 10 times lower than for α2 adrenoceptors and D2-like receptors (stronger negative cooperativity; see Table 2). On the other hand, the affinity of NE for α2 adrenoceptors in the striatum, represented by α2A and α2C adrenoceptors, was significantly higher than for dopamine receptors (Fig. 1). Specifically, NE had an affinity for DA receptors about 60-fold lower than for α2 adrenoceptors (Table 2 and Fig. 1). We can therefore assume that extracellular physiological levels of DA that are able to activate DA receptors are also able to bind α2 adrenoceptors.
Table 2. Competitive inhibition experiments of [3H]SCH 23390, [3H]YM-09151-2 or [3H]RX821002 versus DA and NE in sheep brain striatum.
| RECEPTOR | BINDING PARAMETERS | |
|---|---|---|
|
| ||
| DA | NE | |
| KDB1: 8 ± 3 | KDB1: 53 ± 90## | |
| D1-like | KDB2: 8000 ±1000*** | KDB2: 50000 ± 10000# |
| Dc: -2.4 | Dc: -2.4 | |
|
| ||
| KDB1: 3.5 ± 0.6 | KDB1: 60 ± 40## | |
| D2-like | KDB2: 700 ± 200 | KDB2: 3400 ± 100 |
| Dc: -1.7 | Dc: -1.2 | |
|
| ||
| KDB1: 6 ± 1 | KDB1: 0.8 ± 0.1 | |
| α2 | KDB2: 1000 ± 200 | KDB2: 5000 ± 3000 |
| Dc: -1.6 | Dc: -3.2 | |
Binding parameters from competitive-inhibition experiments of [3H]SCH 23390 (D1-like receptor antagonist), [3H]YM-09151-2 (D2-like receptor antagonist) or [3H]RX821002 (α2 receptor antagonist), versus NE and DA in sheep brain striatum. KDB1, KDB2 and Dc values were obtained according to the two-state dimer model (see Materials and Methods and ref. 22). Values for α2- adrenoceptors are from Table 1. KDB1 and KDB2 (in nM) are expressed as means ± S.E.M. of 3-5 experiments performed in triplicate. Statistical differences between affinity parameters obtained were calculated by one-way ANOVA followed by Dunnett's post hoc test; for DA,
p<0.001 vs. D2-like receptors; for NE,
p<0.05 and
p<0.01 vs. α2 receptors.
α2A and α2C adrenoceptor-mediated G protein activation by DA and synthetic DA receptor ligands
The G protein activation BRET assay (see Materials and Methods) was used to determine the potency and efficacy of the above studied ligands to activate α adrenoceptors in HEK-293T cells transfected with α2A or α2C adrenoceptor and one of the five different RLuc-fused Gαti/o subunits (Gαi1, Gαi2, Gαi3, Gαo1 and Gαo2) with Venus-fused Gγ2 protein as BRET acceptor pair. The amount of Gαi/o subunits transfected produced values between 0.5 and 1 million luminescence units (arbitrary units). Previously, we found that luminescence values between 200,000 and 1.5 million did not produce a significant alteration of the Emax of drug-induced BRET. Moreover, the levels of α2 adrenoceptor were around 2 pmol/mg protein. We also previously reported that, using the same cell line and assay conditions, neither DA or NE produce a significant BRET change when transfected with the same fused G protein subunits but without receptor co-transfection [19]. These transfected receptor levels were only slightly higher than those obtained in binding experiments in sheep brain cortex (see above). A concentration-response of the ligand-induced change in BRET values allows to determine the potency as well as the relative efficacy (to NE) at α2A and α2C adrenoceptor-mediated G protein activation. Results were largely in agreement with the values obtained with binding experiments, considering that cortical values should represent ligand binding parameters of α2A adrenoceptors, while striatal values represent combined ligand binding parameters for both α2A and α2C adrenoceptors. NE was more potent at α2A than at α2C adrenoceptor, except for Gαi2 and Gαi3. On the other hand, DA had similar potencies at both adrenoceptors, except for Gαi2 and Gαo1. At both α2A and α2C adrenoceptors, DA showed high potency and efficacy as compared with NE (Figs. 3 and 4 and Tables 3 and 4), although DA was always less potent than NE. The relative DA/NE potency depended on the α adrenoceptor and on the Gαi/o subtype (see Table 3). Therefore, the potencies of DA at activating α2A adrenoceptor varied from about fifteen-fold lower, for Gαi1, to about thirty-fold lower, for Gαo1. On the other hand, the potencies of DA as compared to NE at activating α2C adrenoceptor were very close to those of NE and they varied from less than two-fold lower, for Gαi1, to about seventy-fold lower, for Gαi3 (Table 3).
Figure 3. G protein activation of α2A by dopaminergic and adrenergic ligands.
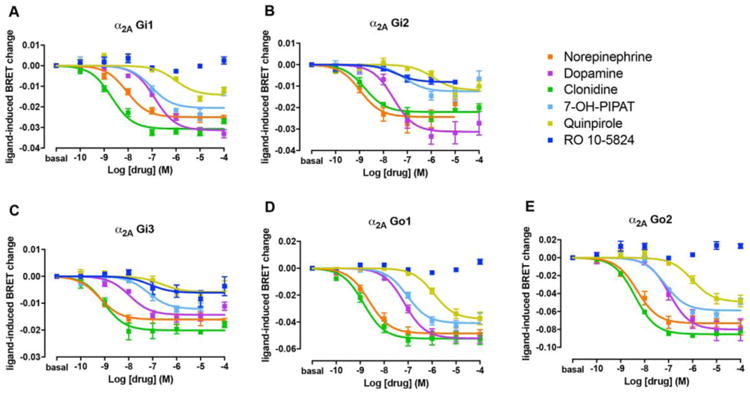
Concentration-response experiments of G protein activation by NE, DA, clonidine and D2-like receptor ligands mediated by α2A in HEK-293T cells transiently transfected with α2A adrenoceptor, the G protein subunits Gαi1-RLuc (A), Gαi2-RLuc (B), Gαi3-RLuc (C), Gαo1-Rluc (D) or Gαo2-RLuc (E), γ2-mVenus and non-fused β1. Cells were treated with Coelenterazine H followed by increasing concentrations of one of the ligands. Ligand-induced changes in BRET values were measured as described in the Materials and Methods section. BRET values in the absence of ligands were subtracted from the BRET values for each agonist concentration. Data were adjusted to a sigmoidal concentration-response function by nonlinear regression analysis and represent means ± S.E.M. of 3 to 11 experiments performed in triplicate (see Table 1 and 2 for EC50 and Emax values and statistical analysis).
Figure 4. G protein activation of α2C by dopaminergic and adrenergic ligands.
