

Abstract
The present study aimed to determine whether an miRNA (miR)-29b inhibitor protected against cerebral ischemia/reperfusion (I/R) injury in vitro and to investigate the underlying mechanisms. As a model for induced cerebral IR injury, N2a cells were exposed to an oxygen-glucose deprivation/reoxygenation (OGD/R) environment. Using this model, it was demonstrated that miR-29b was significantly upregulated compared with cells in a normal environment. The interactions between miR-29b and myeloid cell leukemia sequence (MCL)-1 were then investigated using dual-luciferase assays, revealing a strong regulation of MCL-1 through the 3′untranslated region. Using the OGD/R model, the present study additionally examined the effects of miR-29b and miR-29b inhibitor on cell viability and apoptosis using Cell Counting kit 8 and flow cytometry assays, respectively. miR-29b transfection led to increased N2a cell apoptosis and reduced cell viability under an OGD/R environment. However, this effect was reversed by the miR-29b inhibitor. Finally, the effects of miR-29b on the expression of several Wnt-associating proteins were examined. It was observed that B cell lymphoma-2 was inhibited by miR-29b, as was MCL-1, whereas caspase-3 expression was promoted. The miR-29b inhibitor demonstrated the opposite effect. Overall, miR-29b promoted neurocyte apoptosis by targeting MCL-1 during cerebral I/R injury. The results of the present study suggest a potential novel therapeutic target for the treatment of ischemic stroke.
Keywords: miR-29b, MCL-1, cerebral ischemia-reperfusion injury, neural apoptosis
Introduction
Reperfusion is believed to be important for the recovery of ischemic brain injuries and also limits subsequent infarction development. Cerebral ischemia/reperfusion (I/R) is characterized by an initial restriction of blood supply to the brain, followed by restoration of blood flow and re-oxygenation (1,2). However, IR induced cerebrovascular dysfunction is also thought to be a significant clinical issue due to the neurological damage that can occur, such as in patients that have suffered ischemic strokes (3). Ischemic stroke are one of the most devastating neurological diseases world-wide and the third most common cause of death globally. They can also lead to permanent disability in adults. In China, approximately 2.5 million people are at risk of stroke and 1 million may die from stroke-related consequences annually (4–6). The exact pathogenic mechanisms that lead to cerebral IR injury are not yet completely understood and it is therefore important to find new effective measures to prevent cerebral I/R injuries and avert ischemic strokes.
MicroRNAs (miRNAs) are small (18–25 nt) noncoding single-stranded RNA molecules that act to negatively regulate transcription and post-transcription by modulating the stability and/or translational efficiency of target messenger RNAs (mRNAs) (7). It has previously been established that most mature miRNAs alter mRNA degradation or translation through binding the 3′-untranslated region (3′UTR) of target mRNAs (8,9). Prior research has also revealed that some miRNAs, including miR-29b, miR-21, miR-200, and miR-497, can act as potential therapeutic targets. These miRNAs appear to contribute to cerebral I/R injury by altering key signaling elements that typically have high expression in the cerebral cortex after ischemic injury (10). More recent studies have indicated that miR-29 may also negatively regulate Bcl-2 family members, including pro-survival proteins such as BCL-w (BCL2L2) and MCL-1. Despite these proteins being important regulators of cerebral I/R injury, the mechanisms behind their effects are unknown (11–13).
Based on prior evidence, we investigated the effects of miR-29b on cerebral IR injury and aimed to identify any underlying mechanisms. Our study utilized an OGD/R (Oxygen-glucose deprivation/reoxygenation) environment as an in vitro model of induced cerebral IR injury. After administration of miR-29b or miR-29b inhibitor, we evaluated any differences in neural apoptosis mediated by MCL-1 and other relevant proteins. Our data contributes to a better understanding of cerebral IR injury and will aid efforts to develop new strategies to improve its clinical treatment.
