

Abstract
Objective
To study risk factors for decreasing aβ 1–42 concentrations in cerebrospinal fluid (CSF) in cognitively unimpaired individuals with initially normal amyloid and tau markers, and to investigate whether such aβ1–42 decreases are associated with subsequent decline in cognition and other biomarkers of Alzheimer's disease.
Methods
Cognitively normal subjects (n = 83, 75 ± 5 years, 35(42%) female) with normal CSF aβ 1–42 and tau and repeated CSF sampling were selected from ADNI. Subject level slopes of aβ 1–42 decreases were estimated with mixed models. We tested associations of baseline APP processing markers (BACE1 activity, aβ 1–40, aβ 1–38 and sAPP β) and decreasing aβ 1–42 levels by including an interaction term between time and APP marker. Associations between decreasing aβ 1–42 levels and clinical decline (i.e., progression to mild cognitive impairment or dementia, MMSE, memory functioning) and biological decline (tau, hippocampal volume, glucose processing and amyloid PET) over a time period of 8–10 years were assessed.
Results
Aβ 1–42 levels decreased annually with −4.6 ± 1 pg/mL. Higher baseline BACE1 activity (β(se) = −0.06(0.03), P < 0.05), aβ 1–40 (β(se)= −0.11(.03), P < 0.001), and aβ 1–38 levels (β(se) = −0.11(0.03), P < 0.001) predicted faster decreasing aβ 1–42. The fastest tertile of decreasing aβ 1–42 rates was associated with subsequent pathophysiological processes: 11(14%) subjects developed abnormal amyloid levels after 3 ± 1.7 years, showed increased risk for clinical progression (Hazard Ratio[95CI] = 4.8[1.1–21.0]), decreases in MMSE, glucose metabolism and hippocampal volume, and increased CSF tau and amyloid aggregation on PET (all P < 0.05).
Interpretation
Higher APP processing and fast decreasing aβ 1–42 could be among the earliest, pre‐amyloid, pathological changes in Alzheimer's disease.
Introduction
Alzheimer's disease is a slowly progressive neurodegenerative disorder that is characterized by aggregation of amyloid beta into plaques in the brain. The presence of plaques can be detected by decreased concentrations of amyloid β 1–42 (aβ 1–42) in cerebrospinal fluid (CSF). Cognitively normal individuals without evidence for aggregated amyloid can show decreasing concentrations of aβ 1–42 in CSF over time.1, 2, 3, 4, 5, 6 However, it remains uncertain as to whether such decreases indicate an early, pre‐amyloid stage of Alzheimer's disease. Investigating the risk factors for rate of decrease in aβ 1–42 concentrations, and whether such decreases are associated with subsequent decline in cognition and other biomarkers of Alzheimer's disease over time is critical for understanding the very early pathophysiology of Alzheimer's disease and may enable development of strategies toward prevention of amyloid accumulation.
Aβ 1–42 is produced via amyloidogenic processing of the transmembrane amyloid β precursor protein (APP) through cleavage by beta‐site APP cleaving enzyme 1 (BACE1).7 During this process, other aβ isoforms, aβ 1‐40 and aβ 1‐38, are also produced and these isoforms are less prone to aggregation than aβ 1–42.7, 8 In autosomal‐dominant Alzheimer's disease, it has been shown that several genetic mutations cause increased production of aβ 1–42.9 In sporadic Alzheimer's disease, cross‐sectional studies have reported higher aβ 1‐40 CSF levels with higher amyloid plaque burden as measured with positron emission tomography (PET).10 But, lower aβ 1–42 levels in CSF or amyloid plaques on positron emission tomography (PET) without a clear association with aβ 1‐40 levels have also been reported.11, 12 As such, it is still unclear to what extent increased APP processing is involved in amyloid aggregation in sporadic Alzheimer's disease.