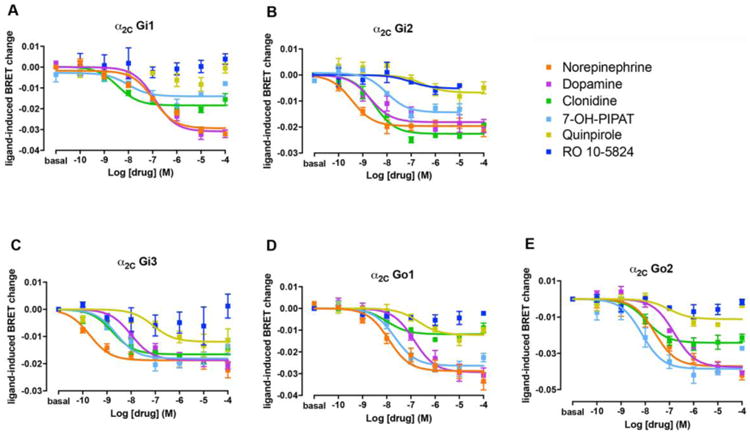
Concentration-response experiments of G protein activation by NE, DA, clonidine and D2-like receptor ligands mediated by α2C in HEK-293T cells transiently transfected with α2C receptor, the G protein subunits Gαi1-RLuc (A), Gαi2-RLuc (B), Gαi3-RLuc (C), Gαo1-RLuc (D) or Gαo2-RLuc (E), γ2-mVenus and non-fused β1. Cells were treated with Coelenterazine H followed by increasing concentrations of one of the ligands. Ligand-induced changes in BRET values were measured as described in the Materials and Methods section. BRET values in the absence of ligands were subtracted from the BRET values for each agonist concentration. Data were adjusted to a sigmoidal concentration-response function by nonlinear regression analysis and represent means ± S.E.M. of 3 to 9 experiments performed in triplicate (see Table 1 and 2 for EC50 and Emax values and statistical analysis).
Table 3. Potency of NE, DA, clonidine, 7-OH-PIPAT and quinpirole obtained from G protein activation experiments mediated by α2A and α2C adrenoceptors coupled to the different Gαi/o subtypes.
| Gα subunit | Receptor | NE | DA | Clonidine | 7-OH-PIPAT | Quinpirole | DA/NE |
|---|---|---|---|---|---|---|---|
| Gαil | α2A | 11±2** | 170 ± 40 | 3±1 | 80 ± 20 * | 700 ±250 | 15 |
| α2C | 90 ±30 | 150 ± 40 | 6±2 | (11 ±4) | ND | 1.7 | |
|
| |||||||
| Gαi2 | α2A | 1.3 ±0.3* | 30 ±3 | 2.0 ±0.8 | 120 ± 6 * | (1000 ±300) | 23 |
| α2C | 0.4 ±0.2 | 5±3 | 3±1 | 10 ±2 | (200 ±100) | 12.5 | |
|
| |||||||
| Gαi3 | α2A | 0.6 ±0.2 | 15 ±5 | 1.0 ±0.2 | 60 ± 20 * | (700 ± 400) | 25 |
| α2C | 0.4 ±0.2 | 30 ±20 | 4±1 | 5±3 | 400 ±100 | 75 | |
|
| |||||||
| Gαol | α2A | 3.0 ±0.5* | 80 ±10 | 2.0 ± 0.4* | 100 ± 20 | 1300 ± 200** | 27 |
| α2C | 19 ±6 | 126 ±8 | (12 ±5) | 20 ±3 | 230 ±50) | 7 | |
|
| |||||||
| Gαo2 | α2A | 6±1* | 100 ± 20 | 4.0 ± 0.2** | 66 ±7* | 820 ± 80 *** | 17 |
| α2C | 50 ±10 | 140 ± 20 | 11 ± 2 | 14 ±7 | (100 ±10) | 2.8 | |
Potency (EC50 values, in nM) of NE, DA, clonidine and D2-like receptor ligands obtained from G protein activation experiments mediated by α2A and α2C coupled to the different Gαi/o subtypes (Figs. 3 and 4). EC50 values were obtained from a sigmoidal concentration-response function adjusted by nonlinear regression analysis and are expressed as means ± S.E.M. of 3 to 11 experiments performed in triplicate. In parenthesis, values corresponding to experiments showing low efficacy, Emax lower than 50% (Table 4). DA/NE: ratio of EC50 values of DA and NE for each receptor and Gαi/o protein subtype. Statistical differences between α2A and α2C adrenoceptors were calculated by non-paired, two-tailed Student's t test;
p<0.05,
p<0.01 and
p<0.001.
Table 4. Efficacy of NE, DA, clonidine, 7-OH-PIPAT and quinpirole obtained from G protein activation experiments mediated by α2A and α2C adrenoceptors coupled to the different Gαi/o subtypes.
| Gα Subunit | Receptor | NE | DA | Clonidine | 7-OH-PIPAT | Quinpirole |
|---|---|---|---|---|---|---|
| Gαi1 | α2A | 100 ± 5 | 120 ± 6 * | 120 ± 5 | 94 ± 6 | 70 ± 2 * |
| α2C | 100 ± 2 | 105 ± 5 | 64 ± 4 ** | 41 ± 1 ** | ND | |
|
| ||||||
| Gαi2 | α2A | 100 ± 10 | 120 ± 20 | 89 ± 6 | 55 ± 2 * | 47 ± 8 * |
| α2C | 100 ± 7 | 80 ± 10 | 107 ± 4 | 90 ± 20 | 46 ± 9 ** | |
|
| ||||||
| Gαi3 | α2A | 100 ± 10 | 90 ± 10 | 112 ± 8 | 71 ± 6 | 40 ± 9 ** |
| α2C | 100 ± 9 | 95 ± 6 | 85 ± 5 | 90 ± 10 | 80 ± 15 | |
|
| ||||||
| Gαo1 | α2A | 100 ± 7 | 108 ± 6 | 110 ± 10 | 81 ± 9 | 78 ± 9 |
| α2C | 100 ± 10 | 100 ± 10 | 41 ± 3 ** | 98 ± 7 | 42 ± 3 ** | |
|
| ||||||
| Gαo2 | α2A | 100 ± 9 | 110 ± 10 | 120 ± 2 | 85 ± 4 | 72 ± 8 |
| α2C | 100 ± 8 | 114 ± 6 | 70 ± 8 * | 106 ± 3 | 34 ± 6 ** | |
Efficacy (Emax values, as percentage of NE values) of NE, DA, clonidine and D2-like receptor ligands obtained from G protein activation experiments mediated by α2A and α2C coupled to the different Gαi/o subtypes (Figures 3 and 4). Emax values were obtained from a sigmoidal concentration-response function adjusted by nonlinear regression analysis and are expressed as means ± S.E.M. of 3 to 11 experiments performed in triplicate. In italics, values of Emax lower than 50%. ND: not detectable. Statistical differences between NE and the other ligands for each receptor and Gαi/o protein subtype were calculated by one-way ANOVA, followed by Dunnett post hoc test;
p<0.05,
p<0.01 and
p <0.001.
The prototypical non-selective α adrenoceptor agonist clonidine only showed a significantly higher potency at α2A than at α2C adrenoceptors for Gαo1 and Gαo2 (Table 3). An additional difference as compared to NE was that clonidine behaved as a full agonist at α2A and as a partial agonist at α2C adrenoceptor, except for Gαi2 and Gαi3 (Figs. 3 and 4 and Table 4). Intriguingly, the level of efficacy of clonidine for α2C adrenoceptor varied significantly with the associated Gαi/o protein subtypes, from no decrease for Gαi2 to a very significant loss of efficacy for Gαo1 (Table 4). Previous studies have already reported a partial agonism of clonidine at α2 adrenoceptors, but with disparate results [37, 38], which, at least for α2C, could be related to the Gαi/o protein subtypes involved. In summary, the differences in the respective potency values of NE, clonidine and DA for α2A and α2C adrenoceptors in the G protein activation BRET experiments, correlates with the higher affinities of NE and clonidine in the cortex and similar affinities of DA in cortex and striatum.
