

Abstract
Neurosurgery always results in neuroinflammation, which may activate microglial cells. Previous studies have demonstrated that fentanyl could be used for the induction or maintenance of anesthesia prior to surgery. However, it is unknown if fentanyl attenuates neuroinflammation prophylactically. Cell viability in groups that were treated with different concentrations of fentanyl (0.01, 0.1, 1 or 5 µmol/l) was analyzed by an MTT assay. BV-2 microglial cells were treated with lipopolysaccharide (LPS) at a concentration of 1 µg/ml to mimic neuroinflammation in vitro. BV-2 cells were pretreated with 5 µmol/l fentanyl prior to stimulation by LPS. The protein levels of tumor necrosis factor (TNF)-α, interleukin (IL)-1β and IL-10 in the culture medium were assessed by ELISA. The mRNA level of toll-like receptor (TLR)4 was evaluated by reverse transcription-quantitative polymerase chain reaction analysis. The protein levels of TLR4, glycogen synthase kinase (GSK)-3β and phosphorylated (p)-GSK-3β in BV-2 cells were assessed by western blot analysis. The MTT assay demonstrated that low concentrations of fentanyl (0.01, 0.1 or 1 µmol/l) did not affect the cell viability of BV-2 cells, while 5 µmol/l fentanyl significantly reduced BV-2 cell viability. The results of ELISA revealed that LPS significantly upregulated the release of TNF-α, IL-1β and IL-10, which were repressed by fentanyl pretreatment. Fentanyl pretreatment significantly reduced the LPS-induced elevation of TLR4 at mRNA and protein levels as well as p-GSK-3β protein levels in BV-2 cells. In conclusion, fentanyl pretreatment protects BV-2 cells from LPS-induced neuroinflammation by inhibiting TLR4 expression and GSK-3β activation. Neuroinflammation induced by surgery serves an important role in the development of postoperative cognitive dysfunction (POCD) and targeting the TLR4 and GSK-3β signaling pathway may provide a novel therapeutic approach for the treatment of POCD.
Keywords: fentanyl, BV-2 cells, toll-like receptor 4, glycogen synthase kinase-3β, neuroinflammation
Introduction
Inflammation is a process that is associated with the adherence and invasion of leukocytes to injured or infected tissues. It is modulated by a number of mediators including cytokines, chemokines, prostaglandins and substance P (1). The central nervous system (CNS) was reported to respond to peripheral inflammatory stimuli by initiating a local inflammatory response, also known as neuroinflammation (2). There are numerous critical features in the development of neuroinflammation, including glial activation, the accumulation of pro-inflammatory cytokines and the expression of adhesion molecules (1,3,4).
Neuroinflammation is inflammation in the peripheral nervous system and the CNS. Excessive inflammatory responses activate microglia, leading to the release of pro-inflammatory factors, including interleukin (IL)-1β, IL-6 and tumor necrosis factor-α (TNF-α). In turn, pro-inflammatory factors can aggravate neuroinflammatory reactions, neuronal degeneration and brain function. A large number of clinical and animal studies have demonstrated that neuroinflammation is closely associated with mental and neurodegenerative diseases (5). Non-steroidal anti-inflammatory drugs are commonly used to treat neuroinflammation (6,7). To the best of the authors' knowledge, no previous studies have investigated the effects of analgesic drugs on neuroinflammation.
As a glycolipid that is a major component of the outer leaflet of the outer membrane in Gram-negative bacteria, lipopolysaccharide (LPS) is commonly applied in the study of neuroinflammation in microglia (8,9). LPS activates several cellular responses, which accelerate the release of pro-inflammatory cytokines (10). The BV-2 microglia cell line exhibits properties associated with inflammation following treatment with 1 µg/ml LPS, although the cell viability is not affected (11,12). Therefore, LPS was adopted as the stimulant of inflammation in BV-2 cells in the present study.
Fentanyl, a high-potency opiate, is widely prescribed to treat acute and chronic pain (13,14). The mechanisms responsible for the analgesic effects of fentanyl have been extensively investigated (15–17). It has been well documented that inflammation results in pain (18,19). It remains unknown whether fentanyl attenuates neuroinflammation in BV-2, therefore the effect of fentanyl on LPS-induced neuroinflammation in BV-2 cells and its molecular mechanisms were investigated in the present study.