In the present study, we investigated whether decreasing amyloid in cognitively normal individuals with initially normal amyloid and tau markers can be considered an early pathophysiological event in Alzheimer's disease, and to this end we studied both upstream and downstream processes associated with decreasing amyloid. We hypothesized that if increased APP processing is related with amyloid aggregation in Alzheimer's disease, then higher CSF levels of proteins associated with APP processing might be associated with faster aβ 1–42 decreases over time. We further hypothesized that if faster decreasing aβ 1–42 levels consequently initiate downstream pathophysiological processes, then such decreases should be associated with subsequent decline in cognition and other brain markers associated with Alzheimer's disease.
Therefore, we had two objectives: First, to investigate whether higher baseline levels of markers involved in APP processing (i.e., BACE1, sAPPβ, aβ 1–40, aβ 1–38) were associated with faster decreases in aβ 1–42 over time in a sample of cognitively unimpaired older adults with initially normal aβ 1–42 and tau CSF biomarkers. Second, to investigate whether these faster rates of decreasing aβ 1–42 levels subsequently lead to other pathophysiological changes in Alzheimer's disease, by studying relationships between rates of decreasing aβ 1–42 levels with declines in cognition and other established biomarkers for Alzheimer's disease (i.e., amyloid PET, CSF tau, Fludeoxyglucose (FDG) PET, and hippocampal volume).
Methods
Subjects
The data analyzed in this study was obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). We selected 83 subjects from ADNI‐1 and ADNI‐2 who at study enrolment had normal cognition, normal CSF levels of aβ 1–42 (>192 pg/mL) and tau (<93 pg/mL)13 and at least two longitudinal aβ 1–42 measurements available. All subjects had baseline and longitudinal diagnosis, MMSE, and memory delayed recall scores on the logical memory subscale II of the Wechsler Memory Scale, the Alzheimer's Disease Assessment Scale‐Cognitive (ADAS) data and the Ray Auditory Verbal Learning (RAVLT) available, which were used as measures for cognitive functioning. ADNI started in 2003 as a public–private collaboration under the supervision of Principle Investigator Michael W. Weiner, MD. The primary goal of ADNI is to study whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological measures can be combined to measure the progression of mild cognitive impairment and early Alzheimer's disease. For up‐to‐date information, please see http://www.adni-info.org. The institutional review boards of all participating institutions approved the procedures for this study. Written informed consent was obtained from all participants or surrogates.
CSF
CSF samples were obtained and analyzed as previously described by Toledo et al. [1] Briefly, aβ 1–42 and tau concentrations were measured with the multiplex xMAP luminex platform (Luminex Corp, Austin, TX) with the INNOBIA AlzBio3 kit (Innogenetics, Ghent, Belgium) at the ADNI Biomarker Core laboratory at the University of Pennsylvania Medical Center. Longitudinal measurements for each participant were obtained from the same batch. ADNI 1 subjects (n = 53, 63%) had at baseline measurements of proteins associated with amyloidogenic APP processing (BACE1, sAPPβ, aβ 1–40 and aβ 1–38). Secreted APP‐β (sAPPβ) concentrations were based on Luminescence counts as measured with LJL‐ Analyst (Molecular Devices, Sunyvale, CA).14 BACE1 enzymatic activity was measured with a two‐step assay as described previously.15 Aβ 1–40 and aβ 1–38 concentrations were measured with 2D‐UPLC‐tandem mass spectometry as previously described.16 More detailed information on protein assessment and quality control is described at http://adni.loni.ucla.edu.
