

Abstract
Introduction
Both Δ9 Tetrahydrocannabidiol (THC) and cannabidiol (CBD) components of cannabis, have been shown to have anticonvulsant effects. Cannabis oils are used to treat seizures in drug‐resistant epilepsy (DRE). Recent trials provide data on dosing, side effects, and efficacy of CBD, yet there is a paucity of information on THC in epilepsy. Primary objective was to establish dosing and tolerability of TIL‐TC150 ‐ a cannabis plant extract produced by Tilray®, containing 100 mg/mL CBD and 2 mg/mL THC‐ in children with Dravet syndrome. Secondary objectives were to assess impact of therapy on seizures, electroencephalogram (EEG) and quality of life.
Methods
Twenty children received add‐on therapy with TIL‐TC150. The dose ranged from 2 to 16 mg/kg/day of CBD and 0.04 to 0.32 mg/kg/day of THC. Patients were monitored for tolerability and adverse events, and secondary objectives.
Results
Nineteen participants completed the 20‐week intervention. Mean dose achieved was 13.3 mg/kg/day of CBD (range 7–16 mg/kg/day) and 0.27 mg/kg/day of THC (range 0.14–0.32 mg/kg/day). Adverse events, common during titration included somnolence, anorexia, and diarrhea. Abnormalities of liver transaminases and platelets were observed with concomitant valproic acid therapy. There was a statistically significant improvement in quality of life, reduction in EEG spike activity, and median motor seizure reduction of 70.6%, with 50% responder rate of 63%.
Conclusions
TIL‐TC150 was safe and well tolerated in our subjects. TIL‐TC150 treatment resulted in a reduction in seizure counts, spike index on EEG, and improved quality of life measures. This study provides safety and dosing information for THC‐containing cannabinoid preparations.
Introduction
Despite the expansion of available antiepileptic drugs (AED), dietary, and surgical therapies in the last 20 years, up to 30% of children with epilepsy continue to have seizures, termed drug‐resistant epilepsy.1 Dravet syndrome is a catastrophic epilepsy syndrome impacting all developmental realms including cognition, behavior, and motor function.2, 3, 4, 5, 6 Typically, a mutation in the SCN1A sodium channel gene is causative.7 The devastation caused by DRE provides impetus to explore new therapies through research.
Recently, there has been growing interest in using cannabis plant extracts in the treatment of DRE.8, 9, 10, 11, 12, 13, 14 Δ9 tetrahydrocannabidiol (THC) and cannabidiol (CBD) have both shown anticonvulsant properties in animal studies.15, 16, 17, 18, 19, 20 THC has also been shown to be pro‐convulsant in some animal models.14 The clinical use of cannabis plant extracts in epilepsy has been limited by concern of the psychotropic effects of THC, but THC‐containing compounds have been studied in other indications.21 It is important to differentiate the actions of THC and CBD within the endocannabinoid system.22, 23 While THC directly binds to the CB1 receptor (widely distributed in the central nervous system) to elicit its effects, CBD does not.24, 25 The mechanism of action of CBD involves intermediary pathways without direct action on CB1. Therefore, it is plausible that THC‐containing cannabinoid preparations may be superior to CBD‐only preparations in their anticonvulsant effect, and that combination CBD‐THC products may increase tolerance of THC by reducing its psychoactive properties26, 27
Primetime attention by media outlets, patient advocacy groups and families concerning a mixed CBD/THC containing cannabis extract [so‐called ‘Charlotte's Web’] to treat seizures in patients with Dravet syndrome, resulted in increasing demands from advocacy groups for cannabinoids to be considered in the treatment of epilepsy.28, 29 This resulted in a massive expansion of the academic literature exploring the potential of CBD (excluding THC due to the aforementioned concern, and due to THC being considered illegal in many jurisdictions) as a therapy in DRE.23, 30, 31 In turn, prospective studies of CBD in DRE were conducted. Specifically, a pharmaceutical‐grade formulation of purified CBD in oil has been studied in epilepsy patients in several clinical trials and has been shown to be more efficacious but less tolerated than placebo in treating seizures.32, 33, 34, 35
The cannabis oils available in Canada contain mixed extract of predominantly CBD and THC.36 While the clinical trials of pure CBD alluded to above have given us an understanding of CBD dosing, side effects and efficacy, there is a paucity of information regarding dosing and safety of cannabinoid products containing THC. However, products vary significantly in their pharmaceutical formulations (purity and constituent component stability) and have not undergone formal clinical study. Clearly, there is a gap in our understanding of these preparations.
The primary objective of this study was to establish safe dosing and tolerability of TIL‐TC150, a cannabis extract containing only CBD and THC, at a ratio of 50:1 CBD: THC in children with Dravet syndrome. Secondary objectives were to assess the impact of CBD/THC therapy on seizure frequency, spike index, cognition, and quality of life.
Material and Methods
This prospective, open‐label, interventional study was conducted at the Hospital for Sick Children (SickKids) in Toronto, Canada. Institutional Research Ethics Board approval was obtained as was Health Canada approval for the study drug. The study was registered on clinicaltrials.gov (Identifier: NCT02983695). Twenty participants with Dravet syndrome with DRE were recruited at a single academic pediatric neurology center in Ontario. Inclusion criteria were: age between 1 and 18 years, clinical and electrographic characteristics of Dravet syndrome, confirmation of the SCN1A gene mutation, a diagnosis of DRE, frequent motor seizures, stable AED therapy for 4 weeks pre‐intervention, and ketogenic diet/vagal nerve stimulation device settings for at least 3 months pre‐intervention, and willingness of caregivers to comply with seizure diary and study drug administration.
Exclusion criteria were comorbid renal, liver or cardiac disease, evidence of an underlying inborn error of metabolism, previous significant drug reaction, any known or suspected family history of schizophrenia, known sensitivity or allergic reaction to cannabinoids, and current therapy with a cannabinoid product. Complete inclusion and exclusion criteria available as supplemental material (Data S1).
The active ingredients in TIL‐TC150 are CBD and THC, present in a 50:1 ratio. TIL‐150 was produced by Tilray, a licensed Canadian producer of medical cannabis, using extracted, purified CBD and THC from their Cannabis sativa L. strains, and suspended in grapeseed oil. Production complied with GMP standards and was subject to Health Canada review prior to study initiation. Given the data for dosing of CBD from previous studies, we calculated that a 50:1 ratio product of clinical trial grade purity would allow us reach doses of CBD similar to those used in the clinical trials of pure CBD, while maintaining the THC dose at <0.5 mg/kg/day.30, 37 This THC dose was a conservative estimated dose, in an attempt to avoid psychoactive effects, based on extrapolated THC dosing from adult studies of smoked cannabis examining psychoactive effects, and published clinical experience with CBD/THC cannabis oils.