Materials and methods
Cell culture and treatment
Mouse neuroblastoma (N2a) cells were purchased from the Chinese Academy of Sciences (Shanghai, China). N2a cells were cultured in growth media containing high glucose-Dulbecco's modified Eagle's medium (Hyclone; GE Healthcare Life Sciences, Logan, UT, USA), supplemented with 10% fetal bovine serum (Hyclone, GE Healthcare Life Sciences), and 1% penicillin/streptomycin (Mediatech, Inc., Manassas, VA, USA) in a humidified atmosphere of 5% CO2 at 37°C.
Following dilution into single cell suspensions, N2a cells were seeded onto 96-well plates (1×104 cells/well). Cells were exposed to an oxygen-glucose deprivation (OGD) environment for 24 h and the culture medium was then replaced with deoxygenated glucose-free Hanks' balanced salt solution (HBSS) for a further 24 h. Cells were then harvested for total RNA isolation.
MiRNA mimics transfection
miR-29b mimics, miR-29b inhibitor, mimics negative control (NC), and inhibitor NC were purchased from Guangzhou RiboBio, Co., Ltd., (Guangzhou, China). The miR-29b mimics sequence (5′-UAGCACCAUUUGAAAUCAGUGUU-3′), miR-29b inhibitor sequence (5′-AACACUGAUUUCAAAUGGUGCUA-3′), miR-29b mimics NC sequence (5′-UUCUCCGAACGUGUCACGUTT-3′); miR-29b inhibitor NC sequence (5′-CAGUACUUUUGUGUAGUACAA-3′). 2×105 N2a cells were cultured in 6-well plates and then transfected with 200 µl mature miR-29b mimics, miR-29b inhibitor, mimics NC, and inhibitor NC (GenePharma Co., Ltd., Shanghai, China) for 72 hrs. All transfections were completed using Lipofectamine™ 3000 (Invitrogen Life Technologies, Carlsbad, CA, USA) according to the manufacturer's protocols.
Cell viability detection
N2a R cells were transfected with 1 µg of miR-29b mimic or an inhibitor using Lipofectamine® 2000 (Invitrogen). Next, 100 µl Cell Counting kit-8 (CCK8) solution (Dojindo Molecular Technologies, Inc., Kumamoto, Japan) was added to each well and incubated for 1 h in an incubator. Absorbance at 450 nm was measured using a microplate reader.
Apoptosis assay
Quantification of apoptotic cells was performed using an Annexin V-propidium iodide (PI) apoptosis kit (Multiscience Biotech, Ltd., Hangzhou, China). N2a cells were collected, washed with phosphate buffered saline (PBS) and re-suspended in 200 µl binding buffer containing 5 µl Annexin V (10 µg/ml). They were then left for 10 min in the dark. Cells were next incubated with 10 µl PI (20 µg/ml) and analyzed by flow cytometry (EPICS® XL™; Beckman Coulter, Inc., Brea, CA, USA). Data acquisition and analysis were performed using CellQuest™ software (BD Biosciences, Franklin Lakes, NJ, USA).