MRI and PET biomarkers
Hippocampal volumes from the adnimerge file were precomputed parcellations from the automated subcortical segmentation atlas in FreeSurfer 4.4 (surfer.nmr.mgh.hardvard.edu)17 using an unbiased within‐subject and average template to align scans from different time points.18 Hippocampal volumes below 6732 mm3 were considered abnormal. Cortical amyloid aggregation was measured with 18F‐AV‐45 (AV45) PET and glucose metabolism with F18‐fluorodeoxyglucose (FDG) PET. Both PET images were coregistered to subject's time‐aligned structural MRIs. For AV45, standard Uptake Values ratios (SUVr) were calculated by dividing the average signal in each of 68 anatomical regions (defined by the Desikan‐Killiany atlas using Freesurfer 5.3.0.) by the average uptake in the cerebellum. FDG SUVr was calculated by dividing the average uptake in five anatomical regions (posterior cingulate, and bilateral temporal and angular gyri) by uptake of the cerebellar vermis and pons. The cut‐point for abnormality was defined as >1.1 for AV45,19 and as <1.21 for FDG PET.20 Repeated FDG PET was available for 65 (78%) subjects, repeated hippocampal volume measures were available for 81 (98%) subjects, and repeated amyloid PET was available for 51 (61%) subjects.
Statistical analyses
For our first objective, we tested whether upstream APP processing markers (aβ 1–40, aβ 1–38, sAPPβ, BACE1) were associated with baseline and decreases over time in aβ 1–42 levels (outcome variable) with linear mixed models that included the main effects time and APP processing markers, and an interaction term of time X APP processing marker, adjusting for age and gender. For our second objective, we tested whether subject‐specific rates in decreasing aβ 1–42 levels over time were associated with subsequent downstream declines in cognitive and biological markers (i.e., CSF tau, amyloid and FDG PET and hippocampal volume). To this end, we first estimated subject‐specific slopes of aβ 1–42 levels over time using a linear mixed model with random slopes and intercepts and time as main effect. Cox proportional Hazard models were used to assess whether faster decreases in aβ 1–42 (predictor variable, continuous subject‐specific slopes) were associated with time to clinical progression to mild cognitive impairment or dementia (outcome variable), adjusting for age and gender. We repeated Cox proportional Hazard models including baseline tau levels (model 2), and baseline MMSE scores (model 3) as additional predictor variables. We further tested whether subject aβ 1–42 slopes were associated with decline in MMSE, delayed recall memory scores and biomarkers (outcome variables) using linear mixed models with main effects time and subject‐specific aβ 1–42 slopes, and the interaction between time X subject slopes. Linear mixed models were adjusted for age and gender, and cognitive measures additionally corrected for years of education. GAMM models were used to test whether a nonlinear or a linear association best described the data: in case of significance nonlinear relationships were defined as having an estimated degrees of freedom to describe an association of ≥2 (i.e., at least quadratic) are reported and linear estimates otherwise. In order to aid interpretation of the results, we repeated analyses taking tertiles of aβ 1–42 slopes as predictor variable. These groups were also compared on baseline clinical and biological characteristics with linear regression, Kruskal–Wallis, or Chi2 tests when appropriate. The R package “emmeans” was used to obtain estimated marginalized mean and slope estimates. All statistical analyses were performed in R (version 3.4.2 “short summer”).
Results
Sample characteristics
Included subjects with normal cognition, normal amyloid and tau CSF levels and longitudinal CSF available were on average 75 years old, 42% female, had an average MMSE score of 29 and the majority of subjects did not carry an APOE ε4 allele (Table 1). Twelve subjects showed clinical progression to MCI (10, 12%) or dementia (2, 2%).
Table 1.