During the 4‐week pre‐intervention period after study enrollment with consent, the caregiver(s) of each participant completed a seizure diary documenting baseline seizure frequency. All enrolled participants also underwent a 24‐h ambulatory EEG to assess ictal and interictal epileptiform activity. The Vineland Adaptive Behaviour Scale, Second Edition (Vineland™‐II), survey interview form was administered to the parent/legal guardian in person or by telephone to assess adaptive behavior of the participant. Concomitant AEDs were monitored with trough drug levels where appropriate. Compliance monitoring was assessed by clinic evaluations which included measuring the amount of drug remaining in the medication bottle, follow up phone calls, and email.
During the 20‐week intervention period, participants were started on TIL‐TC150 at an initial dose of 2 mg/kg/day CBD (0.04 mg/kg/day THC) divided twice daily with weekly titration by 2 mg/kg/day every 7 days up to a maximum dose of 16 mg/kg/day CBD (0.32 mg/kg/day THC). Optimal dose (either maximum targeted or maximal tolerated dose) was then continued for the remainder of the period. The primary end point was analyzed during the final 4 weeks, weeks 17–20 when participants were on a stable optimal dose of study drug. Seizure diaries and 24‐hr ambulatory EEG recordings were assessed and compared with those from the pre‐intervention period from weeks −4 to 0. Safety and tolerability were assessed by clinical evaluations at baseline, every 2 weeks for the first month, and then monthly for 4 months up to 20 weeks. Clinical evaluations consisted of obtaining anthropometric measurements, a physical and neurological examination, review of seizure diary, completion of the Pediatric Epilepsy Side Effect Questionnaire (PESQ) by parents/legal guardians, and blood work to assess for complete blood count, liver and renal function, and trough AED levels. Secondary measures were assessed through the completion of the Quality of Life in Childhood Epilepsy questionnaire (QOLCE)37, 38, 39, 40 and via parent/legal guardian interview using Vineland™‐II.41
Participants who chose to continue the TIL‐TC150 therapy after the initial intervention period entered the longitudinal maintenance period to assess for tolerability and safety. Secondary outcomes were assessed at up to 64 weeks, along with repeat evaluation of QOLCE and Vineland scoring at the end of week 64. A complete schedule of events for study participants is available (Data S2).
Ambulatory EEG
Two 24‐h ambulatory EEG studies were performed for each participant, the first in the pre‐intervention period and the second at the primary endpoint period. All EEGs were recorded with 19 channels, using the 10–20 International System for electrode placement, using Natus® NeuroWorks® software version 8.1 with a sampling rate of 200 Hz, band pass filter between 1 Hz and to 70 Hz. The recordings were referenced to Pz’ with additional EKG monitoring. No video was obtained during the recordings. Recordings were then digitally converted for further analysis using Persyst 13 (Persyst®, Developmental Corporation, Prescott, AZ). The EEGs were read by 2 independent, blinded (to identity of participant and timing of EEG study) reviewers board certified by the Canadian Society of Clinical Neurophysiologists. Persyst® software was used to calculate the spike index using the Spike Index tool. Each EEG was manually inspected for seizure events. The results of the 2 reviewers were then compared for interrater reliability and disagreement was resolved with consensus from a third reviewer.
Quality of life in childhood epilepsy questionnaire (QOLCE)
The QOLCE is a validated questionnaire completed by caregivers to assess impact of epilepsy on aspects of a child's quality of life.38, 39, 40 The questionnaire scores overall quality of life, and discrete domains (physical activity, well‐being, general health, cognition, social activity, and behavior) using Likert rating.
Vineland adaptive behavior scales
All ratings on the Vineland™‐II were converted to age‐normed standardized scores. Domain scores (communication, daily living, socialization, motor skills, maladaptive behavior index) and the adaptive behavior composite (ABC) score were transformed to standard scores (mean = 100, SD = 15), while the subdomains (expressive, receptive, written, personal, domestic, community, interpersonal relations, play and leisure time, coping skills, gross, fine, internalizing and externalizing) were transformed into v‐scores (mean = 15, SD = 3).
Statistical analysis
Statistical software GraphPad Prism, Inc. version 7.03 (California, USA, http://www.graphpad.com) was used for statistical analyses. The study period was divided into 6 blocks of 4 weeks. The changes in seizure response during each block (frequency of seizures, and seizure free days) were compared using one‐way analysis of variance (ANOVA). For analysis of effect of dose on seizure count, we used nonparametric repeated measures ANOVA (Friedman test) and Dunn's multiple comparisons test. For the analysis of effect of dose on seizure free days, we used repeated measures ANOVA and Tukey's multiple comparisons test. To compare pre‐intervention and primary end point intervention, we used Wilcoxon's paired test for most data analyses. The paired t‐test was only used for comparing Vineland scorings as the data were normally distributed.
Previous studies have generated signals suggesting clobazam and valproate may have clinically significant drug interactions with cannabinoid compounds. The effect of Clobazam or VPA on the association between dose and seizure was studied using Spearmans correlation and Mann‐Whitney U test. To determine the effect of age on dose tolerance and seizure response, the study group was dichotomized based on age: age < 10 and age ≥ 10, and were studied using ANOVA (Friedman test) for repeated measures and Dunn's multiple comparisons test. A P < 0.05 was considered statistically significant, r values reported as appropriate for correlation.
Results
Trial population
Twenty children with Dravet syndrome and DRE were enrolled. The characteristics of our population are summarized in Table 1. The mean age was 10.15 years (range 2.1–17.8) with equal sex distribution. Participants were taking a mean of 3 AEDs (range 1–4) at the time of enrollment, with clobazam and valproic acid being the most commonly used medications by 70% and 60% of participants, respectively, followed by stiripentol (50%) and levetiracetam (40%). As Figure 1 shows, 19 participants completed the 20‐week intervention period. One participant died during the primary intervention period due to sudden unexpected death in epilepsy (SUDEP) and was excluded from final analysis of primary and secondary end points. As the last observation period was at 12 weeks for this participant, we performed sensitivity analysis with 20 participants using week 9–12 as primary outcome and it did not change the statistical significance of our results. Of the remaining 19 participants, 14 entered the longitudinal maintenance study after week 20. The five participants who withdrew did not perceive benefit of therapy. This longitudinal observation period is ongoing at this time. At baseline, all participants had motor seizures, while 7 (36.8%) also had significant myoclonic seizures and 3 (15.7%) also had eyelid myoclonus.
Table 1.