Protein isolation and western blotting analysis
In total, 2×106 N2a cells per sample were lysed for western blotting analysis using conventional procedures. The primary antibodies used were anti-mouse polyclonal MCL-1 antibody (1:20,000; Sigma-Aldrich, St. Louis, MO, USA), anti-mouse polyclonal BCL2 antibody (1:1,000, Abcam, Cambridge, UK), anti-mouse polyclonal caspase-3 (CASP3) antibody (1:10,000, Abcam), and anti-mouse polyclonal GAPDH antibody (1:1,000, Santa Cruz Biotechnology, CA, USA). Blots were visualized using chemiluminescence reagent (Meck Millipore, Billerica, MA, USA) in a LAS4000 Luminescent Image Analyzer (GE Healthcare, Tokyo, Japan). TRIzol® reagent (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) was used to extract total RNA from cells following the manufacturer's protocol, although ethanol was used instead of isopropanol for RNA precipitation. RNA quality was determined using a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Inc., Wilmington, DE, USA). A total of 1 µg RNA per sample was reverse-transcribed into cDNA using a DBI Bestar® qPCR RT kit (DBI Bioscience, Ludwigshafen, Germany), according to the manufacturer's protocol.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
RT-qPCR was performed using 96-well optical plates and a 7500 Fast Real-Time PCR System LightCycler (Applied Biosystems; Thermo Fisher Scientific, Inc.). Each 20 µl PCR reaction included 1 µl reverse transcription product (1:5), 0.5 µl sense primer, 0.5 µl Universal reverse primer, and 10 µl mix buffer (DBI Bestar® Sybr-Green qPCR master mix, DBI Bioscience). Reaction conditions were 94°C for 2 min, followed by 40 cycles of 94 and 58°C for 20 sec, and 72°C for 20 sec. All reactions were completed in triplicate. Primer sequences were miR-29b forward: 5′UAGCACCAUUUGAAAUCAGUGUU3′ and reverse: 5′CTCAACTGGTGTCGTGGA3′; and U6 forward: 5′CTCGCTTCGGCAGCACA3′; and reverse: 5′AACGCTTCACGAATTTGCGT3′.
Recombinant plasmid construction
The 3′UTR of mouse MCL-1 mRNA (NM_000286) was retrieved from the GenBank Database and amplified using synthetic primers: MCL-1 forward (5′GCTAGCCGCTACTAGGCTCCCC3′) and MCL-1 reverse (5′CGGGTAGTATATACGCGTCGTTAC3′). The product was cloned into a psiCHECK2 vector (Invitrogen, Carlsbad, CA, USA). Recombinant vector was then amplified in DH5α Escherichia coli and purified using an endotoxin-free plasmid purification kit (QIAGEN, Valencia, CA), according to the manufacturer's instructions. Each segment was amplified by PCR with Takara LA Taq or Primestar (Takara Bio, Inc., Otsu, Japan) and cloned into the vector. Mutation of the miR-29b binding sites in the MCL-1 3′UTR sequence was performed using a KOD-Plus-Mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA), according to the manufacturer's protocol. All constructs were confirmed by restriction enzyme digest and sequencing (Sangon Biotech, Shanghai, China).
Dual-luciferase assay
N2a cells were seeded at a density of 1.0×105 cells/ml in 6-well plates to achieve ~50% confluence the next day. They were first transfected with miR-29b for 24 h and then psiCHECK2- MCL-1-3′UTR-WT/psiCHECK2-MCL-1-3′UTR-MUT plasmids using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) and incubated for an additional 48 h. Cells were collected and assayed for firefly luciferase activity, normalized to the activity of Renilla luciferase, by using a Dual-Luciferase Reporter Assay System (Promega Corp., Madison, WI, USA) and a Biotek Synergy 4 Microplate reader (Biotek, Winooski, USA). These results are presented as the ratio of the luminescence of treated cell samples vs. control samples and are given as the mean ± SD from three individual transfections.
Data analysis
For RT-qPCR data analysis, a relative quantification method was used to determine changes in expression of the target miRNAs. U6 RNA was used to normalize expression and determine any changes in amplification. Fold changes in expression were calculated for each sample using a 2-ΔΔCq method, where ΔΔCq=(Cq target gene-CqU6) PIH-(Cq target gene-CqU6) control. Values for 2-ΔΔCq >1.5 or <0.67 were considered differentially expressed miRNAs. Welch t-tests were used to assess the differential expression of miRNA measured by RT-qPCR.
Other statistical analyses were performed using SPSS, version 17.0 (SPSS, Inc., Chicago, IL, USA). A one-way analysis of variance (ANOVA) was used to compare the log10-transformed relative quantities of target miRNAs between all groups. A Bartlett's post hoc test was used to assess the differences in variance between genes. P<0.05 was considered a statistically significant difference in all experiments.