Sample characteristics of individuals with intact cognition and initially normal CSF amyloid and tau biomarkers
| Characteristic | Total sample (n = 83) | Rates of decreasing aβ 1–42 in tertiles | ||
|---|---|---|---|---|
| Slowest tertile (n = 28) | Intermediate tertile (n = 27) | Fastest tertile (n = 28) | ||
| Age, mean (SD) | 75 (5.4) | 75 (6.2) | 75 (5.5) | 75 (4.7) |
| Female, N (%) | 35 (42%) | 10 (36%) | 13 (48%) | 12 (43%) |
| Years of Education, mean (SD) | 16 (3) | 16 (3) | 16 (3) | 17 (3) |
| MMSE, median (IQR) | 29 (29–30) | 29 (29–30) | 29 (29–30) | 30 (29–30) |
| Logical memory delayed recall, mean (SD) | 13 (3.5) | 12 (3.3) | 13 (3.6) | 14 (3.6) |
| APOE e4, N (%) | 6 (7%) | 2 (7%) | 3 (11%) | 1 (4%) |
| Clinical progression, N (%) | 12 (14%) | 3 (10.71%) | 3 (11.11%) | 6 (21%) |
| Cognitive status at last visit | ||||
| MCI, N (%) | 10 (12%) | 3 (11%) | 3 (11%) | 4 (14%) |
| Dementia, N (%) | 2 (2%) | 0 (0%) | 0 (0%) | 2 (7%) |
| Aβ 1–42 pg/mL, mean (SD) | 243 (28) | 250 (28) | 248 (23) | 230 (28)b , c |
| Max, min annual change in aβ 1–42 pg/mL (i.e., estimated subject slopes) | n.a. | −0.9, −3.9 | −3.91, −4.9a , f | −5, −14.5b , c , f |
| Aβ 1–40 pg/mL, mean (SD) (n = 53) | 7961 (2407) | 7513 (2214) | 7992 (2251) | 8661 (2856) |
| Aβ 1–38 pg/mL, mean (SD) (n = 53) | 1867 (584) | 1766 (558) | 1826 (455) | 2078 (728) |
| BACE1 pg/mL, mean (SD) (n = 51) | 43 (15) | 40.82 (13.01) | 40.44 (10.9) | 49.85 (20) |
| sAPPβ pg/mL, mean (SD) (n = 51) | 4081 (1386) | 3535 (1083) | 4483 (1567)a | 4510 (1388)a |
| Total tau pg/mL, mean (SD) | 57 (15) | 57 (14) | 57 (13) | 58 (17) |
| PET AV45 SUVR, mean (SD) (n = 28 at baseline) | 1.01 (0.06) | 1 (0.04) | 1.01 (0.08) | 1.02 (0.04) |
| AV45 > 1.1, N (%) | 1 (4%) | 0 (0%) | 1 (10%) | 0 (0%) |
| PET FDG, mean (SD) (n = 47 at baseline) | 6.6 (0.6) | 6.5 (0.7) | 6.7 (0.6) | 6.6 (0.6) |
| Hippocampal volume mm3, mean (SD) (n = 75 at baseline) | 7370 (791) | 7536 (800) | 7343 (839) | 7221 (735) |
| Number of CSF samples, median (IQR) | 2 (2–3) | 3 (2–3)a | 2 (2–2) | 2 (2–3) |
| Years between CSF samples median (IQR) | 4 (3–8) | 1.02 (0.99–1.97) | 1.00 (0.99–1.03)a | 1.02 (0.99–1.06) |
| Follow‐up years, median (IQR)[Link] | 4 (3–8) | 7 (4–9) | 4 (3–7)a | 4 (3–9)b |
MMSE is mini‐mental state examination, APOE, Apolipoprotein E; MCI, mild cognitive impairment; PET, positron emission tomography; FDG, Fludeoxyglucose (18F); CSF, cerebrospinal fluid. Groups were based on tertiles of subject‐specific rates of decreasing aβ 1–42 over time. 1Total follow‐up duration was determined as the time between the first assessment and the last time that any of the tested biomarkers was available. ais difference between slow and intermediate groups with P < 0.05; bis difference between slow and fast groups with P < 0.05; cis difference between intermediate and fast groups with P < 0.05; fis different from 0 with P < 0.05.
Rate of decreasing aβ1–42 CSF levels
Across the total group, aβ 1–42 CSF levels decreased with 4.6 ± 1.02 pg/mL annually (P < 0.001). Individuals were grouped according to the lowest, intermediate and highest tertiles of subject‐specific slope values. Across tertiles, subjects had a similar age, gender distribution, level of education and baseline MMSE scores (Table 1). Baseline aβ 1–42 levels were lower in fast decreasing group compared to the other groups.