Baseline characteristics
| Baseline characteristics | |
|---|---|
| Mean age (Range) | 10.15 (2.1–17.8) |
| Female | 10 (50%) |
| Number of current AED | 2.9 (1–4) |
| Current AED – n (%) | |
| Clobazam | 14 (70%) |
| Valproate | 12 (60%) |
| Stiripentol | 10 (50%) |
| Levetiracetam | 8 (40%) |
| Topiramate | 5 (25%) |
| Phenobarbitone | 2 (10%) |
| Lacosamide | 1 (5%) |
| Phenytoin | 1 (5%) |
| Ketogenic diet | 2 (10%) |
| Vagal nerve stimulation | 4 (20%) |
| BMI at baseline, mean(range) | 19.16 (15.8–30.7) |
| Seizure Onset in months, mean(range) | 7.95 (2–36) |
Figure 1.
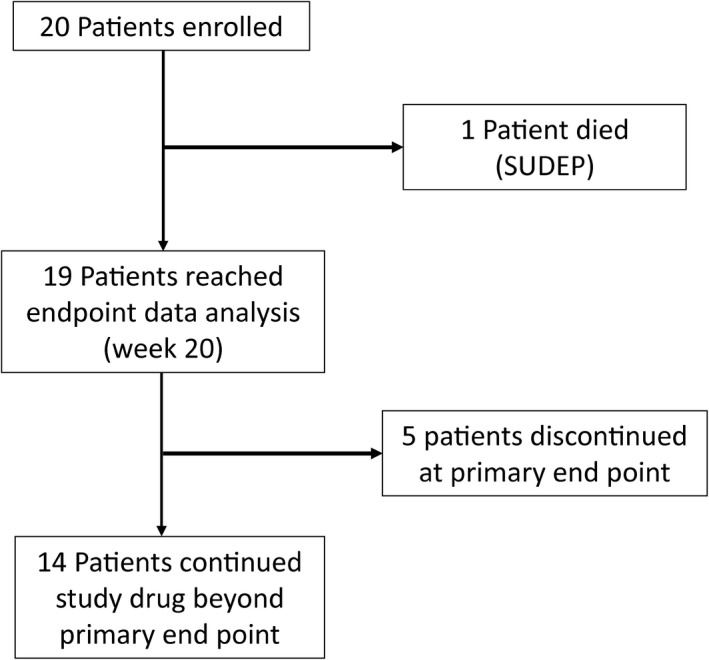
Study participant flow chart.
Dosing and tolerability
Titration to the target dose of 16 mg/kg/day CBD (0.32 mg/kg/day THC) of TIL‐TC150 was achieved in eight participants (42.1%), as shown in Table 2. The mean actual dose attained was 13.3 mg/kg/day (range 7–16 mg/kg/day). The remaining 11 participants did not reach the target dose due to excessive somnolence (81%), anorexia (54.5%), diarrhea (36%), and weight loss >5% (36%). In those who did not achieve target dose (N = 11/19), the mean dose achieved was 11.8 mg/kg/day (range 7–14 mg/kg/day). Figure 2 shows the mean dosing and range of dose achieved by participants for both CBD and THC during the intervention period. The mean CBD dose achieved was 464 mg/day (range 120–840 mg/day). The mean THC dose was 9.28 mg/day (range 2.4–16.8 mg/day). It is important to note that at this THC dosing, there were no reported psychoactive side effects such as hallucinations, however, we acknowledge that assessment of this is challenging in our study population as we often depend on caregiver noted changes in behavior or emotional state, rather than self‐reporting by participants.
Table 2.
Dosing and adverse events
| Dose of TIL‐TC150 |
| Mean dose = 13.3 mg/kg/day (Range 7–16) CBD/0.27 mg/kg/day (range 0.14–0.32) THC |
| Achieved target dose = 9/19(42.1%) |
| Mean CBD maintenance dose = 464.21 mg/day (Range 120–840) |
| Mean THC maintenance dose = 9.28 mg/day (range 2.4–16.8) |
| Adverse events | Number of participants, N = 19 (%) | Correlation, r | P‐Value |
|---|---|---|---|
| Nervous system | |||
| Somnolence/tiredness | 17 (89.5) | −0.975 | 0.034a |
| Increased seizure | 4 (21) | 0.369 | 0.533 |
| Gastrointestinal | |||
| Diarrhea | 6 (31.6) | 0.447 | 0.500 |
| Constipation | 5 (26.3) | −0.667 | 0.267 |
| Gagging | 3 (15.8) | 0.633 | 0.333 |
| General | |||
| Poor balance | 8 (42.1) | −0.738 | 0.200 |
| Sleep disturbance | 4 (21) | 0.224 | 0.995 |
| Mood change/Hyperactivity | 4 (21) | −0.359 | 0.633 |
| Infections | |||
| Upper respiratory tract infection | 4 (21%) | −0.894 | 0.100 |
| Urinary tract infection | 2 (10.5%) | −0.866 | 0.200 |
| Metabolism | |||
| Anorexia | 10 (52.6%) | 0.4 | 0.517 |
| Increased appetite | 3 (15.8%) | −0.707 | 0.400 |
| Side effects Reported in 1 participant (5.3%) | |||
| Gastrointestinal Disturbances (unspecified); Screaming; Bruxism; Hiccups; Facial Acne; Alopecia; Pneumonia; Hyperhidrosis; Hand tremor; Oily Urine; Conjunctivitis; Slowed processing. | |||
Data for adverse events are n (%). Adverse events reported only in one participant were grouped together. Thirteen (68%) of 19 participants that experienced somnolence were on concomitant Clobazam.
Signifies statistical significance (P < 0.05), r value for correlation.
Figure 2.
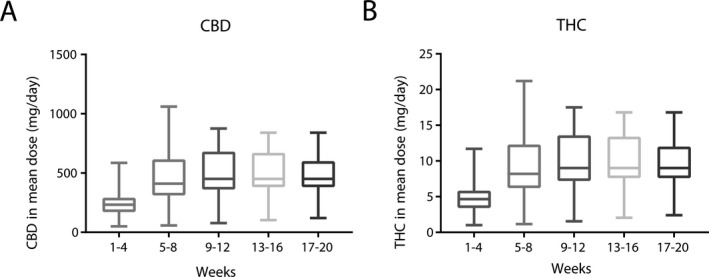
The doses of CBD (A) and THC (B) achieved for all participants during the intervention period. The Boxplots show mean values, with 25th and 75th percentiles. The whiskers denote the minimum and maximum doses.
Laboratory abnormalities were seen in 8 (42.1%) participants. These were predominantly transient and observed during the 8‐week titration phase. Repeat bloodwork at the primary endpoint showed these values had normalized. Specifically, transiently elevated liver enzymes (AST mean 82.8, range 59–165; reference range:<31 U/L:; ALT mean 68.8, range 59–102, reference range:<44 U/L; and GGT mean 125.9, range 56–229, reference range: 0–45 U L) were seen in 8 participants, and 5 of the 8 had concomitant elevated valproate trough levels (mean elevated level 787, range 717–920; reference range 350–700 μmol L). Eight participants had transient, predominantly mild reduction in platelet count (mean 134.3, range 55–193; normal reference range 194–345 x 10^9/L), and five of these participants also had elevated valproate trough levels and liver enzymes. The remainder of laboratory parameters assessed was normal.