Results
Interaction between miR-29b and MCL-1
Based on prior research, overexpression of miR-29b and its effect on Mcl-1 transcript were already described (14). In our study, we chose to use miR-29b and its target mRNA as candidates for our experiments. To investigate the effects of miR-29b, two psiCHECK2 luciferase plasmids containing MCL-1-3′UTR-WT and MCL-1-3′UTR-MUT segments were co-transfected with miR-29b into N2a cells. Treatment with both miR-29b and psiCHECK2-MCL-1-3′UTR-WT decreased luciferase activity in N2a cells compared to cells transfected with psiCHECK2-MCL-1-3′UTR-MUT (Fig. 1). As previous studies have shown miR-29b has a complementary site with the 3′ UTR of Mcl-1, and miR-29b can downregulate the expression level of Mcl-1 (14). Therefore, we suggested that miR-29b negatively regulated Mcl-1.
Figure 1.
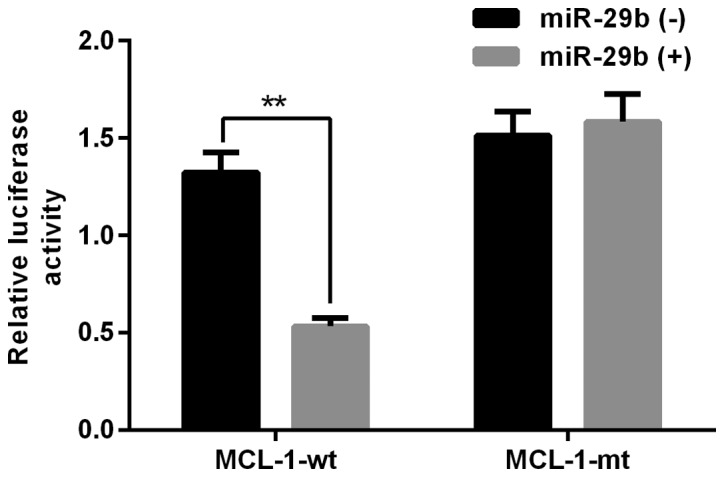
Validation of the interaction between miR-29b and the potential miR-29b-binding fragment located in the 3′UTR of MCL-1, detected by dual-luciferase assay. All experiments were repeated independently three times. **Indicates P<0.01 vs. data from a negative control group at same time point.
Effect of exogenous miR-29b mimics and miR-29b inhibitor in N2a cells
We next explored the effect of miR-29b in vitro. We found that miR-29b levels were significantly higher under an OGD/R environment (Fig. 2). Increased miRNA detection after 48 h confirmed successful transfection with miR-29b mimics.
Figure 2.
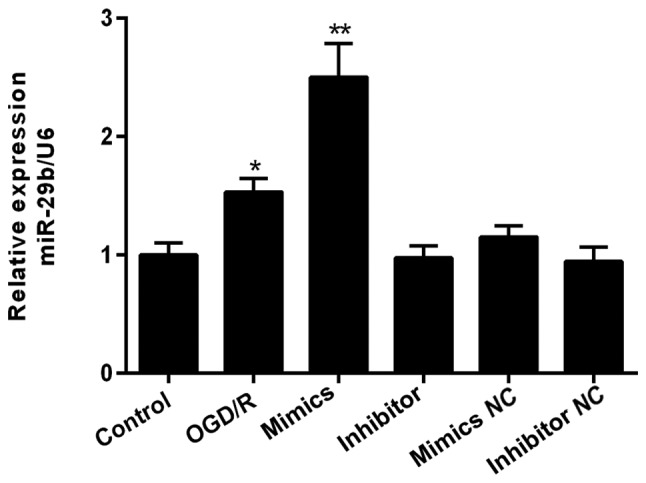
Effect of exogenous miR-29b mimics and inhibitor in N2a cells under an OGD/R environment. miR-29b levels were assessed after transfection for 48 h. *Indicates P<0.05, **indicates P<0.01 vs. data from the OGD/R only group at same time point. All experiments were independently repeated three times. OGD/R, oxygen-glucose deprivation/reoxygenation
Effects of miR-29b mimics and inhibitor on cell proliferation
The effects of miR-29b mimics and inhibitor on N2a cell viability were subsequently assessed. In OGD/R pretreatment N2a cells, cellular viability was significantly reduced by OGD/R, and then further decreased by miR-29b mimic compared to negative control (NC) groups. An opposite effect was found for cells treated with miR-29b inhibitor (Fig. 3).