Upstream processes from amyloid aggregation
Linear mixed models showed no effects of APP processing‐related protein levels with baseline aβ 1–42 levels (Table 2). Steeper annual decreases in aβ 1–42 concentrations were associated with higher baseline levels of aβ 1–40, aβ 1–38 and BACE1 (all P < 0.05; Table 2), and with higher sAPPβ levels at trend level (P = 0.08).
Table 2.
Baseline and annual change effects for amyloid precursor protein (APP) processing markers measured in cerebrospinal fluid (CSF) as predictor and aβ 1–42 CSF levels as outcome variable
| Baseline aβ 1–42 | Annual change aβ 1–42 | |
|---|---|---|
| Predictor variable | β (SE) | β (SE) |
| Aβ 1–40 | 0.19 (0.11) | −0.11 (0.03b |
| Aβ 1–38 | 0.17 (0.11) | −0.11 (0.03)b |
| BACE1 | −0.04 (0.12) | −0.06 (0.03)a |
| sAPPβ | −0.03 (0.12) | −0.05 (0.03) |
All variables were Z‐transformed in order to aid comparability of β estimates. β's from interaction effects with time are interpreted as follows: one standard deviation increase in predictor variable is associated with a standard deviation increase in slope of annual change in amyloid β 1–42 values. All analyses were adjusted for age and gender. ais P < 0.05; bis P < 0.0001.
Downstream consequences of aggregating amyloid
Next, we studied whether initial decreases in aβ 1–42 were followed by decline in cognitive functioning and other biomarkers over time. Cox proportional Hazard models showed that faster rates of decreasing aβ 1–42 levels predicted clinical progression to MCI or dementia (Table 3), which seemed to be specific for the fast tertile of decreasing aβ 1–42 subjects who had an almost fivefold increased risk to clinically progress to MCI or dementia during a median (IQR) follow‐up time of 4 (2.3–7.0) years (Fig. 1A; Table 3). Results remained similar after additionally adjusting for baseline tau levels, and MMSE scores. In those models, higher but still normal baseline tau levels were also associated with clinical progression. Subjects with fast decreasing aβ 1–42 also showed decline in MMSE and in delayed recall memory scores on the Wechsler's Memory and ADAS scales (Table), and tended to decline on the RAVTL delayed memory test, but this was not significant (Fig. 1B‐E). Subjects in the slow tertile showed improvement in logical delayed recall memory scores over time, and similar but not significant trends on the other memory tests (Fig. 1C).
Table 3.
Hazard ratios (95% confidence interval) for clinical progression to mild cognitive impairment or dementia
| Predictor | Model 1 | P value | Model 2 | P value | Model 3 | P value |
|---|---|---|---|---|---|---|
| Aβ 1–42 slope continuous | 0.81 (0.67, 0.99) | 0.037 | 0.81 (0.67, 0.99) | 0.0497 | 0.82 (0.66, 1.01) | 0.068 |
| Aβ 1–42 slope categorical in tertiles (n progressing/total n) | ||||||
| Slow (3/28) | Reference | Reference | Reference | |||
| Intermediate (3/27) | 2.1 (0.4, 10.4) | 0.388 | 2.6 (0.4, 13.9) | 0.275 | 2.6 (0.5, 13.9) | 0.274 |
| Fast (3/27) | 4.8 (1.1, 21.0) | 0.037 | 8.0 (1.1, 43.0) | 0.016 | 7.8 (1.4, 43.1) | 0.019 |
| Baseline tau levels | n.e. | n.e. | 1.1 (1.0, 1.1) | 0.014 | 1.1 (1.0, 1.1) | 0.014 |
| Baseline MMSE | n.e. | n.e. | n.e. | n.e. | 1.04 (0.6, 1.7) | 0.877 |
MMSE is mini‐mental state examination, n.e. is not estimated. Model 1 included as predictor aβ 1–42 slope either continuously or as categorical variable, Model 2 is Model 1 + baseline tau CSF levels as continuous variable, Model 3 is Model 2 + MMSE as continuous variable. All models are adjusted for age and gender.