Adverse events were reported in 18 participants (95%), see Table 2. The majority of these were transient in nature, resolving during the first 8‐week titration phase. No major adverse events were encountered, and no participants withdrew from the trial prior to the end point due to adverse events. Of the participants who reached the target dose of 16 mg/kg/day, six required a subsequent dose reduction due to side effects, and 5 participants did not continue dose escalation to the target of 16 mg/kg/day due to side effects. Somnolence was the most common adverse event affecting 17 participants (89.5%) in at least one 4 week block. This was assessed by caregiver reporting. However, as shown in Figure 3, there was a significant negative correlation (P = 0.03; r = −0.97) between the number of participants where somnolence was reported and intervention weeks, showing that somnolence subsided gradually. A significant weight loss of more than 5% from baseline was seen in five participants (26.3%). Other commonly encountered side effects are shown in Table 2.
Figure 3.
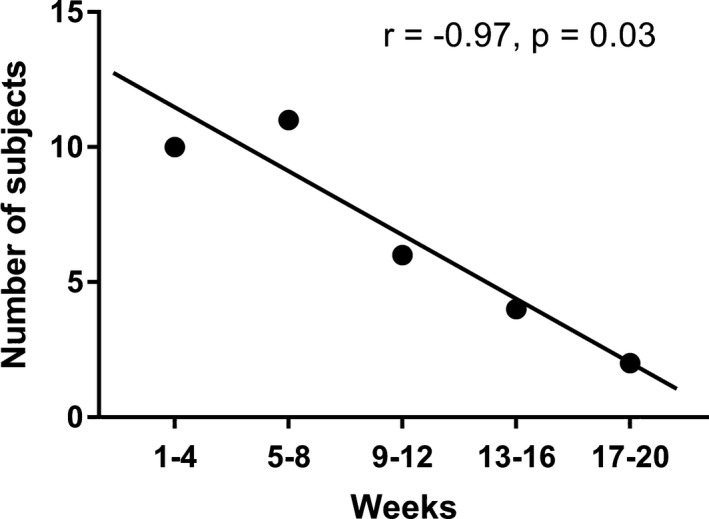
Change in reported somnolence during intervention period, in 4‐week blocks. Rates of reported somnolence were high, and increased in the first 8 weeks during TIL‐TC150 titration, subsequently dropping during the remaining weeks. There was a reduction in somnolence during the last 4‐week period (weeks 17–20) when compared with the first 8 weeks (r = −0.97).
Seizure outcomes
The impact of TIL‐TC150 on seizures was measured by seizure diary recordings, by objective data from the 24‐h EEG, and are displayed in Table 3.
Table 3.
Summary of secondary outcomes (N = 19)
| Pre‐intervention | Primary end point | P‐Value | |
|---|---|---|---|
| Seizure Diary – median. (IQR) or mean (SD)b | |||
| Seizure count (motor) | 17.0 (31) | 5.0 (26) | 0.006a |
| Myoclonic jerks (days) | 5.0 (16) | 0.5 (5) | 0.031a |
| Seizure free days, mean (SD) | 11.89 (9.48) | 18.32 (8.78) | 0.008a |
| 24‐h EEG data – median. (IQR)c | |||
| Spike‐per‐Second | 0.09 (0.11) | 0.06 (0.05) | 0.022a |
| Number of seizure/24 h | 0 (1) | 0 (2) | 0.953 |
| PESQ – median (IQR) | |||
| Total PESQ Score | 4.20 (14.7) | 6.3 (27.3) | 0.38 |
| Quality of Life Childhood Epilepsy (QOLCE) questionnaire – mean (SD) | |||
| Quality of Life | 31.58 (20.14) | 50.00 (22.05) | 0.001a |
| Physical activity | 40.80 (14.29) | 42.44 (17.57) | 0.75 |
| Well‐being | 62.69 (15.39) | 66.81 (15.88) | 0.38 |
| General health | 35.53 (25.43) | 42.11 (25.07) | 0.24 |
| Cognition | 39.60 (21.98) | 50.79 (25.50) | 0.06 |
| Social activity | 17.82 (13.0) | 20.68 (14.14) | 0.28 |
| Behavior | 51.11 (10.80) | 53.80 (10.25) | 0.27 |
| Total score | 39.60 (11.13) | 46.02 (14.20) | 0.045a |
| Vineland ™ ‐II | |||
| Domain Scores – mean of standard score (SD) | |||
| Communication | 53.16 (18.24) | 56.67 (18.89) | 0.02a |
| Daily living skills | 54.32 (20.33) | 55.67 (19.74) | 0.91 |
| Socialization | 59.68 (18.14) | 63.61 (19.70) | 0.06 |
| Motor skills | 63.53 (17.52) | 63.17 (15.54) | 0.95 |
| Adaptive behavior composite | 54.00 (18.69) | 56.61 (18.83) | 0.16 |
| Maladaptive behavior index | 18.83 (1.20) | 17.94 (1.89) | 0.02a |
| Subdomains – mean of v‐score (SD) | |||
| Receptive | 6.53 (3.67) | 7.61 (3.81) | 0.01a |
| Expressive | 6.16 (4.38) | 6.67 (4.46) | 0.31 |
| Written | 6.78 (2.53) | 7.06 (2.49) | 0.33 |
| Personal | 6.00 (4.33) | 6.06 (3.81) | 0.66 |
| Domestic | 7.63 (3.79) | 8.28 (3.66) | 0.33 |
| Community | 6.37 (4.40) | 6.67 (4.58) | 0.71 |
| Interpersonal relations | 7.32 (3.65) | 8.33 (4.26) | 0.03 |
| Play and leisure time | 6.21 (4.14) | 6.72 (4.24) | 0.41 |
| Coping skills | 8.79 (3.19) | 9.61 (3.71) | 0.09 |
| Gross motor skills | 8.95 (2.22) | 9.28 (1.87) | 0.23 |
| Fine motor skills | 8.63 (3.72) | 8.28 (3.50) | 0.40 |
| Internalizing | 18.33 (4.35) | 18.59 (2.29) | 0.12 |
| Externalizing | 16.17 (3.93) | 16.88 (1.58) | 0.68 |
Signifies P < 0.05.
All 19 participants were included in seizure count, seizure free days. seven participants had myoclonic seizures, calculated as days participants experienced them. As seizure count data was not normally distributed we report median and IQR.
16 participants were included in the final EEG analysis.