Materials and methods
Cell culture
BV-2 cells, purchased from the American Type Culture Collection (Manassas, VA, USA), were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml streptomycin in an incubator at the temperature of 37°C with 95% humidity and 5% CO2. Fentanyl (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was dissolved in 0.1% dimethyl sulfoxide (DMSO) to avoid toxicity in BV-2 microglial cells and a corresponding amount of 0.1% DMSO without fentanyl was added to the cells of the control group. The cells were also kept in the dark during each experiment.
MTT assay
BV-2 microglial cells were seeded in 96-well plates at a density of 5×103 cells/well. Following treatment with various concentrations of fentanyl (0.01, 0.1, 1 and 5 µmol/l) for 24 h, cells were cultured in fresh DMEM supplemented with 10% FBS, 100 U/ml penicillin, 100 µg/ml streptomycin and 5 mg/ml MTT for another 4 h. The blue formazan products in cells were dissolved in DMSO. The optical density of the reaction medium was determined at 570 nm using a microplate reader.
ELISA
Levels of tumor necrosis factor (TNF)-α, interleukin (IL)-1β and IL-10 in the supernatant of the BV-2 cell culture, obtained by centrifugation (15 min; 1,000 × g; 2–8°C) were measured using TNF-α (MTA00B), IL-1β (MLB00C) and IL-10 (M1000B) ELISA kits (all R&D Systems, Inc., Minneapolis, MN, USA) in accordance with the manufacturer's protocol. BV-2 microglial cells were seeded in 96-well cell culture plates and fentanyl pretreatment was added to the medium with or without 1 µg/ml LPS (from Escherichia coli strain 055:B5; Sigma-Aldrich; Merck KGaA). The TNF-α level was determined 6 h after LPS stimulation (20), while IL-1β and IL-10 levels were determined 24 h after LPS stimulation (21).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis
The toll-like receptor (TLR)4 mRNA level in BV-2 cells reaches the highest level at 4 h after LPS stimulation, as previously reported (22). Thus, in the current study, the same time point was selected for RT-qPCR. RNA was isolated from BV2 cells using TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.). Analysis was performed using the Maxima First Strand cDNA Synthesis kit for RT-qPCR with dsDNase (Invitrogen; Thermo Fisher Scientific, Inc.). For the RT reaction 10X dsDNase Buffer (1 µl), dsDNase (1 µl), total RNA (1 µg) and nuclease-free water to a total volume of 10 µl were incubated for 2 min at 37°C. Then, 5X Reaction mix (4 µl), Maxima Enzyme Mix (2 µl) and nuclease-free water (4 µl) were added and the mixture was incubated for 10 min at 25°C followed by 15 min at 50°C. Following RT, qPCR was performed in 96-well plates with the ABI PRISM® 7000 Sequence Detection system (Applied Biosystems; Thermo Fisher Scientific, Inc.). The qPCR reaction mixture contained the following: cDNA (0.5 µg), forward primer (0.5 µM), reverse primer (0.5 µM), 50X ROX reference dye (0.5 µl), 2X master mix (25 µl) and water to 50 µl. The thermocycling conditions were as follows: Initial denaturation 95°C for 15 min; followed by 40 cycles of denaturation at 94°C for 10 sec, annealing for GAPDH at 62°C and for TLR4 at 55°C for 30 sec, extension at 72°C for 30 sec; and a final extension at 72°C for 10 min. The relative amount of TLR4 was measured using the 2−∆∆Cq method (23). The primers were as follows: TLR4, 5′-TGGTTGCTGTTCTTATTCTGATTTG-3′ (forward) and 5′-GACCCATGAAATTGGCACTCAT-3′ (reverse); GAPDH, 5′-TCCTGCACCACCAACTGCTTAGCC-3′ (forward) and 5′-GTTCAGCTCTGGGATGACCTTGCC-3′ (reverse). GAPDH was used as the endogenous control to normalize TLR4.