Figure 1.
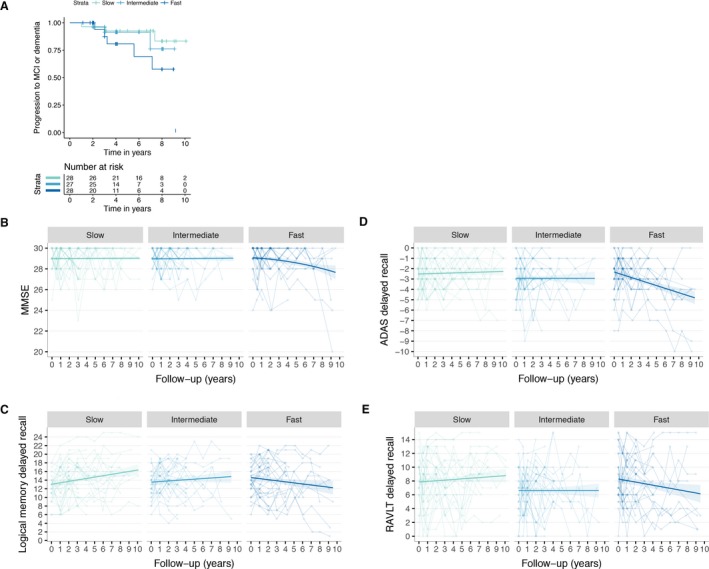
(A) Kaplan–Meier for time to clinical progression to mild cognitive impairment or dementia according to rate of decreasing Aβ 1–42 levels (fast, intermediate or slow). Changes over time on: MMSE (B); Logical Memory Delayed Recall (C); ADAS‐cog delayed recall (D; please note that this measure was inverted so for all cognitive tests lower scores indicate worse function); and RAVLT delayed recall.
We further investigated which downstream biological processes were associated with decreasing aβ 1–42 (Fig. 2; Table 4). Figure 2A illustrates combined biomarker trajectories after they were scaled (and inverted for amyloid PET and tau) such that all positive values indicate normal ranges and negative values abnormal ranges. This figure shows that subjects with fast decreasing aβ 1–42, first developed abnormal CSF aβ 1–42 levels, after which about a year later amyloid aggregation and hippocampal atrophy reached abnormal values, and about 3 years later glucose hypometabolism on PET reached abnormality. Only subjects with fast decreasing aβ 1–42 showed amyloid aggregation on amyloid PET, and decreases on FDG PET (Figures 2C and D). Increases in CSF tau levels were also observed in subjects with intermediate decreasing aβ 1–42 (Fig. 2E). Hippocampal volume decreased independently of the rate of decreasing aβ 1–42 levels (Fig. 2F).
Figure 2.

(A) Trajectories of biomarkers and MMSE combined from normal (positive values) to abnormal (negative values). Amyloid PET (AV45) and tau were inverted, all variables were Z‐transformed according to baseline levels and centered according to marker specific cut‐points such that 0 indicates threshold for abnormality for all markers (see next descriptions for cut‐points biomarkers, for MMSE a score below 26 was considered abnormal). (B) CSF aβ 1–42 changes over time (dotted line indicates cut‐point of 192); (C) Amyloid PET standardized uptake value ratio (AV45 SUVr) over time (dotted line indicates cut‐point of 1.1); (D) FDG PET SUVr changes over time (dotted line indicates cut‐point of 1.21); (E) Tau CSF changes over time (dotted line indicates cut‐point of 93); (F) Hippocampal volume changes over time (dotted line indicates cut‐point of 6732). All plots are stratified for intermediate and fast rates of aβ 1–42 cerebrospinal fluid (CSF) decreasing levels over time.
Table 4.