The non-selective D2-D3-D4 receptor agonist quinpirole and the selective D3 receptor agonist 7-OH-PIPAT also activated α2A and α2C adrenoceptors, but with very different profiles (see Figs. 3 and 4 and Tables 3 and 4). 7-OH-PIPAT behaved as a low efficacious agonist at α2A adrenoceptors for the Gαi2 subtype. On the other hand, 7-OH-PIPAT behaved as a partial agonist at α2C- Gαi1 complexes but as a full agonist with the other α2C-Gαi/o complexes. This D3 receptor agonist, at α2A adrenoceptor, was, in general, as potent as DA and, for both Gαo subtypes, was as potent α2C adrenoceptor agonist as NE. In contrast, quinpirole showed a weak potency (submicromolar range) but also behaved as a partial or full agonist depending on the Gαi/o subtype. At α2A, quinpirole behaved as a partial agonist for Gi1, Gi2 and Gi3 and full agonist for Go1 and Go2, whereas at α2C it behaved as a partial agonist for all G protein subtypes except for Gi3 (full agonist) and showed no activity when coupled with Gαi1. As shown in Fig. 5, yohimbine, a non-selective α2 adrenoceptor antagonist, completely blocked the effect of 7-OH-PIPAT and quinpirole at both α2A and α2C adrenoceptors (for Gαo1), demonstrating the specificity of the α2 adrenoceptor signal produced by both agonists. The potency values of 7-OH-PIPAT and quinpirole in G protein activation BRET experiments correlates with the nanomolar and submicromolar affinities, respectively, seen in binding assays with brain membranes. Moreover, the higher potencies of 7-OH-PIPAT for α2C versus α2A adrenoceptors also correlate with our binding results due to the fact that 7-OH-PIPAT showed negative cooperativity in cortex but not in striatum. For quinpirole, however, it would be difficult to establish correlations with results from binding assays due to its low efficacy in BRET assays, which might lead to inaccurate values (Table 3, in parenthesis, and Table 4, in italics). Finally, and unexpectedly, the selective D4 receptor agonist RO-105824 (with subnanomolar affinity for α adrenoceptors) did not produce a significant activation of α2A or α2C adrenoceptors coupled to any of the Gαi/o subtypes, except for a small efficacy at α2A for Gαi2 and Gαi3 (Figs. 3 and 4). To confirm the binding of this putative selective D4 receptor ligand to α2A and α2C adrenoceptors, they were tested for their ability to modify the effect of clonidine. RO-105824 did counteract the respective full and partial agonistic effect of clonidine (1 μM) at the α2A and α2C adrenoceptors coupled to Gαo1 (Fig. 5C-D). Therefore, the results of BRET and radioligand-binding experiments disclosed a previously unknown additional role of the D4 receptor agonist RO-105824, as a very potent and low-efficacious ligand for α2 adrenoceptors at Gαi/o activation.
Figure 5. Specificity of the effect of dopaminergic ligands on α2A and α2C adrenoceptors.
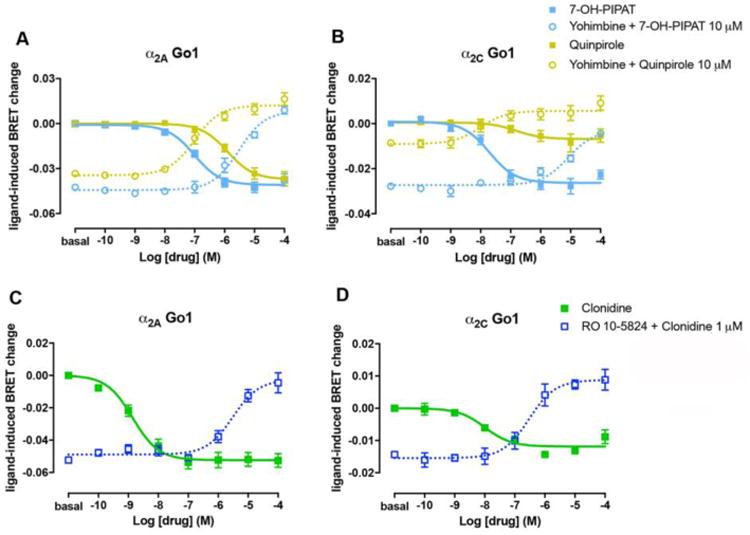
(A, B) Dose-dependent inhibition by increasing concentrations of the non-selective α2 receptor antagonist yohimbine of Gαo1 protein activation induced by 10 μM of the D2-like receptor agonists 7-OH-PIPAT (dotted blue) or 10 μM of quinpirole (dotted yellow) in HEK-293T cells transfected with α2A (A) or α2C (B) adrenoceptors, Gαo1-RLuc, γ2-mVenus and non-fused β1. As controls, concentration-response curves of Gαo1 protein activation by 7-OH-PIPAT (full blue) or quinpirole (full yellow) from Fig. 3D and Fig. 4D are showed. Cells were treated with Coelenterazine H followed by the addition of 7-OH-PIPAT or quinpirole. Ligand-induced changes in BRET values were measured as described in the Materials and Methods section. (C, D) Dose-dependent inhibition of the agonistic effect of clonidine at 1 μM by increasing concentrations of RO-105824 (dark blue) in cells transfected with α2A (C) or α2C (D) adrenoceptors, Gαo1-RLuc, γ2-mVenus and non-fused β1. As controls, concentration-response curves of Gαo1 protein activation by clonidine (full green) from Fig. 3D and Fig. 4D are showed. Data were adjusted to a sigmoidal concentration-response function by nonlinear regression analysis and represent means ± S.E.M. of 3 to 6 experiments performed in triplicate.
We also determined the effect of DA and synthetic DA receptor ligands on a DMR label-free assay in CHO transfected cells. This approach detects changes in local optical density due to cellular mass movements induced upon receptor activation (see Materials and Methods section), and DMR responses primarily reflect G protein-dependent signaling in living cells, since it can be abrogated by toxins or inhibitors of the G proteins involved [39]. DA was as capable as NE at activating cellular signaling in CHO cells transfected with α2A-RLuc8 receptor (Fig. 6A). The amount of α2A-RLuc8 receptor expressed was about 0.3 pmol/mg protein. DA and NE activation decreased by adding the α2AR antagonist BRL 44408 indicating the specificity of the cell activation through α2A receptor. The synthetic DA receptor ligands 7-OH-PIPAT, quinpirole and RO-105824 at 300 nM were also able to produce a significant response (Fig. 6B), substantially lower for RO-105824, which correlates with the G protein activation BRET assays (Fig. 3). These results indicate that DA and synthetic DA receptor ligands are also α2 adrenoceptor ligands able to activate Gαi/o proteins, which correlate with their efficacy with DMR.
Figure 6. DMR induced by NE, DA and synthetic DA receptor ligands using label-free assay.