Figure 3.
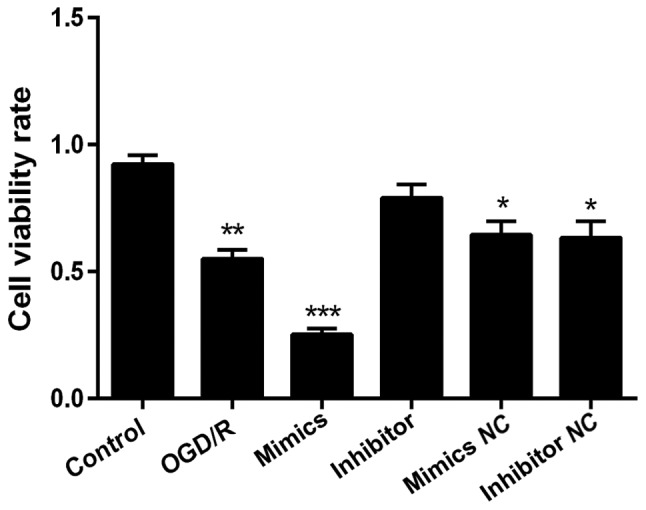
Effect of exogenous miR-29b mimics and inhibitor on the viability of N2a cells. Cell viability rates were measured 24 h after transfection using a CCK8 assay. Data are shown as the means ± standard deviation. All tests were repeated independently three times. *Indicates P<0.05; **indicates P<0.01; ***indicates P<0.001 vs. data from the OGD/R only group at the same time point.
Effects of miR-29b mimics and inhibitor on cell apoptosis
We also assessed rates of apoptosis following transfection with miR-29b mimics or inhibitor following OGD/R pre-treatment for 48 h using flow cytometry. Compared to N2a cells transfected with miR-29b mimics, cells exposed to miR-29b inhibitor had less cells exhibiting apoptotic morphology, such as nuclear fragmentation, cell shrinkage, and cellular rupture debris. Conversely, apoptosis occurred at a significantly higher rate in cells treated with the miR-29b mimics, compared to the group treated with OGD/R only (P<0.01; Fig. 4A). The percentage differences in N2a cell apoptosis rates are shown in Fig. 4B.
Figure 4.

Effect of exogenous miR-29b mimics and inhibitor on apoptosis of N2a cells in a cerebral I/R injury model. (A) Dot plots and (B) histogram of apoptosis assays. All experiments were independently repeated three times. *Indicates P<0.05; **indicates P<0.01; ***indicates P<0.001 vs. the data from the OGD/R only group at the same time point.
Effect of miR-29b mimics and inhibitor on Wnt-associated proteins in N2a cells
In addition to MCL-1, we also investigated BCL2 and caspase-3 (CASP3) expression across the same groups. We found that BCL2 levels (Fig. 5A-B) followed a similar trend to that of MCL-1 (Fig. 5A and C) and was downregulated in an OGD/R environment. Expression was inhibited further in cells transfected with miR-29b mimics and but higher in cells exposed to miR-29b inhibitor. Caspase-3 (CASP3) levels showed an opposite effect (Fig. 5A and D).
Figure 5.

Effect of exogenous miR-29b mimics and inhibitor on Wnt-associated proteins in N2a cells, semi-quantified by western blots. (A) Representative images of western blots of BCL2, MCL-1, and caspase-3 (CASP3). Histogram of band intensities for (B) BCL2, (C) MCL-1, and (D) CASP3. All experiments were independently repeated three times. *Indicates P<0.05; **indicates P<0.01 vs. the OGD/R only group at the same time point.