Baseline and annual change effects of outcome markers with rates of decreasing aβ 1–42 as continuous and as categorical predictors
| Outcome variable | Aβ 1–42 slopes as continuous predictor | Aβ 1–42 slopes as categorical predictor (in tertiles) | ||||||
|---|---|---|---|---|---|---|---|---|
| Baseline | Annual change | Baseline | Annual change | |||||
| Continuous | Continuous | Slow | Intermediate | Fast | Slow | Intermediate | Fast | |
| MMSE | −0.02 (0.05) | 0.01 (0.01) | 29 (0.2) | 29 (0.2) | 29 (0.2) | −0.01 (0.04) | −0.05 (0.05) | −0.11 (0.05)a , e |
| Logical memory delayed recall | −0.25 (0.18) | 0.08 (0.03)b | 14 (0.6) | 14.1 (0.7) | 14 (0.7) | 0.34 (0.11)c | 0.11 (0.14) | −0.02 (0.13)d |
| ADAS delayed recall1 | −0.02 (0.07) | 0.04 (0.01)b | −2.3 (0.3) | −2.9 (0.3) | −2.9 (0.3) | 0.02 (0.05) | 0.05 (0.07) | −0.13 (0.06)a |
| RAVLT delayed recall | 0.02 (0.16) | 0.02 (0.02) | 8.1 (0.6) | 6.7 (0.6) | 7.8 (0.6) | −0.01 (0.09) | 0 (0.11) | −0.11 (0.1) |
| PET AV45 SUVr | −0.01 (0.006)a | −0.002 (0.001)a | 0.96 (0.02) | 0.99 (0.02) | 1.02 (0.01)d | 0.002 (0.004) | 0.002 (0.004) | 0.011 (0.003)c , e |
| PET FDG SUVr | −0.002 (0.008) | 0.003 (0.001)b | 1.3 (0.03) | 1.3 (0.03) | 1.3 (0.03) | 0.005 (0.003) | −0.002 (0.004) | −0.012 (0.006)a , d |
| Tau pg/mL | −1.6 (0.9) | 0.06 (0.15) | 58 (3) | 56 (3) | 58 (3) | 0.6 (0.58) | 2.4 (1.2)a | 1.5 (0.6)a |
| Hippocampal volume mm3 | 24 (43) | 3.9 (2.8) | 7715 (151) | 7422 (167) | 7462 (160) | −75 (10)c | −72 (12)c | −97 (12)c |
Unstandardized beta's (SE) are reported. MMSE is mini‐mental state examination, PET is positron emission tomography, AV45 is florbetapir, CSF is cerebrospinal fluid, FDG is Fludeoxyglucose. Cognitive markers were adjusted for age, gender, and years of education. Biomarkers were adjusted for age and gender. ais P < 0.05; bis P < 0.01; cis P < 0.001; dis P < 0.05 compared to reference group; eis P < 0.05 nonlinear relationship (linear beta reported to aid comparison with other groups). 1ADAS delayed recall scores were reversed, so that for all clinical measures lower values indicate worse memory functioning.
Discussion
This study in cognitively unimpaired older individuals shows that a pre‐amyloid stage can be identified during which initially normal aβ 1–42 levels start aggregating. Upstream from amyloid aggregation, higher baseline CSF levels of BACE1 activity, aβ 1–40 and aβ 1–38 were associated with subsequent faster decreases in aβ 1–42 levels, suggesting that increased levels of proteins associated with APP processing might play a role in the initial phases of amyloid aggregation in Alzheimer's disease. Furthermore, our results suggest that fast decreasing aβ 1–42 CSF concentrations initiate downstream pathophysiological processes that are known to be involved in AD, as fast decreasing aβ 1–42 was associated with subsequent clinical and cognitive decline, as well as amyloid aggregation on PET, glucose hypometabolism, increase in CSF tau and hippocampal atrophy. Together our findings suggest that initially higher levels of APP processing‐associated proteins and subsequent decreasing aβ 1–42 levels indicate the earliest, pre‐amyloid, pathophysiological stage in Alzheimer's disease.