Western blot analysis
Cells were lysed with 0.5 ml radioimmunoprecipitation assay buffer (Beyotime Institute of Biotechnology, Haimen, China) and the concentration of protein was determined using an Enhanced BCA Protein Assay kit (P0010; Beyotime Institute of Biotechnology). Protein samples (20 µg) from the lysates were separated by 10% SDS-PAGE and blotted onto polyvinylidene fluoride membranes. Membranes were blocked with 5% skimmed milk or 5% bovine serum albumin (Beyotime Institute of Biotechnology) for phosphorylated (p-) protein for 1 h at room temperature. Membranes were then treated with primary antibodies against TLR4 (1:1,000; sc-293072; Sigma-Aldrich; Merck KGaA), GSK-3β (1:1,000; sc-71186; Sigma-Aldrich; Merck KGaA), p-GSK-3β (1:1,000; sc-373800; Sigma-Aldrich; Merck KGaA) and β-actin (1:1,000; sc-58673; Sigma-Aldrich; Merck KGaA) at 4°C overnight. Following three rinses with Tris-buffered saline and Tween-20, membranes were treated with horseradish peroxidase (HRP)-conjugated secondary antibody (mouse anti-Armenian hamster IgG-HRP; 1:10,000; sc-2789; Sigma-Aldrich; Merck KGaA) at room temperature for 1 h. Bands were visualized by an enhanced chemiluminescence kit (Sigma-Aldrich; Merck KGaA). β-actin was used as an endogenous control. Image pro plus 6.0 (Media Cybernetics, Inc., Rockville, MD, USA) was used for densitometry.
Statistical analysis
Data are presented as the mean ± standard deviation. Data were analyzed by one-way analysis of variance followed by the Schefffe post-hoc test. Analyses were conducted using SPSS 12.0 software (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Low concentrations of fentanyl do not induce cytotoxicity in BV-2 cells
The cell viability of BV-2 cells was assessed by an MTT assay. BV-2 cells that were treated with 0.01, 0.1 or 1 µmol/l fentanyl demonstrated no significant reduction in cell viability compared with the control group. However, 5 µmol/l fentanyl significantly decreased cell viability by 36% compared with the control group (P<0.05; Fig. 1). Consequently, 5 µmol/l fentanyl was selected for subsequent experiments.
Figure 1.
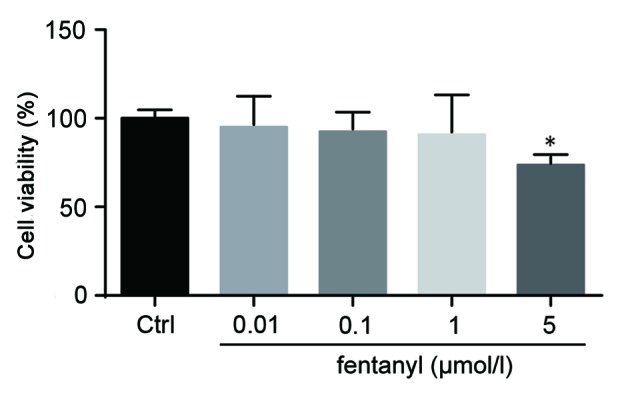
Low concentrations of fentanyl did not induce cytotoxicity in BV-2 cells. The cell viability of BV-2 cells that were treated with 0.01, 0.1, 1 and 5 µmol/l fentanyl was measured using an MTT assay. *P<0.05 vs. control group. Ctrl, control.
Fentanyl pretreatment suppresses the LPS-induced elevation of IL-1β, TNF-α and IL-10 in BV-2 cells
The levels of IL-1β, TNF-α and IL-10 that were released from BV-2 cells were assessed by ELISA (Fig. 2). Released cytokines were not affected by treatment with 5 µmol/l fentanyl alone in BV-2 cells. Compared with the control group, 1 µg/ml LPS significantly elevated the release of IL-1β, TNF-α and IL-10 (P<0.001; Fig. 2). Pretreatment with 5 µmol/l fentanyl significantly decreased the LPS-upregulated release of IL-1β, TNF-α and IL-10 compared with the LPS stimulated group (P<0.01; Fig. 2).
Figure 2.