Comparing the two 4 week periods, weeks −4–0 (pre‐intervention) with weeks 17–20 (primary end‐point) diary data, we noted a statistically significant reduction in motor seizures of 70.6%, from a median (IQR) of 17.0 (31) seizures noted in the pre‐intervention period to a median (IQR) of 5.0 (26) seizures observed in week 17–20 (P = 0.006). In our cohort, we had a number of children with severe disabling myoclonic seizures, manifesting as myoclonic jerks (seven participants) or eyelid myoclonus (three participants), which would occur typically hundreds of time throughout the day. Caregivers were not asked to count these but did record as days with or without myoclonic jerks. These children had a statistically significant reduction in ‘days with myoclonic jerks’ from a median of 5.0 days in the 4‐week period pre‐intervention, to 0.5 days during weeks 17–20 (P = 0.031).
The 50% responder rate was 63% overall, with a seizure reduction rate of 50–90% was seen in nine participants (47%), while three participants (16%) had a reduction >90% and two participants became seizure free. Seizure reduction of <50% was seen in seven participants (37%). Four participants reported an increase in seizure frequency. When comparing pre‐intervention with primary end point time periods, Figure 4 displays the percentage change in frequency of motor seizures, comparing both the pre‐intervention 4‐week period with the primary end point 4‐week period (weeks 17–20). Two participants were seizure free during the last 4 weeks. Also illustrated is the percentage change from first 4 weeks compared with the mean changes across the entire 20‐week intervention period. In the participants who reached the target treatment dose of 16 mg/kg/day of TIL‐TC150, a statistically significant reduction in motor seizures, and an increase in seizure free days was seen at primary end point versus pre‐intervention, compared to those who did not reach the target dose. The median motor seizure count was reduced from 16.5 to 3.0 per 4 week block in those who reached the target dose as compared to 22.0–5.0 per 4 week block in those who did not (P = 0.031 vs. P = 0.23). The group that reached the target dose also had an increase in seizure free days from a mean of 11.38 to 19.3 days in a 4‐week period versus 12.27–17.55 days in those who did not (P = 0.045 vs. P = 0.097). The overall impact of TIL‐TC150 on seizure free days during the 4‐week periods from pre‐intervention to primary end point, is shown in Figure 5. The mean number of seizure free days increased from 11.89 in weeks −4–0 to 18.31 in weeks 17–20 (P = 0.0075).
Figure 4.
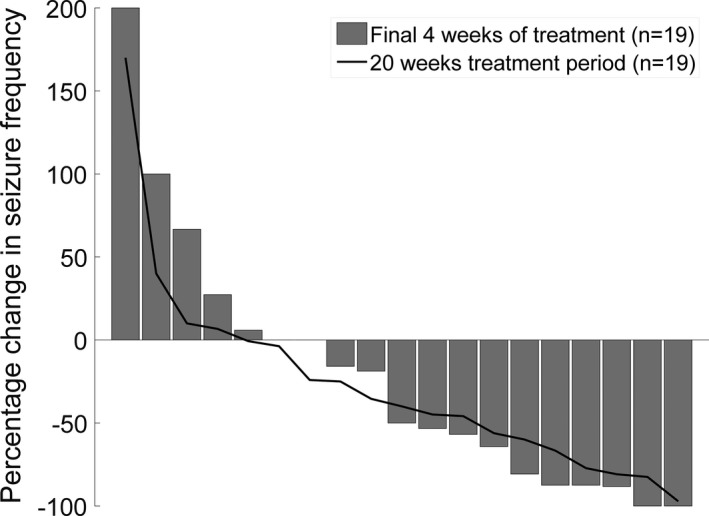
Percentage change in Seizure frequency. Each bar represents a study participant. Bars represent the percentage change in frequency of motor seizures during the primary end point period (17–20 weeks), compared with the pre‐intervention period (−4–0 weeks) measured by seizure diary. The red line represents the mean percentage change across the entire 20 weeks. Percentage changes for each participant are sorted from highest to lowest values. 2 participants were seizure free during the last 4 weeks.
Figure 5.
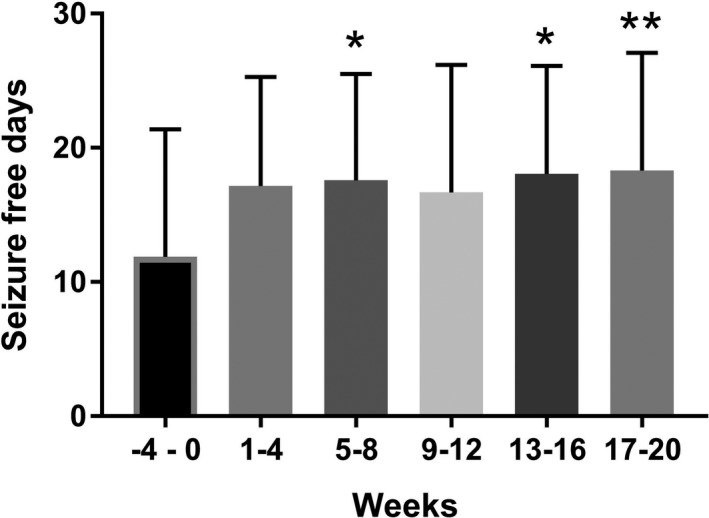
Mean number of seizure free days in each 4‐week period comparing pre‐intervention (week −4–0) with each 4 week block of intervention period (weeks 1–20). The mean (bar) and standard deviation (tail) are shown below for all 19 participants. *P < 0.05; **P < 0.01
EEG outcomes
Twenty‐four hour ambulatory EEG studies were completed by 16 of 19 participants at two time points, pre‐intervention and at primary endpoint. Two participants declined the second EEG, and a third participant had a technical fault in their second recording, which did not allow analysis of their EEG. Of the three participants who were not included in the EEG analysis, two had a statistically significant improvement in their seizures and one did not. There was interrater reliability of 87% among reviewers and consensus was reached with the remaining 13%. When analyzing all studies, the median (IQR) spike per second calculation showed a statistically significant reduction from 0.09 (0.11) at pre‐intervention period, to 0.06 (0.05) at the primary endpoint (P = 0.022). The spike‐per‐second reduction was also statistically significant in participants who had more than 50% seizure reduction by seizure diary report (P = 0.02) when compared to those with a <50% seizure reduction (P = 0.6).
Although not statistically significant, the mean number of electrographic seizures decreased from 1.25 seizures/day (median (IQR): 0(1)) to 1 seizure/day (median (IQR): 0(2)). Of 10 participants who showed a reduction in their spike indices, nine showed comparable reductions in seizure frequencies. No increase in baseline spike indices was seen in three participants while seizure frequency increased in one patient and decreased in two participants. A simultaneous increase in seizure frequency and spike indices was seen in two participants while one patient had an increased spike index and decreased seizure frequency. There was no clear correlation between rate of baseline spike activity and seizure reduction at primary end point.