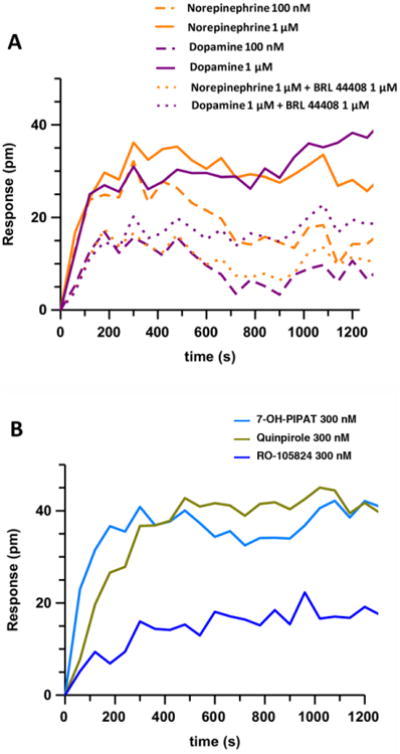
DMR assay was performed in CHO cells transfected with α2A adrenoceptor. (A) Cells were pretreated (dotted lines) or not (full lines) with the α2A adrenoceptor antagonist BRL 44408 at 1 μM for 30 minutes before adding the endogenous agonists DA or NE at 300 nM or 1 μM. (B) Cells were treated with 100 nM of several synthetic DA receptor ligands. The resulting shifts of reflected light wavelength (pm) were monitored over time. Each panel is a representative experiment of n = 3 different experiments. Each curve is the mean of a representative optical trace experiment carried out in triplicates.
α2A and α2C adrenoceptor-mediated effects of NE and DA on adenylyl cyclase activity
NE and DA-induced changes in adenylyl cyclase activity were also analyzed by measuring cAMP levels in intact cells transiently transfected with α2A or α2C adrenoceptor, using the CAMYEL BRET biosensor (see Materials and Methods section and ref. 21). HEK-293T cells have been reported to endogenously express β adrenoceptors [40]. Accordingly, we recently reported that NE, in non-transfected HEK-293T cells, stimulated Gs-mediated cAMP increase, which could be completely inhibited by the selective β adrenergic blocker propranolol (10 μM) (ref. 19; Suppl. Fig. 1; same website address as above). Therefore, the β adrenoceptor antagonist propranolol was added throughout the cAMP detection experiments. As shown in Fig. 7, NE and DA produced an increase in BRET, corresponding to a decrease in forskolin-induced cAMP accumulation for both α2A and α2C adrenoceptor-transfected cells. The decrease in adenylyl cyclase activity by NE and DA, provided apparent EC50 values that were qualitatively and quantitatively close to those observed with the Gi/o activation BRET assays, as NE was more potent than DA at α2A and α2C (1.4 ± 0.2 and 7 ± 4 nM for NE and 140 ± 40 and 90 ± 20 nM for DA, respectively). The putative Gi/o-dependent effects mediated by NE and DA were blocked by the non-selective α2 adrenoceptor antagonist yohimbine, confirming the receptor specificity of the signal (Fig 7). In addition, cells were treated with pertussis toxin which catalyzes the ADP-ribosylation of the αi subunits of the heterotrimeric G protein preventing its interaction with the receptor. As expected, pertussis toxin treatment selectively eliminated the initial, therefore Gi-dependent, component of the NE- and DA-mediated effects (Fig. 7). Surprisingly, NE and DA showed inverted U-shaped concentration-response curves with a putative Gs-dependent profile at high ligand concentrations for both α2A and α2C adrenoceptor-transfected cells (Fig. 7). These results could be explained by considering previous studies showing that α2 adrenoceptors functionally couple not only to Gi/o proteins but also to Gs [41-45]. Typically, the agonist concentrations necessary to elicit detectable stimulation of adenylyl cyclase are significantly higher than those for inhibition. Equivocal results were published by Zhang et al. [16] when comparing the effect of NE and DA on forskolin-induced adenylyl cyclase activation. In their cell systems, NE seemed to predominantly activate Gs with α2A and Gi with α2C, while DA would predominantly activate Gi with both receptors, but at high micromolar concentrations. In contrast, our experiments show that NE and DA follow the same differential concentration-dependent effects on Gi/o- and Gs- activation and, at least, at 10 and 100 μM, DA and NE promoted Gs activation mediated by both α2A and α2C adrenoceptors (Fig. 7).
Figure 7. Effect of NE and DA on the modulation of adenylyl cyclase activity by activation of α2A and α2C adrenoceptors.
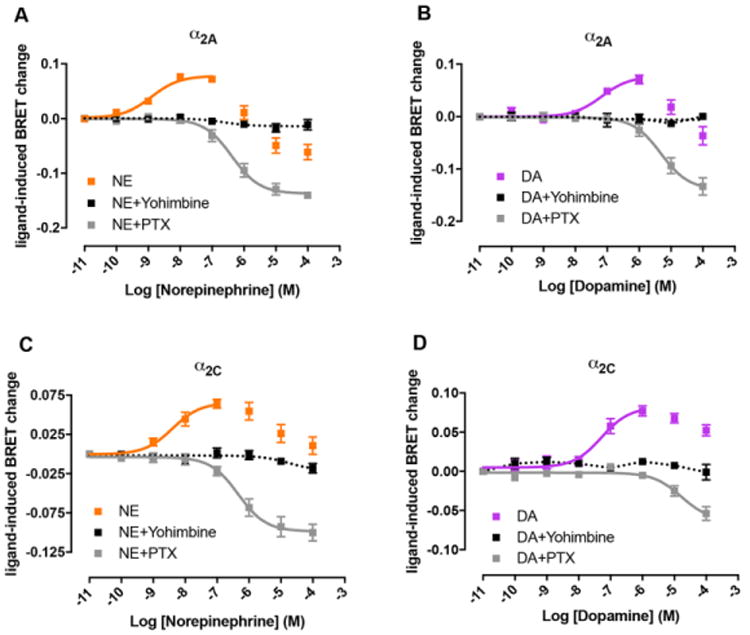
Concentration-response experiments of inhibition of forskolin-induced adenylyl cyclase activity by NE (orange) or DA (purple) mediated by α2A (A, B) or α2C adrenoceptors (C, D) in HEK-293T cells transiently transfected with the CAMYEL sensor and one of the receptors. Cells were treated with forskolin (1 μM) for 10 minutes with or without the selective α2 antagonist yohimbine (10 μM) followed by the addition of Coelenterazine H and increasing concentrations of NE or DA. After 10 minutes, BRET was measured as described in the Materials and Methods section. In grey, cells were treated with 100 ng/ml pertussis toxin (PTX) for 16-18 h previous to the experiment. Values obtained with forskolin alone were subtracted from BRET values for each agonist concentration. Data represent the mean ± S.E.M. of 3 to 7 experiments performed in triplicate.
α2A and α2C adrenoceptor-mediated effects of NE, DA and synthetic DA receptor ligands on ERK1/2 phosphorylation
Finally, we studied the ability of DA and synthetic DA receptor ligands to produce MAPK activation. First, we analyzed the increase on ERK1/2 phosphorylation produced by 300 nM of NE in CHO cells transfected with α2A-RLuc8 receptor. This NE concentration increased ERK1/2 phosphorylation levels 3-fold over basal and this effect was similar to that produced by 1 μM of DA (Fig. 8). Next, we demonstrated that the synthetic DA receptor ligands 7-OH-PIPAT, quinpirole and RO-105824, at 1 μM, were also able to produce MAPK activation (Fig. 8). At this concentration, the efficacy of RO-105824 was similar to that of the other ligands. Together with its very low efficacy disclosed on G protein activation and DMR assays, these results indicate that RO-105824 is a biased agonist of α2A adrenoceptors with functional selectivity for G protein-independent signaling. In summary, adenylyl cyclase activity and ERK1/2 phosphorylation experiments confirm the results from radioligand-binding and G protein activation assays indicating that DA and synthetic DA receptor ligands are efficacious α2 adrenoceptor agonists.
Figure 8. NE, DA and synthetic DA receptor ligands signaling via ERK 1/2 phosphorylation.