Fentanyl pretreatment suppressed the LPS-induced elevation of IL-1β, TNF-α and IL-10 in BV-2 cells. IL-1β, TNF-α and IL-10 protein levels in BV-2 cells were assessed using ELISA kits. Cells were either untreated, or treated with 5 µmol/l fentanyl, 1 µg/ml LPS or fentanyl followed by LPS. ***P<0.001 vs. control group; ##P<0.01 vs. LPS group. Ctrl, control; IL, interleukin; TNF, tumor necrosis factor; LPS, lipopolysaccharide.
LPS-induced upregulation of TLR4 mRNA level is suppressed by fentanyl
An RT-qPCR analysis was conducted to detect the mRNA level of TLR4 in BV-2 cells. No significant differences in the mRNA level of TLR4 were identified between the control group and the fentanyl alone treatment group (Fig. 3). Compared with the control group, the mRNA level of TLR4 was significantly elevated by LPS stimulation (P<0.01; Fig. 3). Fentanyl pretreatment significantly decreased the LPS-induced upregulation of TLR4 mRNA by 38% compared with the LPS stimulated group (P<0.05; Fig. 3).
Figure 3.
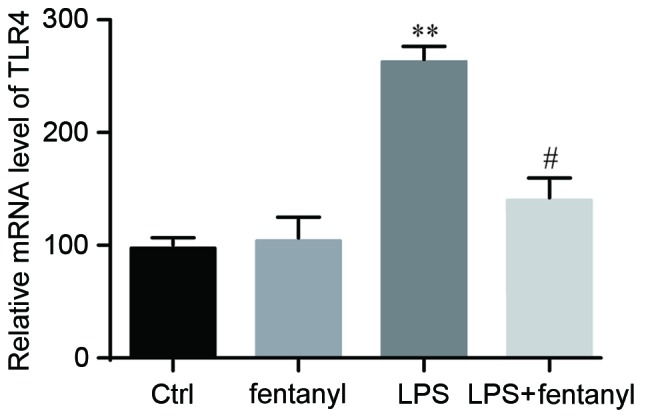
LPS-induced upregulation of TLR4 mRNA level was suppressed by fentanyl. TLR4 mRNA levels were assessed using reverse transcription-quantitative polymerase chain reaction analysis. GAPDH was used as the endogenous control. BV-2 cells were either untreated, or treated with 5 µmol/l fentanyl, 1 µg/ml LPS or fentanyl followed by LPS. **P<0.01 vs. control group; #P<0.05 vs. LPS group. Ctrl, control; TLR, toll-like receptor; LPS, lipopolysaccharide.
LPS-induced upregulation of the TLR4 protein level is repressed by fentanyl
A western blot analysis was performed to detect the TLR4 protein level in BV-2 cells (Fig. 4A). No significant difference in the TLR4 protein level was identified between the control group and the fentanyl alone treatment group (Fig. 4B). The TLR4 protein level was significantly elevated by LPS stimulation compared with the control group (P<0.01; Fig. 4B). Fentanyl pretreatment significantly downregulated the LPS-induced increase of TLR4 protein level compared with the LPS stimulated group (P<0.05; Fig. 4B).
Figure 4.