Discussion
There is an increasing volume of interdisciplinary research linking cancer and neurodegenerative diseases. In the present study, we have demonstrated that apoptosis is clearly increased after exposure to an OGD/R environment. We have also shown in vitro the neuroprotective effects of miR-29b inhibitor on I/R injury through promoting cell viability and suppressing apoptosis. Our data suggests that these effects are mediated through the targeting of MCL-1. Transfection with miR-29b mimics led to up-regulated expression of MCL-1 and BCL2 proteins and a correlating down-regulation of expression for cleaved caspase-3 (CASP3) proteins in our cerebral I/R injury cell model. We therefore conclude that the miR-29b inhibitor can act as a protective regulator against cell apoptosis in cell models that mimic cerebral I/R injury.
A considerable amount of previous research has focused on the regulatory role of miR-29 in carcinoma. However, miR-29b is also significantly up-regulated in neural cells under an OGD/R environment and in patients with cerebral I/R injury (10,15). Activated miR-29b has also been reported to promote neural cell apoptosis targeting BH3 protein during neuronal maturation (16). Supporting previous studies, our results indicate that miR-29 binds the 3′UTR of MCL-1, an important Bcl-2 family protein. Proteins in the Bcl-2 family are defined by the presence of Bcl-2 homology (BH) domains but are divided into anti- or pro-apoptotic regulators. They act through modifying mitochondrial membrane integrity and function, and also affect apoptotic signaling. The Bcl-2 family consists of three subgroups, pro-survival proteins (BCL2, BCLxl [BCL2L1], BCLw [BCL2L2], MCL-1, and A1), multi-domain pro-apoptotic proteins (BAX and BAK), and BH3 domain-only pro-apoptotic proteins (BIM, PUMA [BBC3], BID, BAD, BIK, BMF, HRK, and NOXA [PMAIP1]) (17–19). As an important anti-apoptotic protein in the Bcl-2 family, inhibition of MCL-1 promotes cell death through the mitochondrial pathway and there is some data suggesting it is also involved in the apoptosis that occurs during cerebral ischemic reperfusion (20). Based on these data and our results, we hypothesize that the interaction between miR-29b and MCL-1 is a critical factor during neural cell apoptosis. Finally, recent research has also shown that several miRNAs directly target the 3′UTR of Bcl-2 family proteins. For example, miR-15b is highly expressed following permanent middle cerebral artery occlusion (MCAO) and may directly target BCL2 (21). Another miRNA, miR-491-5p, has also been suggested to bind Bcl-xL (BCL2L1) mRNA, an anti-apoptotic member of the Bcl-2 family (22). Additionally, up-regulation of miR-29b promotes neuronal cell death by inhibiting BCL2 after ischemic brain injury. As many chemotherapeutic drugs induce apoptosis through down-regulation of MCL-1 expression in tumor cells, our results suggest that targeting miR-29b may also be a therapeutic target in cerebral I/R injury by inhibiting MCL-1 expression.
Our experiments have confirmed our initial hypothesis that miR-29b affects neurocyte apoptosis during cerebral I/R injury through targeting MCL-1. We have shown this at both the protein and histological level. However, there were certain limitations in our experiments. For example, our data was obtained using miR-29b mimics in an in vitro environment, and therefore, may not accurately reflect in vivo effects. We therefore plan to conduct further experiments in MCAO animal models to explore the relationships between reperfusion and treatment with miR-29b inhibitor or overexpression of MCL-1. In addition, further studies are needed to validate the neurodamaging and neuroprotective effects of miR-29b by targeting MCL-1 during cerebral ischemia/reperfusion injury. Moreover, it will be necessary to further explore the functions and mechanisms of miR-29b in neuronal cells during cerebral ischemia/reperfusion injury. This will lead to better clinical outcomes for patients that suffer an ischemic stroke.