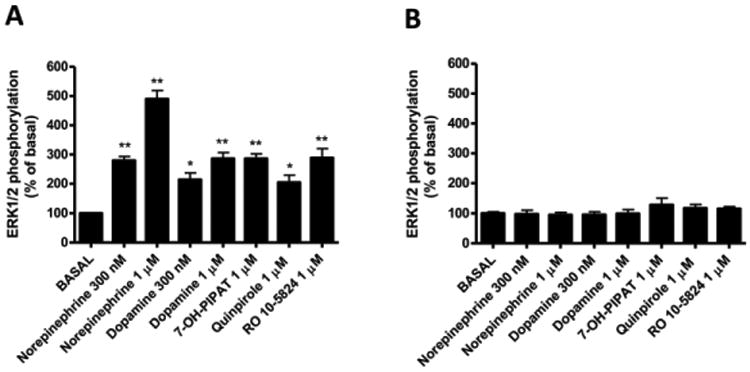
ERK 1/2 phosphorylation was determined in CHO cells transfected with the α2A adrenoceptor (A) and non-transfected control cells (B), treated with 300 nM or 1 μM of the tested ligands for 5 minutes at 37°C. Values are expressed as mean ± S.E.M. (n = 6) of percentage of phosphorylation relative to basal levels in not treated cells. Statistical differences vs. basal conditions were calculated by one-way ANOVA followed by Dunnett's post hoc test; *p<0.05 and **p<0.01.
Structural basis for DA at α2A and α2C adrenoceptors
An examination of the binding mode of DA to the adrenoceptors was undertaken to model the activity seen in the biological assays. To generate models of α2A and α2C adrenoceptors we used the RosettaCM [25] application within the Rosetta suite of macromolecular modeling tools [26, 46]. This method relies on optimal alignment of a target sequence with multiple template structures obtained from the PDB which are hybridized together to generate novel models. We submitted the sequence of α2A and α2C to BLAST-P and found the top five crystal templates by sequence identity to be the DA D3 receptor [27], β1 adrenoceptor [28], β2 adrenoceptor [29], serotonin 5-HT1B receptor [30], and serotonin 5-HT2B receptor [31]. Interestingly, the α2-adrenoceptors have more sequence identity in the transmembrane helical bundle with the D3 receptor than with the related β-adrenoceptors (Table S1). RosettaCM yielded an ensemble of low-energy models of the receptors, which were clustered by structural similarity. The top five cluster centers were included in the docking studies to account for structural diversity and uncertainty in homology modeling. To understand DA activation, we first examined the binding of DA to the D3 receptor, the only crystal structure of a DA receptor to date. As there is not a co-crystal structure of DA/D3 receptor, we first docked DA to the D3 receptor using RosettaLigand [34]. The starting coordinates of epinephrine bound to the β2 adrenoceptor [33] were used to place DA for docking. Binding pocket analysis identified residue D3.32 interacting with the primary amine in DA and the catechol hydroxyls interacting with S5.42 and S5.46. Important hydrophobic packing against the central portion of DA was achieved by V3.33, H6.55, and F6.51. These are the same interactions that were previously identified in a molecular dynamics simulation of DA binding at D3 receptor [47]. Comparing the residues at these positions to those in α2A and α2C adrenoceptors revealed identity at all residues except position 6.55, in which the His has been replaced with a Tyr in both α2A and α2C adrenoceptors (Fig. S1). Docking results of DA at either α2Aor α2C also identified many of these same residues as critical for binding. Particularly D3.32, V3.33, S5.42 and F6.51 were present in all receptor models contributing more than -0.4 Rosetta Energy Units each to the binding of DA (Fig. 9). Residues S5.46 and Y6.55 were also present in all receptor binding modes though contributions varied depending on which receptor type. These results coupled with those from the biological assays provide a strong structural reasoning behind the activity of DA at the α2A or α2C adrenoceptors.
Figure 9. Binding mode of DA at each receptor type.
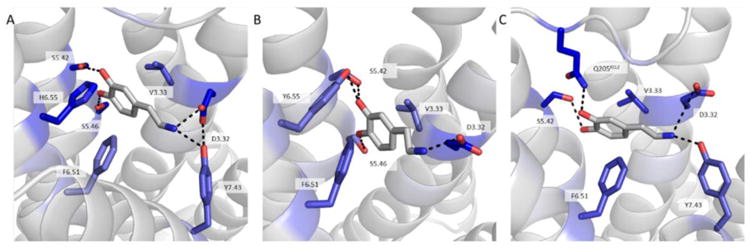
Shown is the docking orientation of DA at the D3 receptor (A) and α2A (B), and α2C adrenoceptors (C). Residues were scored for binding energy ΔΔG and those most strongly contributing to the binding of DA are shown in stick representation. The strength of binding interaction is colored by the depth of blue with dark blue being the most strongly contributing. Important hydrogen bonds to the amine group and catechol hydroxyls are formed in all binding poses.
Discussion
Previous studies reported DA as a potential α2 adrenoceptor ligand on the basis of radioligand binding experiments in transfected mammalian and insect cell lines [16,17] and in bird and rat brains (18) and also from autoradiographic experiments in tissues [18]. Furthermore, DA has been reported to decrease cAMP intracellular levels in transfected mammalian cell lines but only throughout α2C adrenoceptors, not α2A adrenoceptors, and at concentrations much higher than NE (EC50 in the micromolar range) [18].
In our study, we show that α2A and α2C adrenoceptors can bind DA at concentrations in the same order than NE suggesting that they could be activated by DA at in vivo concentrations. First, our results demonstrate that endogenous DA, and also common synthetic DA-receptor ligands, bind to α2 adrenoceptors with moderate to high affinity in the mammalian brain. Second, the affinity of DA for α2 adrenoceptors is in the same range as for D1-like and D2-like receptors, suggesting that endogenous levels of DA can activate both α2 adrenoceptors and dopamine receptors. Third, DA and synthetic DA receptor ligands can activate G protein and induce cell DMR through α2 adrenoceptors. Finally, DA and NE show the same cell signaling pattern, being both capable to modulate adenylyl cyclase activity and ERK1/2 phosphorylation at nanomolar concentrations.
The most conclusive demonstration that DA is an α2 adrenoceptor ligand comes from the results obtained with binding and G protein activation BRET assays, where the affinities or potencies of DA for α2 adrenoceptors were found to be very similar or even higher than for D1-like and some subtypes of D2-like receptors [19]. Particularly the EC50 values of DA for α2A and α2C adrenoceptors (5-170 nM) were consistently lower across all Gi/o protein subtypes as compared with the EC50 values (130-400 nM) for the predominant striatal D2-like receptor D2L [19]. Taking into account that the levels of tonic extracellular DA are 20-30 nM (with peaks of 500 nM) [48], DA could reach sufficient extracellular concentration to activate α2A and α2C adrenoceptors in the striatum, irrespective of the maximal concentration of extracellular NE. In fact, striatal DA release sites are designed for transmitter spillover [49] and most striatal DA receptors are primarily extrasynaptic [50, 51], as well as striatal adrenoceptors, based on the mismatched low NE innervation [7-11]. Although the specific functional role of the DA-sensitive α2 adrenoceptors in neuronal striatal function remains to be established, a previous study suggests that they might mediate an inhibitory modulatory role of the Gs/olf-coupled striatal adenosine A2A and DA D1 receptors [14].