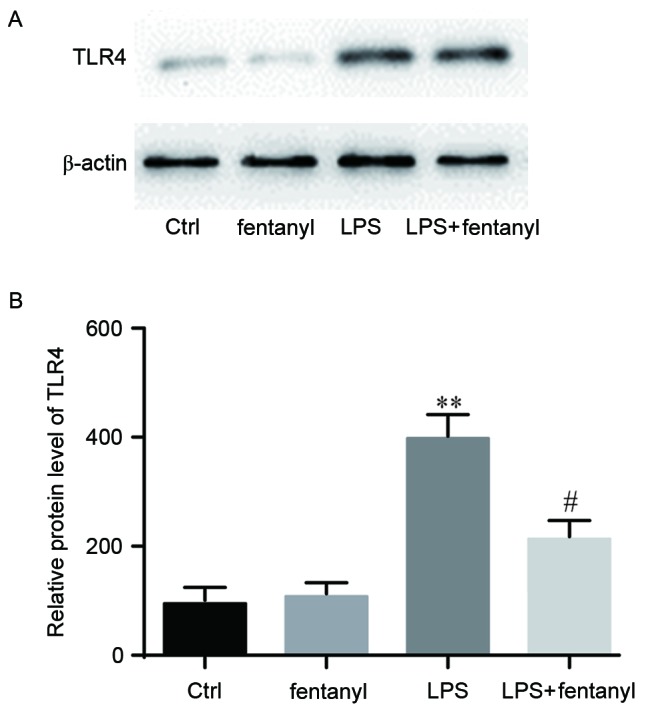
LPS-induced upregulation of the TLR4 protein level was repressed by fentanyl. BV-2 cells were either untreated, or treated with 5 µmol/l fentanyl, 1 µg/ml LPS or fentanyl followed by LPS. β-actin was used as the endogenous control. TLR4 protein levels were (A) measured by western blotting, then (B) quantified and statistically analyzed. **P<0.01 vs. control group; #P<0.05 vs. LPS group. Ctrl, control; TLR, toll-like receptor; LPS, lipopolysaccharide.
Fentanyl pretreatment further promotes LPS-induced inactivation of GSK-3β
p-GSK-3β and total GSK-3β expression levels were evaluated by western blot analysis (Fig. 5). No obvious differences in the total GSK-3β protein level were identified between the four groups. No significant differences in the p-GSK-3β protein level were identified between the control group and the fentanyl alone treatment group. Compared with the control group, LPS significantly increased the level of p-GSK-3β protein (P<0.01; Fig. 5B), which was further and significantly promoted by fentanyl pretreatment compared with the LPS stimulated group (P<0.05; Fig. 5B).
Figure 5.
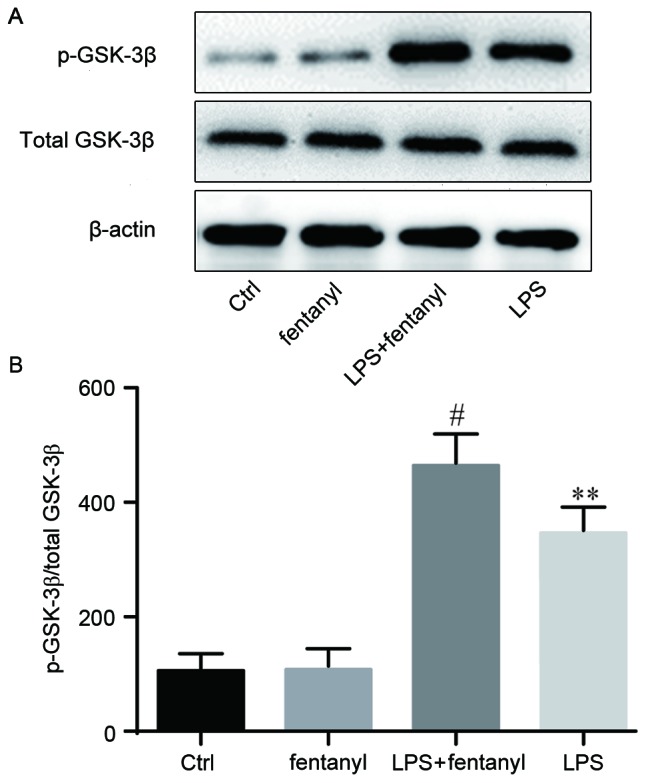
Fentanyl pretreatment further promoted LPS-induced inactivation of GSK-3β. BV-2 cells were either untreated, or treated with 5 µmol/l fentanyl, 1 µg/ml LPS or fentanyl followed by LPS. GSK-3β and p-GSK-3β protein levels were (A) measured by western blotting, then (B) quantified and statistically analyzed. **P<0.01 vs. control group; #P<0.05 vs. LPS group. Ctrl, control; GSK, glycogen synthase kinase; p-, phosphorylated; LPS, lipopolysaccharide.
Discussion
The current study revealed that pretreatment of 5 µmol/l fentanyl inhibited the LPS-induced release of IL-1β, TNF-α and IL-10, as well as the mRNA and protein levels of TLR4 in BV-2 cells. This process was associated with an increased p-GSK-3β protein level.