Acknowledgements
The present study was supported by funds from the Natural Science Foundation of China (no. 81460276), the Science and Technology Development Funds of Guizhou (no. J. [2015] 2090).
Funding
This study was supported by funds from the Natural Science Foundation of China (no. 81460276), the Science and Technology Development Funds of Guizhou (no. J. [2015] 2090).
Availability of data and materials
All data generated or analysed during this study are included in this article.
Authors' contributions
ZH, LL, SAZ and JL designed the study. TPJ, SAZ, YPS and ZZ performed the experiments. ZH wrote the paper. ASZ, RG and RL helped perform the analysis with constructive discussions. SZ revised the manuscript. All authors read and approved the manuscript.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
Not applicable.
References
- 1.Zhai WW, Sun L, Yu ZQ, Chen G. Hyperbaric oxygen therapy in experimental and clinical stroke. Med Gas Res. 2016;6:111–118. doi: 10.4103/2045-9912.184721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang W, Zhao L, Bai F, Zhang T, Dong H, Liu L. The protective effect of dopamine against OGD/R injury-induced cell death in HT22 mouse hippocampal cells. Environ Toxicol Pharmacol. 2016;42:176–182. doi: 10.1016/j.etap.2016.01.020. [DOI] [PubMed] [Google Scholar]
- 3.Fluri F, Schuhmann MK, Kleinschnitz C. Animal models of ischemic stroke and their application in clinical research. Drug Des Devel Ther. 2015;9:3445–3454. doi: 10.2147/DDDT.S56071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ni J, Wang X, Chen S, Liu H, Wang Y, Xu X, Cheng J, Jia J, Zhen X. MicroRNA let-7c-5p protects against cerebral ischemia injury via mechanisms involving the inhibition of microglia activation. Brain Behav Immun. 2015;49:75–85. doi: 10.1016/j.bbi.2015.04.014. [DOI] [PubMed] [Google Scholar]
- 5.Zuo XL, Deng HL, Wu P, Xu E. Do different reperfusion methods affect the outcomes of stroke induced by MCAO in adult rats? Int J Neurosci. 2016;126:850–855. doi: 10.3109/00207454.2015.1074903. [DOI] [PubMed] [Google Scholar]
- 6.Li F, Shi W, Zhao EY, Geng X, Li X, Peng C, Shen J, Wang S, Ding Y. Enhanced apoptosis from early physical exercise rehabilitation following ischemic stroke. J Neurosci Res. 2017;95:1017–1024. doi: 10.1002/jnr.23890. [DOI] [PubMed] [Google Scholar]
- 7.De Gasperi R, Graham ZA, Harlow LM, Bauman WA, Qin W, Cardozo CP. The signature of microRNA dysregulation in muscle paralyzed by spinal cord injury includes downregulation of microRNAs that target myostatin signaling. PLoS One. 2016;11:e0166189. doi: 10.1371/journal.pone.0166189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xia HF, Jin XH, Cao ZF, Hu Y, Ma X. MicroRNA expression and regulation in the uterus during embryo implantation in rat. FEBS J. 2014;281:1872–1891. doi: 10.1111/febs.12751. [DOI] [PubMed] [Google Scholar]
- 9.Floris I, Kraft JD, Altosaar I. Roles of MicroRNA across prenatal and postnatal periods. Int J Mol Sci. 2016;17:E1994. doi: 10.3390/ijms17121994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Di Y, Lei Y, Yu F, Changfeng F, Song W, Xuming M. MicroRNAs expression and function in cerebral ischemia reperfusion injury. J Mol Neurosci. 2014;53:242–250. doi: 10.1007/s12031-014-0293-8. [DOI] [PubMed] [Google Scholar]