The possibility of DA-mediated activation of α2A and α2C adrenoceptors in extrastriatal areas should not however, be underestimated. Cortical α2A adrenoceptors are most probably able to be activated by DA, particularly in the prefrontal cortex, which receives a rather dense DA innervation [52]. In fact, there is recent evidence for the localization of α2A adrenoceptors in the cortical terminals from mesencephalic DA neurons [53], which could play a role as “DA autoreceptors”. But there is also evidence for the localization of both α2A and α2C adrenoceptors in the soma and dendrites of the mesencephalic DA cells of both substantia nigra and ventral tegmental area [53, 54]. Apart from the NE input, these α2A and α2C adrenoceptors should be able to act as “DA autoreceptors” that control the non-synaptic somatodendritic DA release [49]. Adding the present results to our recent study that also indicates a significant role of NE as a Gi/o-coupled D2-like receptor agonist [19], we could state that Gi/o-coupled adrenoceptors and DA receptors should probably be considered as members of one ‘functional’ family of catecholamine receptors. A general consideration from the DA and D2-like receptor ligand sensitivity of cortical α2A adrenoceptors is that it should also be involved in the cognitive-enhancing effects associated with their activation, with possible implications for attention deficit hyperactivity disorder [55].
Molecular modeling of DA binding to the various receptors provides a likely binding hypothesis for the results obtained in the biological assays. Of note is the striking similarity between the ligand binding pocket of the D3 receptor and that of α2A and α2C adrenoceptors. Many of the residues that line the binding pocket are identical or chemically well conserved. Given this similarity it was perhaps unsurprising that the docking of DA at α2A and α2C was nearly identical to DA binding to the D3 receptor. The lower potency of DA at α2A and α2C adrenoceptors compared to NE seems to depend on a lower number of strong interactions as compared to those between DA and D3 receptors. The pocket may have evolved to bind the slightly bulkier NE and therefore is not of an ideal size for DA. However, the differences may also be due to the lower resolution of binding predictions for a comparative model as opposed to a crystal structure. Despite this, the structural model strongly mimics the results of the binding and activation experiments and therefore provides further evidence of DA acting as a ligand at these receptors.
Another major finding of the present study is that α2A and α2C adrenoceptors are also common targets for compounds previously characterized as D2-like receptor ligands. Particularly striking was the ability of prototypical D3 and D4 receptor agonists 7-OH-PIPAT and RO-105824 to bind with high affinity to α2A and α2C adrenoceptors, which might call for revisiting results of previous studies using these compounds. Furthermore, these two compounds and the other DA-synthetic ligands assayed, as well as NE, were able to activate ERK1/2 phosphorylation by binding to α2 adrenoceptors. The final pharmacological profile of RO-105824 was that of a potent biased agonist for α2A adrenoceptor with functional selectivity for a G protein-independent signaling. On the other hand, based on BRET experiments, both potency and efficacy dependence on the receptor and the Gαi/o protein subtype was the norm for all ligands, including the endogenous neurotransmitters. We already described that NE and DA show different receptor- and Gαi/o subtype-dependent potencies of D2-like receptor-mediated G protein activation [19]. The present results extend these findings to other receptors and to non-endogenous ligands, as well as to differences in efficacy. Even though G proteins of the Gαs-Gαolf family do show contrasting brain expression pattern [56], to our knowledge no clear region-specific pattern of mRNA expression for Gαi/o protein subtypes has been reported. Detailed characterization of the expression patterns for Gαi/o protein subtypes would then be central to determine their role in α2A and α2C adrenoceptor-activation and thus their possible specific targeting with Gαi/o subtype functionally selective compounds.
In conclusion, DA is a potent and efficacious ligand at α2 adrenoceptors, which modulates forskolin-induced adenylyl cyclase activity and ERK1/2 phosphorylation. The concentration required for these effects is in the range of that for activating D2-like and D1-like receptors, indicating that these receptors are members of one “functional” family of catecholamine receptors. Our results provide a clear answer to the mismatch between the low striatal NE innervation and the high density of striatal α2 adrenoceptors, which behave as functional DA receptors.
Acknowledgments
The authors would like to thank Dr. Céline Galés (INSERM) for generously sharing various Gi-like plasmid constructs for BRET experiments. This research was supported by “Ministerio de Economía y Competitividad” and European Regional Development Funds of the European Union Grant SAF2014-54840-R, “Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas” Grant CB06/05/0064, the “Fundació La Marató de TV3” Grant 20140610, the fellowship from the Japan Society for the Promotion of Science, and intramural funds of the National Institute on Drug Abuse. Work in the Meiler laboratory is supported through NIH (R01 GM080403, R01 GM099842, R01 DK097376) and NSF (CHE 1305874).
Footnotes
Conflict of interests: The authors declare no conflicts of interest
Author contributions: M. S.-S., V. C.-A., H. Y., N.-S. C, E. M., A. C., and B. J. B. performed the experiments and analyzed the data. H. Y., B. J. B., J. M., V. C. and S. F., designed the experiments. M. S.-S., V. C.-A., H. Y., B. J. B., J. M., E. I. C., V. C. and S. F., wrote the manuscript.
References
- 1.Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(1):S1–324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.MacDonald E, Kobilka BK, Scheinin M. Gene targeting--homing in on alpha 2-adrenoceptor-subtype function. Trends Pharmacol Sci. 1997;18(6):211–219. doi: 10.1016/s0165-6147(97)01063-8. [DOI] [PubMed] [Google Scholar]
- 3.Nicholas AP, Pieribone V, Hokfelt T. Distributions of mRNAs for alpha-2 adrenergic receptor subtypes in rat brain: an in situ hybridization study. J Comp Neurol. 1993;328(4):575–594. doi: 10.1002/cne.903280409. [DOI] [PubMed] [Google Scholar]
- 4.Scheinin M, Lomasney JW, Hayden-Hixson DM, Schambra UB, Caron MG, Lefkowitz RJ, Fremeau RT., Jr Distribution of alpha 2-adrenergic receptor subtype gene expression in rat brain. Brain Res Mol Brain Res. 1994;21(1-2):133–149. doi: 10.1016/0169-328x(94)90386-7. [DOI] [PubMed] [Google Scholar]
- 5.Fagerholm V, Rokka J, Nyman L, Sallinen J, Tiihonen J, Tupala E, Haaparanta M, Hietala J. Autoradiographic characterization of alpha(2C)-adrenoceptors in the human striatum. Synapse. 2008;62(7):508–515. doi: 10.1002/syn.20520. [DOI] [PubMed] [Google Scholar]