Everyday adaptive functioning
Vineland™‐II was administered at pre‐intervention and at the primary end point (17–20 weeks). Statistically significant changes were seen in mean domain scores for both communication (P = 0.02) and the maladaptive behavior index (P = 0.02). Within subdomains, statistically significant changes were seen across the study in receptive communication skills (P = 0.01), as shown in Table 3. There were no consistent relationships between seizure reduction and scores on the Vineland™‐II. For example, one participant showed a clinically significant decrease in adaptive functioning on nine scales, and experienced an 80% reduction in seizures over time, while another showed improvement in seven areas of functioning yet had an increase in seizures at the primary end point.
Quality of life outcomes
A statistically significant improvement in quality of life was seen in the total QOLCE score from a mean of 39.9 at baseline to 46.02 at the primary endpoint (P = 0.045). The sub‐domain scores in Table 3, showed variable improvement from the baseline scores, and identified a statistically significant improvement in the overall quality of life item (P = 0.001).
Discussion
The primary objective of this study was to establish dosing and tolerability of TIL‐TC150 ‐ a cannabis plant extract containing 100 mg/mL CBD and 2 mg/mL THC‐ in children with DRE due to Dravet syndrome. Our secondary objectives were to assess the impact of therapy on seizures and quality of life. The data from our study indicate that TIL‐TC150 was safe and well tolerated in our subjects with DRE due to Dravet Syndrome. TIL‐TC150 treatment resulted in a reduction in seizure counts and spike index on EEG, and improvements in quality of life measures.
A major limitation in our study concerns the small number of participants, the majority of whom were on concomitant clobazam. An overestimation of the impact of cannabinoids on seizures is possible as cannabidiol exerts an inhibitory effect on CYP3A4 and CYP2C19 which lead to increase in clobazam exposure.43 It is important not to discount media influence and the rising popularity of cannabis enriched drugs, which may impact family perception and increase their expectations of success, and thus subconsciously impact their objectivity. The open‐label nature of this study can influence subjects/caregivers also.
Our study adds to a growing body of evidence that cannabinoids exert antiseizure effects and are safe and tolerable in treating pediatric DRE. Much of the evidence to date has focused on research with a single CBD‐only product. Our study used a preparation containing both THC and CBD, which is more comparable with the initial publicized mixed CBD/THC compound,29 and with the cannabinoid extracts available in Canada.36
Earlier studies include an open‐label interventional trial32 of 214 pediatric and adult patients with intractable epilepsy, where CBD was reported to have an adequate safety profile and potential efficacy in seizure reduction. A subsequent double‐blinded, placebo controlled trial33 of 120 patients with Dravet syndrome demonstrated that the CBD‐ only product was more efficacious than placebo in seizure reduction. Moreover, a recent double‐blind randomized controlled trial34 that included 171 patients with Lennox‐Gastaut syndrome showed that this same CBD oil was efficacious as an add‐on therapy in reducing drop seizures, and it was generally well tolerated. While CBD dosing ranged across the studies (up to 50 mg/kg/day), most targeted 20 mg/kg/day of CBD, while side effects increased with doses above 15 mg/kg/day32
Our study demonstrated a satisfactory safety and tolerability profile of TIL‐TC150 in children with DRE due to Dravet Syndrome. In addition, we observed promising clinically beneficial effects including a reduction in seizure frequency and spike index with improvements in certain aspects of adaptive functioning and quality of life measures. As well, TIL‐TC150 produced comparable outcomes to other published studies that used purified Cannabidiol.32, 33, 34, 35 Despite the addition of TIL‐TC150 to regimens of multiple antiepileptic drugs in our cohort, there were no participant withdrawals or serious adverse events observed. This is in contrast to previous studies which demonstrated a withdrawal rate of 3–16% in the cannabidiol treated groups. We speculate that the lower rate of withdrawal in our study could be related to a number of factors including: a slower titration approach, lower target doses, and the potential development of tolerance over time.37
While adverse events were common, most were mild and self‐limited and resolved either by nonescalation or reduction in the TIL‐TC150 dose. Most adverse events were observed during initiation of the TIL‐TC150, with the majority of participants reporting improved tolerability over time. For example, somnolence, the most common side effect seen in 17 participants (89.5%), showed significant reduction over time as participants developed tolerability. This can be attributed to a dose and region‐dependent downregulation and desensitization of cannabinoid receptors. For example, in vivo studies have shown that CB1 endocannabinoid receptor (CB1Rs) desensitization can occur with prolonged and repeated doses.42 In addition, previous studies have described the development of increased somnolence with the simultaneous use of clobazam and cannabinoids, likely due to increased concentration of N‐desmethylclobazam due to cannabinoid inhibition of CYP3A4 and CYP2C19.43, 44
Other side effects that led to an inability to achieve the target dose were anorexia, diarrhea, (though these were not statistically significant based on correlation analysis), and weight loss. A significant weight loss of more than 5% from baseline was seen in five participants (26.3%) and more commonly seen in participants above 10 years of age, although this was not statistically significant (P = 0.125). Weight loss and appetite suppression is the opposite of what is typically associated with cannabis use, both from reports of recreational marijuana use and from the clinical experience of appetite stimulation in oncology patients.45, 46 It has been postulated by producers that terpenes, the compounds responsible for the characteristic odor of cannabis, may be responsible for the appetite stimulant properties of the cannabis plant, or perhaps that there is synergism between THC and terpenes which might mediate this, a so‐called entourage effect.47 Due to the paucity of evidence for any antiseizure effect of terpenes, our study drug was not formulated with these compounds – although many artisanal and recreational cannabis products typically are.
Interestingly, elevated hepatic aminotransferases levels were observed in 5 of 8 participants taking valproic acid with concomitant increase in trough valproic acid levels. This increase was observed early and was transient and disappeared after reduction in the valproic acid dose or reduction or nonescalation of TIL‐TC150 dose. This interaction can be explained by the stress caused by cannabinoids on the CYP‐450 metabolic pathway shared with valproic acid.44 This suggests that patients using cannabis oils alongside other hepatically metabolized drugs should be closely monitored for liver function through blood work.
Four participants reported an increase in seizure frequency. As our cohort consisted of participants with DRE, this is likely best attributed to fluctuation in disease severity and is in keeping with rates seen in other clinical trials for those with DRE. Among the recent clinical trials evaluating CBD, increased seizures were seen in 7–11%.32, 33, 34, 35 Although we were very conservative in our dosing of THC in this study, it is plausible that treatment may could lead to increase in seizures.
SUDEP is reported in clinical trials in those with DRE.48, 49 Dravet syndrome is associated with higher rates of SUDEP, reportedly 9.3/1000 patient years in a recent study, almost double the rate quoted for adults with DRE.50 One participant died of SUDEP unrelated to TIL‐TC150 use. This serves as a sad reminder of the realities and risks of severe DRE and the fears that families live with on a daily basis, that drive us to find new therapies.