- 11.Jafarinejad-Farsangi S, Farazmand A, Mahmoudi M, Gharibdoost F, Karimizadeh E, Noorbakhsh F, Faridani H, Jamshidi AR. MicroRNA-29a induces apoptosis via increasing the Bax: Bcl-2 ratio in dermal fibroblasts of patients with systemic sclerosis. Autoimmunity. 2015;48:369–378. doi: 10.3109/08916934.2015.1030616. [DOI] [PubMed] [Google Scholar]
- 12.Xu L, Xu Y, Jing Z, Wang X, Zha X, Zeng C, Chen S, Yang L, Luo G, Li B, Li Y. Altered expression pattern of miR-29a, miR-29b and the target genes in myeloid leukemia. Exp Hematol Oncol. 2014;3:17. doi: 10.1186/2162-3619-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mott JL, Kobayashi S, Bronk SF, Gores GJ. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene. 2007;26:6133–6140. doi: 10.1038/sj.onc.1210436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang YK, Wang H, Leng Y, Li ZL, Yang YF, Xiao FJ, Li QF, Chen XQ, Wang LS. Overexpression of microRNA-29b induces apoptosis of multiple myeloma cells through down regulating Mcl-1. Biochem Biophys Res Commun. 2011;414:233–239. doi: 10.1016/j.bbrc.2011.09.063. [DOI] [PubMed] [Google Scholar]
- 15.Altintas O, Ozgen Altintas M, Kumas M, Asil T. Neuroprotective effect of ischemic preconditioning via modulating the expression of cerebral miRNAs against transient cerebral ischemia in diabetic rats. Neurol Res. 2016;38:1003–1011. doi: 10.1080/01616412.2016.1232013. [DOI] [PubMed] [Google Scholar]
- 16.Annis RP, Swahari V, Nakamura A, Xie AX, Hammond SM, Deshmukh M. Mature neurons dynamically restrict apoptosis via redundant premitochondrial brakes. Febs J. 2016;283:4569–4582. doi: 10.1111/febs.13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delgado-Soler L, Del Mar Orzaez M, Rubio-Martinez J. Structure-based approach to the design of BakBH3 mimetic peptides with increased helical propensity. J Mol Model. 2013;19:4305–4318. doi: 10.1007/s00894-013-1944-3. [DOI] [PubMed] [Google Scholar]
- 18.Santiveri CM, Sborgi L, de Alba E. Nuclear magnetic resonance study of protein-protein interactions involving apoptosis regulator Diva (Boo) and the BH3 domain of proapoptotic Bcl-2 members. J Mol Recognit. 2012;25:665–673. doi: 10.1002/jmr.2240. [DOI] [PubMed] [Google Scholar]
- 19.Smits C, Czabotar PE, Hinds MG, Day CL. Structural plasticity underpins promiscuous binding of the prosurvival protein A1. Structure. 2008;16:818–829. doi: 10.1016/j.str.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 20.Dewson G. Characterizing Bcl-2 family protein conformation and oligomerization using cross-linking and antibody gel-shift in conjunction with native PAGE. Methods Mol Biol. 2016;1419:185–196. doi: 10.1007/978-1-4939-3581-9_14. [DOI] [PubMed] [Google Scholar]
- 21.Shi H, Sun BL, Zhang J, Lu S, Zhang P, Wang H, Yu Q, Stetler RA, Vosler PS, Chen J, Gao Y. miR-15b suppression of Bcl-2 contributes to cerebral ischemic injury and is reversed by sevoflurane preconditioning. CNS Neurol Disord Drug Targets. 2013;12:381–391. doi: 10.2174/1871527311312030011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Denoyelle C, Lambert B, Meryet-Figuière M, Vigneron N, Brotin E, Lecerf C, Abeilard E, Giffard F, Louis MH, Gauduchon P, et al. miR-491-5p-induced apoptosis in ovarian carcinoma depends on the direct inhibition of both BCL-XL and EGFR leading to BIM activation. Cell Death Dis. 2014;5:e1445. doi: 10.1038/cddis.2014.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analysed during this study are included in this article.