- 6.Lehto J, Virta JR, Oikonen V, Roivainen A, Luoto P, Arponen E, Helin S, Hietamaki J, Holopainen A, Kailajarvi M, Peltonen JM, Rouru J, Sallinen J, Virtanen K, Volanen I, Scheinin M, Rinne JO. Test-retest reliability of (11)C-ORM-13070 in PET imaging of alpha2C-adrenoceptors in vivo in the human brain. Eur J Nucl Med Mol Imaging. 2015;42(1):120–127. doi: 10.1007/s00259-014-2899-z. [DOI] [PubMed] [Google Scholar]
- 7.Ordway GA, Jaconetta SM, Halaris AE. Characterization of subtypes of alpha-2 adrenoceptors in the human brain. J Pharmacol Exp Ther. 1993;264(2):967–976. [PubMed] [Google Scholar]
- 8.Uhlen S, Lindblom J, Tiger G, Wikberg JE. Quantification of alpha2A and alpha2C adrenoceptors in the rat striatum and in different regions of the spinal cord. Acta Physiol Scand. 1997;160(4):407–412. doi: 10.1046/j.1365-201X.1997.00175.x. [DOI] [PubMed] [Google Scholar]
- 9.Lindvall O, Bjorklund A. The organization of the ascending catecholamine neuron systems in the rat brain as revealed by the glyoxylic acid fluorescence method. Acta Physiol Scand Suppl. 1974;412:1–48. [PubMed] [Google Scholar]
- 10.Swanson LW, Hartman BK. The central adrenergic system. An immunofluorescence study of the location of cell bodies and their efferent connections in the rat utilizing dopamine-beta-hydroxylase as a marker. J Comp Neurol. 1975;163(4):467–505. doi: 10.1002/cne.901630406. [DOI] [PubMed] [Google Scholar]
- 11.Aston-Jones G. Locus coeruleus, A5 and A7 noradrenergic cell groups. In: Paxinos G, editor. The Rat Nervous System. Academic Press; San Diego: 2004. pp. 259–294. [Google Scholar]
- 12.Gobert A, Billiras R, Cistarelli L, Millan MJ. Quantification and pharmacological characterization of dialysate levels of noradrenaline in the striatum of freely-moving rats: release from adrenergic terminals and modulation by alpha2-autoreceptors. J Neurosci Methods. 2004;140(1-2):141–152. doi: 10.1016/j.jneumeth.2004.04.040. [DOI] [PubMed] [Google Scholar]
- 13.Holmberg M, Scheinin M, Kurose H, Miettinen R. Adrenergic alpha2C-receptors reside in rat striatal GABAergic projection neurons: comparison of radioligand binding and immunohistochemistry. Neuroscience. 1999;93(4):1323–1333. doi: 10.1016/s0306-4522(99)00260-2. [DOI] [PubMed] [Google Scholar]
- 14.Hara M, Fukui R, Hieda E, Kuroiwa M, Bateup HS, Kano T, Greengard P, Nishi A. Role of adrenoceptors in the regulation of dopamine/DARPP-32 signaling in neostriatal neurons. J Neurochem. 2010;113(4):1046–1059. doi: 10.1111/j.1471-4159.2010.06668.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ihalainen JA, Tanila H. In vivo regulation of dopamine and noradrenaline release by alpha2A-adrenoceptors in the mouse nucleus accumbens. J Neurochem. 2004;91(1):49–56. doi: 10.1111/j.1471-4159.2004.02691.x. [DOI] [PubMed] [Google Scholar]
- 16.Zhang W, Klimek V, Farley JT, Zhu MY, Ordway GA. alpha2C adrenoceptors inhibit adenylyl cyclase in mouse striatum: potential activation by dopamine. J Pharmacol Exp Ther. 1999;289(3):1286–1292. [PubMed] [Google Scholar]
- 17.Alachkar A, Brotchie JM, Jones OT. Binding of dopamine and 3-methoxytyramine as l-DOPA metabolites to human alpha(2)-adrenergic and dopaminergic receptors. Neurosci Res. 2010;67(3):245–249. doi: 10.1016/j.neures.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 18.Cornil CA, Ball GF. Interplay among catecholamine systems: dopamine binds to alpha2-adrenergic receptors in birds and mammals. J Comp Neurol. 2008;511(5):610–627. doi: 10.1002/cne.21861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sánchez-Soto M, Bonifazi A, Cai NS, Ellenberger MP, Newman AH, Ferré S, Yano H. Evidence for Noncanonical Neurotransmitter Activation: Norepinephrine as a Dopamine D2-Like Receptor Agonist. Mol Pharmacol. 2016;89(4):457–466. doi: 10.1124/mol.115.101808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Urizar E, Yano H, Kolster R, Gales C, Lambert N, Javitch JA. CODA-RET reveals functional selectivity as a result of GPCR heteromerization. Nat Chem Biol. 2011;7(9):624–630. doi: 10.1038/nchembio.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang LI, Collins J, Davis R, Lin KM, DeCamp D, Roach T, Hsueh R, Rebres RA, Ross EM, Taussig R, Fraser I, Sternweis PC. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J Biol Chem. 2007;282(14):10576–10584. doi: 10.1074/jbc.M609695200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Casadó V, Cortés A, Ciruela F, Mallol J, Ferré S, Lluis C, Canela EI, Franco R. Old and new ways to calculate the affinity of agonists and antagonists interacting with G-protein-coupled monomeric and dimeric receptors: the receptor-dimer cooperativity index. Pharmacol Ther. 2007;116(3):343–354. doi: 10.1016/j.pharmthera.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 23.Ferré S, Casadó V, Devi LA, Filizola M, Jockers R, Lohse MJ, Milligan G, Pin JP, Guitart X. G protein-coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol Rev. 2014;66(2):413–434. doi: 10.1124/pr.113.008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Casadó V, Ferrada C, Bonaventura J, Gracia E, Mallol J, Canela EI, Lluis C, Cortés A, Franco R. Useful pharmacological parameters for G-protein-coupled receptor homodimers obtained from competition experiments. Agonist-antagonist binding modulation. Biochem Pharmacol. 2009;78(12):1456–1463. doi: 10.1016/j.bcp.2009.07.012. [DOI] [PubMed] [Google Scholar]
- 25.Song Y, DiMaio F, Wang RY, Kim D, Miles C, Brunette T, Thompson J, Baker D. High-resolution comparative modeling with RosettaCM. Structure. 2013;21(10):1735–1742. doi: 10.1016/j.str.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bender BJ, Cisneros A, 3rd, Duran AM, Finn JA, Fu D, Lokits AD, Mueller BK, Sangha AK, Sauer MF, Sevy AM, Sliwoski G, Sheehan JH, DiMaio F, Meiler J, Moretti R. Protocols for Molecular Modeling with Rosetta3 and RosettaScripts. Biochemistry. 2016;55(34):4748–4763. doi: 10.1021/acs.biochem.6b00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330(6007):1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller-Gallacher JL, Nehme R, Warne T, Edwards PC, Schertler GF, Leslie AG, Tate CG. The 2.1 A resolution structure of cyanopindolol-bound beta1-adrenoceptor identifies an intramembrane Na+ ion that stabilises the ligand-free receptor. PLoS One. 2014;9(3):e92727. doi: 10.1371/journal.pone.0092727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318(5854):1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang C, Jiang Y, Ma J, Wu H, Wacker D, Katritch V, Han GW, Liu W, Huang XP, Vardy E, McCorvy JD, Gao X, Zhou XE, Melcher K, Zhang C, Bai F, Yang H, Yang L, Jiang H, Roth BL, Cherezov V, Stevens RC, Xu HE. Structural basis for molecular recognition at serotonin receptors. Science. 2013;340(6132):610–614. doi: 10.1126/science.1232807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wacker D, Wang C, Katritch V, Han GW, Huang XP, Vardy E, McCorvy JD, Jiang Y, Chu M, Siu FY, Liu W, Xu HE, Cherezov V, Roth BL, Stevens RC. Structural features for functional selectivity at serotonin receptors. Science. 2013;340(6132):615–619. doi: 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kothiwale S, Mendenhall JL, Meiler J. BCL∷Conf: small molecule conformational sampling using a knowledge based rotamer library. J Cheminform. 2015;7:47. doi: 10.1186/s13321-015-0095-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ring AM, Manglik A, Kruse AC, Enos MD, Weis WI, Garcia KC, Kobilka BK. Adrenaline-activated structure of beta2-adrenoceptor stabilized by an engineered nanobody. Nature. 2013;502(7472):575–579. doi: 10.1038/nature12572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meiler J, Baker D. ROSETTALIGAND: protein-small molecule docking with full side-chain flexibility. Proteins. 2006;65(3):538–548. doi: 10.1002/prot.21086. [DOI] [PubMed] [Google Scholar]