Our study demonstrated that a meaningful clinical reduction in seizures rates is achievable in pharmaco‐resistant participants by the addition of TIL‐TC150 to their concomitant antiepileptic regimens. This reduction was appreciated early in the first weeks of dose escalation with broad spectrum effects on multiple seizure types. Many participants had more than a 50% reduction in motor seizures with few achieving seizure freedom. Participants had a significant increase in the mean number of seizure free days with the addition of TIL‐TC150. For children with DRE and frequent seizures, an increase in the number of days per month without seizures is clinically meaningful, allowing them to participate in school and engage with their peers and families. Interestingly, seizure control seemed to be dose‐dependent as participants who reached the target dose had higher rates of seizure reduction. Moreover, this reduction in clinical seizure frequency was also mirrored by a statistically significant reduction in the spike index on EEG recording.
Subsequent investigation should be directed toward the demonstration of efficacy of CBD/THC in a wider population of children with drug‐resistant epilepsy. Cannabinoid compounds are complex due to their many chemically active components, and the possibility of mitigating anorexia is compelling. Further studies are required to identify which component of the cannabinoids mediates appetite.
To our knowledge, this study is the first of its kind to examine with rigor the dosing and tolerability of a mixed cannabinoid product containing both CBD and THC in children with DRE. We have provided dosing information for a THC containing product, which was not previously available. In addition, we have added to evidence that cannabinoids have a role in the treatment of children with DRE and highlighted the importance of close monitoring during titration including laboratory blood work.
Author Contributions
BM, OCS, LW, KS, and MZ contributed to the conception and design of the study; all authors contributed to the acquisition and/or analysis of data; BM, OCS, LW, SA, MZ, and KS contributed to drafting the text. All authors contributed to final edits of the manuscript.
Conflict of Interest
This study was funded in majority by Tilray ® who produced the study drug used in this study. They were not involved in any aspect of the study design, running of the clinical trial, or interpretation of the results. None of the investigators received any honorarium from Tilray.
Supporting information
Data S1. Inclusion and exclusion criteria for enrollment.
Data S2. Schedule of events.
Acknowledgment
This study was supported by funding from Tilray® and The Little Rocky Project. We would like to thank our EEG technologist colleagues at the Hospital for Sick Children for assisting with the prolonged EEG studies. Most importantly we are very grateful to the children and their families who participated in our study.
Funding Information
This study was supported by funding from Tilray ® and The Little Rocky Project.
Funding Statement
This work was funded by Tilray grant R_2016_0011967; Little Rocky Project grant .
References
- 1. Kwan P, Arzimanoglou A, Berg AT, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc task force of the ILAE commission on therapeutic strategies. Epilepsia 2010;51:1069–1077. [DOI] [PubMed] [Google Scholar]
- 2. Yakoub M, Dulac O, Jambaqué I, et al. Early diagnosis of severe myoclonic epilepsy in infancy. Brain Dev. 1992;14:299–303. [DOI] [PubMed] [Google Scholar]
- 3. Sakauchi M, Oguni H, Kato I, et al. Retrospective multi‐institutional study of the prevalence of early death in Dravet syndrome. Epilepsia 2011;52:1144–1149. [DOI] [PubMed] [Google Scholar]
- 4. Akiyama M, Kobayashi K, Yoshinaga H, Ohtsuka Y. A long‐term follow‐up study of Dravet syndrome up to adulthood. Epilepsia 2010;51:1043–1052. [DOI] [PubMed] [Google Scholar]
- 5. Dravet C. Dravet syndrome history. Dev Med Child Neurol 2011;53:1–6. [DOI] [PubMed] [Google Scholar]
- 6. Catarino CB, Liu JYW, Liagkouras I, et al. Dravet syndrome as epileptic encephalopathy: evidence from long‐term course and neuropathology. Brain 2011;134(Pt 10):2982–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Steel D, Symonds JD, Zuberi SM, Brunklaus A. Dravet syndrome and its mimics: beyond SCN1A. Epilepsia 2017;58:1807–1816. [DOI] [PubMed] [Google Scholar]
- 8. Gloss D, Vickrey B. Cannabinoids for epilepsy. Cochrane Database Syst Rev 2012;6:CD009270. [DOI] [PubMed] [Google Scholar]
- 9. Mechoulam R, Carlini EA. Toward drugs derived from cannabis. Naturwissenschaften 1978;65:174–179. [DOI] [PubMed] [Google Scholar]
- 10. Cunha JM, Carlini EA, Pereira AE, et al. Chronic administration of cannabidiol to healthy volunteers and epileptic patients. Pharmacology 1980;21:175–185. [DOI] [PubMed] [Google Scholar]
- 11. Ames FR, Cridland S. Anticonvulsant effect of cannabidiol. S Afr Med J 1986;69:14. [PubMed] [Google Scholar]
- 12. Trumbly B, Sherman M. Double‐blind clinical study of cannabidiol as a secondary anticonvulsant. Presented at Marijuana ‘90 International Conference on Cannabis and Cannabinoids, Kolympari (Crete), July 8–11, 1990.