The upregulation of IL-1β and TNF-α is reported to be stimulated by LPS, thus they are involved in the progression of systemic inflammation and organ failure (24,25). Conversely, IL-10 may have the ability to repress the synthesis of multiple pro-inflammatory cytokines (26). The released levels of IL-1β, TNF-α and IL-10 were detected in different treatment groups. The results demonstrated that, compared with the control group, LPS elevated the release of IL-1β, TNF-α and IL-10, which was inhibited by fentanyl pretreatment. Emerging evidence demonstrated that fentanyl serves an important role in regulating the levels of IL-1β, TNF-α and IL-10 in LPS-induced neuroinflammation. However, the corresponding molecular mechanisms that were responsible for the aforementioned effects remained unclear. Consequently, further experiments were performed to establish the molecular mechanisms.
TLRs are primarily expressed by immune and immune-like cells, including granulocytes, monocyte macrophages, NK, T and B cells, and evolved to detect danger signals. TLR1, TLR2 and TLR4 are reported to respond to nerve injury, they are upregulated in the CNS, and are positively associated with the production of pro-inflammatory cytokines TNF-α and IL-1β (27). TLR4 knockout mice and rats that were intrathecally administered with TLR4 antisense oligonucleotides exhibited downregulated spinal microglia activation and spinal pro-inflammatory cytokines, and reduced neuropathic pain (28). TLR4 is expressed specifically by microglia (29), while LPS is an agonist to TLR4 (30). In a previous study, internalization of TLR4 generated microglial cells that were less sensitive to LPS, resulting in decreased TNF-α production in BV-2 cells (31). Elevated TLR4 expression was deemed to take part in the progression of inflammation-associated organ injury (32). Thus, the mRNA and protein levels of TLR4 in BV-2 cells were assessed in the current study. The results demonstrated that, in comparison with the control group, TLR4 levels were higher in the LPS stimulated group. Fentanyl pre-administration reduced the LPS-induced elevation of TLR4 levels compared with the LPS stimulated group. Nonetheless, the downstream molecule of TLR4 remained unknown.
Inactivation of GSK-3β was demonstrated to negatively affect the release of pro-inflammatory cytokines in response to TLR4 stimulation (33,34). The protein levels of p-GSK-3β and total GSK-3β were examined in the current study. The results revealed that the protein level of total GSK-3β was not obviously different between the four groups. However, the protein level of p-GSK-3β was higher in the LPS stimulated group compared with the control group, and pre-administration of fentanyl led to further elevation of p-GSK-3β compared with the LPS stimulated group.
In summary, a possible novel treatment for neuroinflammation was proposed. The results of the current study suggested that fentanyl pre-treatment exhibits a protective role during LPS-induced inflammation, via the targeting of the TLR4/p-GSK-3β signaling pathway, ultimately inhibiting pro-inflammatory cytokines. Neuroinflammation induced by surgery serves an important role in the development of postoperative cognitive dysfunction (POCD) and targeting the TLR4/p-GSK-3β signaling pathway, which may provide a novel therapeutic approach for the treatment of POCD.
Acknowledgements
The present study was supported by the commercial sponsorship of SINCH Pharmaceuticals Tech. Co., Ltd..