- 35.Lemmon G, Meiler J. Rosetta Ligand docking with flexible XML protocols. Methods Mol Biol. 2012;819:143–155. doi: 10.1007/978-1-61779-465-0_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meador-Woodruff JH, Damask SP, Wang J, Haroutunian V, Davis KL, Watson SJ. Dopamine receptor mRNA expression in human striatum and neocortex. Neuropsychopharmacology. 1996;15:17–29. doi: 10.1016/0893-133X(95)00150-C. [DOI] [PubMed] [Google Scholar]
- 37.Pohjanoksa K, Jansson CC, Luomala K, Marjamaki A, Savola JM, Scheinin M. Alpha2-adrenoceptor regulation of adenylyl cyclase in CHO cells: dependence on receptor density, receptor subtype and current activity of adenylyl cyclase. Eur J Pharmacol. 1997;335(1):53–63. doi: 10.1016/s0014-2999(97)01154-0. [DOI] [PubMed] [Google Scholar]
- 38.Kukkonen JP, Renvaktar A, Shariatmadari R, Akerman KE. Ligand- and subtype-selective coupling of human alpha-2 adrenoceptors to Ca++ elevation in Chinese hamster ovary cells. J Pharmacol Exp Ther. 1998;287(2):667–671. [PubMed] [Google Scholar]
- 39.Schröder R, Schmidt J, Blättermann S, Peters L, Janssen N, Grundmann M, Seemann W, Kaufel D, Merten N, Drewke C, Gomeza J, Milligan G, Mohr K, Kostenis E. Applying label-free dynamic mass redistribution technology to frame signaling of G protein-coupled receptors noninvasively in living cells. Nat Protoc. 2011;6(11):1748–1760. doi: 10.1038/nprot.2011.386. [DOI] [PubMed] [Google Scholar]
- 40.Atwood BK, Lopez J, Wager-Miller J, Mackie K, Straiker A. Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC Genomics. 2011;12:14. doi: 10.1186/1471-2164-12-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fraser CM, Arakawa S, McCombie WR, Venter JC. Cloning, sequence analysis, and permanent expression of a human alpha 2-adrenergic receptor in Chinese hamster ovary cells. Evidence for independent pathways of receptor coupling to adenylate cyclase attenuation and activation. J Biol Chem. 1989;264(20):11754–11761. [PubMed] [Google Scholar]
- 42.Jones SB, Halenda SP, Bylund DB. Alpha 2-adrenergic receptor stimulation of phospholipase A2 and of adenylate cyclase in transfected Chinese hamster ovary cells is mediated by different mechanisms. Mol Pharmacol. 1991;39(2):239–245. [PubMed] [Google Scholar]
- 43.Eason MG, Jacinto MT, Liggett SB. Contribution of ligand structure to activation of alpha 2-adrenergic receptor subtype coupling to Gs. Mol Pharmacol. 1994;45(4):696–702. [PubMed] [Google Scholar]
- 44.Eason MG, Kurose H, Holt BD, Raymond JR, Liggett SB. Simultaneous coupling of alpha 2-adrenergic receptors to two G-proteins with opposing effects. Subtype-selective coupling of alpha 2C10, alpha 2C4, and alpha 2C2 adrenergic receptors to Gi and Gs. J Biol Chem. 1992;267(22):15795–1580. [PubMed] [Google Scholar]
- 45.Eason MG, Liggett SB. Identification of a Gs coupling domain in the amino terminus of the third intracellular loop of the alpha 2A-adrenergic receptor. Evidence for distinct structural determinants that confer Gs versus Gi coupling. J Biol Chem. 1995;270(42):24753–24760. doi: 10.1074/jbc.270.42.24753. [DOI] [PubMed] [Google Scholar]
- 46.Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, Davis IW, Cooper S, Treuille A, Mandell DJ, Richter F, Ban YE, Fleishman SJ, Corn JE, Kim DE, Lyskov S, Berrondo M, Mentzer S, Popovic Z, Havranek JJ, Karanicolas J, Das R, Meiler J, Kortemme T, Gray JJ, Kuhlman B, Baker D, Bradley P. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feng Z, Hou T, Li Y. Selectivity and activation of dopamine D3R from molecular dynamics. J Mol Model. 2012;18(12):5051–5063. doi: 10.1007/s00894-012-1509-x. [DOI] [PubMed] [Google Scholar]
- 48.Owesson-White CA, Roitman MF, Sombers LA, Belle AM, Keithley RB, Peele JL, Carelli RM, Wightman RM. Sources contributing to the average extracellular concentration of dopamine in the nucleus accumbens. J Neurochem. 2012;121(2):252–262. doi: 10.1111/j.1471-4159.2012.07677.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rice ME, Patel JC, Cragg SJ. Dopamine release in the basal ganglia. Neuroscience. 2011;198:112–137. doi: 10.1016/j.neuroscience.2011.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hersch SM, Ciliax BJ, Gutekunst CA, Rees HD, Heilman CJ, Yung KK, Bolam JP, Ince E, Yi H, Levey AI. Electron microscopic analysis of D1 and D2 dopamine receptor proteins in the dorsal striatum and their synaptic relationships with motor corticostriatal afferents. J Neurosci. 1995;15(7 Pt 2):5222–5237. doi: 10.1016/b978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yung KK, Bolam JP, Smith AD, Hersch SM, Ciliax BJ, Levey AI. Immunocytochemical localization of D1 and D2 dopamine receptors in the basal ganglia of the rat: light and electron microscopy. Neuroscience. 1995;65(3):709–730. doi: 10.1016/0306-4522(94)00536-e. [DOI] [PubMed] [Google Scholar]
- 52.Goldman-Rakic PS, Lidow MS, Smiley JF, Williams MS. The anatomy of dopamine in monkey and human prefrontal cortex. J Neural Transm Suppl. 1992;36:163–177. doi: 10.1007/978-3-7091-9211-5_8. [DOI] [PubMed] [Google Scholar]
- 53.Castelli MP, Spiga S, Perra A, Madeddu C, Mulas G, Ennas MG, Gessa GL. alpha2A adrenergic receptors highly expressed in mesoprefrontal dopamine neurons. Neuroscience. 2016;332:130–139. doi: 10.1016/j.neuroscience.2016.06.037. [DOI] [PubMed] [Google Scholar]
- 54.Lee A, Wissekerke AE, Rosin DL, Lynch KR. Localization of alpha2C-adrenergic receptor immunoreactivity in catecholaminergic neurons in the rat central nervous system. Neuroscience. 1998;84(4):1085–1096. doi: 10.1016/s0306-4522(97)00578-2. [DOI] [PubMed] [Google Scholar]
- 55.Brennan AR, Arnsten AF. Neuronal mechanisms underlying attention deficit hyperactivity disorder: the influence of arousal on prefrontal cortical function. Ann N Y Acad Sci. 2008;1129:236–245. doi: 10.1196/annals.1417.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herve D. Identification of a specific assembly of the g protein golf as a critical and regulated module of dopamine and adenosine-activated cAMP pathways in the striatum. Front Neuroanat. 2011;5:48. doi: 10.3389/fnana.2011.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]