- 13. Murphy L, Bartke A. Marijuana/cannabinoids neurobiology and neurophysiology. 1st ed. Boca Raton, Florida: CRC Press, 1992. [Google Scholar]
- 14. Rosenberg EC, Tsien RW, Whalley BJ, Devinsky O. Cannabinoids and epilepsy. Neurotherapeutics 2015;12:747–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Izquierdo I, Tannhauser M. Letter: the effect of cannabidiol on maximal electroshock seizures in rats. J Pharm Pharmacol 1973;25:916–917. [DOI] [PubMed] [Google Scholar]
- 16. Chesher GB, Jackson DM. Anticonvulsant effects of cannabinoids in mice: drug interactions within cannabinoids and cannabinoid interactions with phenytoin. Psychopharmacologia 1974;37:255–264. [DOI] [PubMed] [Google Scholar]
- 17. Chesher GB, Jackson DM, Malor RM. Interaction of delta9‐tetrahydrocannabinol and cannabidiol with phenobarbitone in protecting mice from electrically induced convulsions. J Pharm Pharmacol 1975;27:608–609. [DOI] [PubMed] [Google Scholar]
- 18. Karler R, Turkanis SA. Subacute cannabinoid treatment: anticonvulsant activity and withdrawal excitability in mice. Br J Pharmacol 1980;68:479–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Turkanis SA, Smiley KA, Borys HK, et al. An electrophysiological analysis of the anticonvulsant action of cannabidiol on limbic seizures in conscious rats. Epilepsia 1979;20:351–363. [DOI] [PubMed] [Google Scholar]
- 20. Consroe P, Benedito MA, Leite JR, et al. Effects of cannabidiol on behavioral seizures caused by convulsant drugs or current in mice. Eur J Pharmacol 1982;83(3–4):293–298. [DOI] [PubMed] [Google Scholar]
- 21. Koppel BS, Brust JCM, Fife T, et al. Systematic review: efficacy and safety of medical marijuana in selected neurologic disorders. Neurology 2014;82:1556–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Elsohly MA, Slade D. Chemical constituents of marijuana: the complex mixture of natural cannabinoids. Life Sci 2005;78:539–548. [DOI] [PubMed] [Google Scholar]
- 23. Devinsky O, Cilio MR, Cross H, et al. Cannabidiol: pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia 2014;55:791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mechoulam R, Parker LA. The endocannabinoid system and the brain. Annu Rev Psychol 2013;64:21–47. [DOI] [PubMed] [Google Scholar]
- 25. Kaur R, Ambwani SR, Singh S. Endocannabinoid system: a multi‐facet therapeutic target. Curr. Clin. Pharmacol. 2016;11:110–117. [DOI] [PubMed] [Google Scholar]
- 26. Karniol IG, Carlini EA. Pharmacological interaction between cannabidiol and delta 9‐tetrahydrocannabinol. Psychopharmacologia 1973;33:53–70. [DOI] [PubMed] [Google Scholar]
- 27. Englund A, Morrison PD, Nottage J, et al. Cannabidiol inhibits THC‐elicited paranoid symptoms and hippocampal‐dependent memory impairment. J. Psychopharmacol. (Oxford) 2013;27:19–27. [DOI] [PubMed] [Google Scholar]
- 28. Maa E, Figi P. The case for medical marijuana in epilepsy. Epilepsia 2014;55:783–786. [DOI] [PubMed] [Google Scholar]
- 29. Porter BE, Jacobson C. Report of a parent survey of cannabidiol‐enriched cannabis use in pediatric treatment‐resistant epilepsy. Epilepsy Behav 2013;29:574–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tzadok M, Uliel‐Siboni S, Linder I, et al. CBD‐enriched medical cannabis for intractable pediatric epilepsy: the current Israeli experience. Seizure 2016;35:41–44. [DOI] [PubMed] [Google Scholar]
- 31. Brodie MJ, Ben‐Menachem E. Cannabinoids for epilepsy: what do we know and where do we go? Epilepsia 2018;59:291–296. [DOI] [PubMed] [Google Scholar]
- 32. Devinsky O, Marsh E, Friedman D, et al. Cannabidiol in patients with treatment‐resistant epilepsy: an open‐label interventional trial. Lancet Neurol. 2016;15:270–278. [DOI] [PubMed] [Google Scholar]
- 33. Devinsky O, Cross JH, Laux L, et al. Trial of cannabidiol for drug‐resistant seizures in the Dravet syndrome. N Engl J Med 2017;376:2011–2020. [DOI] [PubMed] [Google Scholar]
- 34. Thiele EA, Marsh ED, French JA, et al. Cannabidiol in patients with seizures associated with Lennox‐Gastaut syndrome (GWPCARE4): a randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet 2018;391(10125):1085–1096. [DOI] [PubMed] [Google Scholar]
- 35. Devinsky O, Patel AD, Thiele EA, et al. Randomized, dose‐ranging safety trial of cannabidiol in Dravet syndrome. Neurology 2018;90:e1204–e1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Canada H. Authorized Licensed Producers of Cannabis for Medical Purposes ‐ Canada.ca. Canada.ca 2018;[cited 2018 May 2] Available from: https://www.canada.ca/en/health-canada/services/drugs-health-products/medical-use-marijuana/licensed-producers/authorized-licensed-producers-medical-purposes.html
- 37. MacCallum CA, Russo EB. Practical considerations in medical cannabis administration and dosing. Eur J Intern Med 2018;49:12–19. [DOI] [PubMed] [Google Scholar]
- 38. Cairns DR, Sabaz M, Lawson JA, et al. Development, use and planned use of the QOLCE: the quality of life in childhood epilepsy questionnaire. Quality Life Newsletter 2001;2001:17–18. [Google Scholar]
- 39. Sabaz M, Cairns DR, Lawson JA, et al. Validation of a new quality of life measure for children with epilepsy. Epilepsia 2000;41:765–774. [DOI] [PubMed] [Google Scholar]
- 40. Sabaz M, Lawson JA, Cairns DR, et al. Validation of the quality of life in childhood epilepsy questionnaire in American epilepsy patients. Epilepsy Behav 2003;4:680–691. [DOI] [PubMed] [Google Scholar]
- 41. Sparrow S, Balla D, Cicchetti D. Vineland adaptive behavior scales. 2nd ed. Circle Pines, MN: AGS Publ; 2005. [Google Scholar]
- 42. Bloomfield MAP, Ashok AH, Volkow ND, Howes OD. The effects of Δ9‐tetrahydrocannabinol on the dopamine system. Nature 2016;539:369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Geffrey AL, Pollack SF, Bruno PL, Thiele EA. Drug‐drug interaction between clobazam and cannabidiol in children with refractory epilepsy. Epilepsia 2015;56:1246–1251. [DOI] [PubMed] [Google Scholar]
- 44. Gaston T, Bebin E, Cutter G, et al. Interactions between cannabidiol and commonly used antiepileptic drugs. Epilepsia 2017;58:1586–1592. [DOI] [PubMed] [Google Scholar]
- 45. Brisbois TD, de Kock IH, Watanabe SM, et al. Delta‐9‐tetrahydrocannabinol may palliate altered chemosensory perception in cancer patients: results of a randomized, double‐blind, placebo‐controlled pilot trial. Ann Oncol 2011;22:2086–2093. [DOI] [PubMed] [Google Scholar]
- 46. Kirkham TC. Cannabinoids and appetite: food craving and food pleasure. Int. Rev. Psychiatry 2009;21:163–171. [DOI] [PubMed] [Google Scholar]
- 47. Russo EB. Taming THC: potential cannabis synergy and phytocannabinoid‐terpenoid entourage effects. Br J Pharmacol 2011;163:1344–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ryvlin P, Cucherat M, Rheims S. Risk of sudden unexpected death in epilepsy in patients given adjunctive antiepileptic treatment for refractory seizures: a meta‐analysis of placebo‐controlled randomised trials. Lancet Neurol. 2011;10:961–968. [DOI] [PubMed] [Google Scholar]
- 49. Tomson T, Hirsch LJ, Friedman D, et al. Sudden unexpected death in epilepsy in lamotrigine randomized‐controlled trials. Epilepsia 2013;54:135–140. [DOI] [PubMed] [Google Scholar]
- 50. Cooper MS, Mcintosh A, Crompton DE, et al. Mortality in Dravet syndrome. Epilepsy Res 2016;128:43–47. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Inclusion and exclusion criteria for enrollment.
Data S2. Schedule of events.