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
JW, YJ and JL made substantial contributions to the conception and design of the study, performed the experiments and analyzed the data. JW, YJ and JL managed the literature searches and figure preparations. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Allan SM, Rothwell NJ. Inflammation in central nervous system injury. Phil Trans R Soc Lond B Biol Sci. 2003;358:1669–1677. doi: 10.1098/rstb.2003.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yan A, Zhang Y, Lin J, Song L, Wang X, Liu Z. Partial depletion of peripheral M1 macrophages reverses motor deficits in MPTP-treated mouse by suppressing neuroinflammation and dopaminergic neurodegeneration. Front Aging Neurosci. 2018;10:160. doi: 10.3389/fnagi.2018.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baltuch GS. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–240. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- 4.Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. 2006;147(Suppl 1):S232–S240. doi: 10.1038/sj.bjp.0706400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Theoharides TC, Asadi S, Patel AB. Focal brain inflammation and autism. Neuroinflamm. 2013;10:46. doi: 10.1186/1742-2094-10-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Auriel E, Regev K, Korczyn AD. Nonsteroidal anti-inflammatory drugs exposure and the central nervous system. Handb Clin Neurol. 2014;119:577–584. doi: 10.1016/B978-0-7020-4086-3.00038-2. [DOI] [PubMed] [Google Scholar]
- 7.Bacchi S, Palumbo P, Sponta A, Coppolino MF. Clinical pharmacology of non-steroidal anti-inflammatory drugs: A review. Antiinflamm Antiallergy Agents Med Chem. 2012;11:52–64. doi: 10.2174/187152312803476255. [DOI] [PubMed] [Google Scholar]
- 8.Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilms H, Sievers J, Rickert U, Rostami-Yazdi M, Mrowietz U, Lucius R. Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL-1beta, TNF-alpha and IL-6 in an in-vitro model of brain inflammation. J Neuroinflamm. 2010;7:30. doi: 10.1186/1742-2094-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi Y, Lee MK, Lim SY, Sung SH, Kim YC. Inhibition of inducible NO synthase, cyclooxygenase-2 and interleukin-1beta by torilin is mediated by mitogen-activated protein kinases in microglial BV2 cells. British J Phamrmacol. 2009;156:933–940. doi: 10.1111/j.1476-5381.2009.00022.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol. 1990;27:229–237. doi: 10.1016/0165-5728(90)90073-V. [DOI] [PubMed] [Google Scholar]
- 12.Ooi YY, Ramasamy R, Rahmat Z, Subramaiam H, Tan SW, Abdullah M, Israf DA, Vidyadaran S. Bone marrow derived mesenchymal stem cells modulate BV2 microglia responses to lipopolysaccharide. Int Immunopharmacol. 2010;10:1532–1540. doi: 10.1016/j.intimp.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 13.Nelson L, Schwaner R. Transdermal fentanyl: Pharmacology and toxicology. J Med Toxicol. 2009;5:230–241. doi: 10.1007/BF03178274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnston KD. The potential for mu-opioid receptor agonists to be anti-emetic in humans: A review of clinical data. Acta Anaesthesiol Scand. 2010;54:132–140. doi: 10.1111/j.1399-6576.2009.02115.x. [DOI] [PubMed] [Google Scholar]
- 15.Dahan A, Sarton E, Teppema L, Olievier C. Sex-related differences in the influence of morphine on ventilatory control in humans. Anesthesiology. 1998;88:903–913. doi: 10.1097/00000542-199804000-00009. [DOI] [PubMed] [Google Scholar]
- 16.Sarton E, Teppema L, Dahan A. Sex differences in morphine-induced ventilatory depression reside within the peripheral chemoreflex loop. Anesthesiology. 1999;90:1329–1338. doi: 10.1097/00000542-199905000-00017. [DOI] [PubMed] [Google Scholar]
- 17.Stein C, Clark JD, Oh U, Vasko MR, Wilcox GL, Overland AC, Vanderah TW, Spencer RH. Peripheral mechanisms of pain and analgesia. Brain Res Rev. 2009;60:90–113. doi: 10.1016/j.brainresrev.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watkins LR, Maier SF, Goehler LE. Immune activation: The role of pro-inflammatory cytokines in inflammation, illness responses and pathological pain states. Pain. 1995;63:289–302. doi: 10.1016/0304-3959(95)00186-7. [DOI] [PubMed] [Google Scholar]
- 19.McNally L, Bhagwagar Z, Hannestad J. Inflammation, glutamate, and glia in depression: A literature review. CNS Spectr. 2008;13:501–510. doi: 10.1017/S1092852900016734. [DOI] [PubMed] [Google Scholar]
- 20.Tan SW, Ramasamy R, Abdullah M, Vidyadaran S. Inhibitory effects of palm α-, γ- and δ-tocotrienol on lipopolysaccharide-induced nitric oxide production in BV2 microglia. Cell Immunol. 2011;271:205–209. doi: 10.1016/j.cellimm.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 21.Łabuzek K, Liber S, Gabryel B, Okopień B. AICAR (5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside) increases the production of toxic molecules and affects the profile of cytokines release in LPS-stimulated rat primary microglial cultures. Neurotoxicology. 2010;31:134–146. doi: 10.1016/j.neuro.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 22.Gui B, Su M, Chen J, Jin L, Wan R, Qian Y. Neuroprotective effects of pretreatment with propofol in LPS induced BV-2 microglia cells: Role of TLR4 and GSK-3β. Inflammation. 2012;35:1632–1640. doi: 10.1007/s10753-012-9478-x. [DOI] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Crozier TA, Müller JE, Quittkatt D, Weyland W, Sydow M, Wuttke W, Kettler D. Interleukin-1 beta and interleukin-6-plasma concentrations in laparotomies. Interaction with neuroendocrine secretion and postoperative temperature regulation? Anaesthetist. 1993;42:343–349. (In German) [PubMed] [Google Scholar]
- 25.Grivennikov SI, Tumanov AV, Liepinsh DJ, Kruglov AA, Marakusha BI, Shakhov AN, Murakami T, Drutskaya LN, Förster I, Clausen BE, et al. Distinct and nonredundant in vivo functions of TNF produced by t cells and macrophages/neutrophils: Protective and deleterious effects. Immunity. 2005;22:93–104. doi: 10.1016/j.immuni.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 26.Marie C, Pitton C, Fitting C, Cavaillon JM. IL-10 and IL-4 synergize with TNF-alpha to induce IL-1ra production by human neutrophils. Cytokine. 1996;8:147–151. doi: 10.1006/cyto.1996.0021. [DOI] [PubMed] [Google Scholar]
- 27.Owens T, Babcock AA, Millward JM, Toft-Hansen H. Cytokine and chemokine inter-regulation in the inflamed or injured CNS. Brain Res Brain Res Rev. 2005;48:178–184. doi: 10.1016/j.brainresrev.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 28.Morzaev D, Nicholson JD, Caspi T, Weiss S, Hochhauser E, Goldenberg-Cohen N. Toll-like receptor-4 knockout mice are more resistant to optic nerve crush damage than wild-type mice. Clin Exp Ophthalmol. 2015;43:655–665. doi: 10.1111/ceo.12521. [DOI] [PubMed] [Google Scholar]
- 29.Lehnardt S, Lachance C, Patrizi S, Lefebvre S, Follett PL, Jensen FE, Rosenberg PA, Volpe JJ, Vartanian T. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J Neurosci. 2002;22:2478–2486. doi: 10.1523/JNEUROSCI.22-07-02478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hwang D. Modulation of the expression of cyclooxygenase-2 by fatty acids mediated through toll-like receptor 4-derived signaling pathways. FASEB J. 2001;15:2556–2564. doi: 10.1096/fj.01-0432com. [DOI] [PubMed] [Google Scholar]
- 31.Willis LM, Bielinski DF, Fisher DR, Matthan NR, Joseph JA. Walnut extract inhibits LPS-induced activation of BV-2 microglia via internalization of TLR4: Possible involvement of phospholipase D2. Inflamm. 2010;33:325–333. doi: 10.1007/s10753-010-9189-0. [DOI] [PubMed] [Google Scholar]
- 32.Barsness KA, Arcaroli J, Harken AH, Abraham E, Banerjee A, Reznikov L, McIntyre RC. Hemorrhage-induced acute lung injury is TLR-4 dependent. Am J Physiol Regul Integr Comp Physiol. 2004;287:R592–R599. doi: 10.1152/ajpregu.00412.2003. [DOI] [PubMed] [Google Scholar]
- 33.Hu X, Paik PK, Chen J, Yarilina A, Kockeritz L, Lu TT, Woodgett JR, Ivashkiv LB. IFN-gamma suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity. 2006;24:563–574. doi: 10.1016/j.immuni.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 34.Rodionova E, Conzelmann M, Maraskovsky E, Hess M, Kirsch M, Giese T, Ho AD, Zöller M, Dreger P, Luft T. GSK-3 mediates differentiation and activation of proinflammatory dendritic cells. Blood. 2007;109:1584–1592. doi: 10.1182/blood-2006-06-028951. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.