Aβ 1–42 is produced when APP is initially cleaved by BACE1.7 Alzheimer's disease causing genetic mutations in APP, and also in PSEN1 and PSEN2, can promote aβ 1–42 production, the isoform that is particularly prone to aggregation.9, 21, 22, 23 So far, it has been challenging to study the involvement of APP processing in the initial stages of aβ 1–42 aggregation in sporadic Alzheimer's disease, because the onset of amyloid aggregation is difficult to estimate. The results from our longitudinal analyses suggest that increased APP processing may also play a role in sporadic Alzheimer's disease, as higher levels of BACE1, aβ 1–40 and aβ 1–38 were associated with subsequent faster decreases in aβ 1–42. These findings converge with observations in autosomal‐dominant AD of higher aβ 1–40 levels in mutation carriers compared to controls 15 years before estimated year of symptom onset, which was followed by decreases in aβ 1–42 at 10 years before estimated symptom onset.24 A recent rodent study suggested that BACE1 inhibition strategies might be most effective during the initial phase of plaque formation.25 Our results indicate that it might be possible to identify individuals who are in such a pre‐amyloid stage of the disease, when they have higher levels of APP markers, in combination with fast decreasing aβ 1–42 levels. However, these APP‐associated protein levels may change over time in a nonlinear way,24, 26 and so more longitudinal studies are required to study whether it is possible to develop approaches for identifying subjects at risk for incipient amyloidosis based on these markers. Another question that remains is what biological process causes APP processing to increase. One explanation is that APP processing increases with aging, as a cross‐sectional study observed higher BACE1 levels with older age,15 which may explain increasing prevalences of amyloid abnormality for older ages.27 Increased APP processing has also been associated with higher neuronal activation in rodent models, with highly active brain areas showing increased vulnerability for amyloid plaque formation.28, 29, 30, 31 Support for the involvement of a similar mechanism in human beings has recently been reported, with brain areas showing relatively higher activation during nontask conditions were also among the regions showing the first signs of amyloid aggregation as measured with amyloid PET.32
Most individuals with normal amyloid and tau CSF markers remain cognitively stable,33, 34 but still some of these individuals show cognitive decline. It is this subset of individuals that provides the opportunity to study the earliest pathogenic changes involved in Alzheimer's disease. Previously, we showed that low amyloid concentrations within the normal range were associated with clinical progression in nondemented individuals, suggesting that a dynamical range exists during which amyloid starts to decrease.35 We now further extend on those findings and show that faster decreasing aβ 1–42 of initially normal levels are associated with subsequent downstream declines in clinical, cognitive, and biological markers for Alzheimer's disease in cognitively normal individuals. Previous studies have demonstrated that normal aβ 1–42 CSF levels in nondemented individuals can decrease below the cut‐point for abnormality over time.1, 2, 3, 4, 5, 6 However, those previous studies have reported conflicting results as to whether decreases in aβ 1–42 levels are associated with clinical progression, as one study did not observe an association with decline on cognitive test scores when grouping subjects according to whether or not subjects developed abnormal aβ 1–42 levels within 3 years,4 while another study observed a trend for decline on MMSE scores when subjects were grouped according to the median slope.5 With our approach, we compared subjects who had initially both normal amyloid and tau biomarkers based on whether they showed slow, intermediate or fast decreasing aβ 1–42 levels, and this enabled us to show that fast decreasing aβ 1–42 levels were associated with clinical progression to mild cognitive impairment or dementia, and showed faster decline on the MMSE and on neuropsychological memory tests that measure delayed recall, which is sensitive to AD‐related cognitive decline. This suggests that decreasing aβ 1–42 levels trigger other downstream pathophysiological processes involved in Alzheimer's disease. Still, the close correspondence in time for amyloid to become abnormal and clinical progression may seem to be at odds with earlier observations in autosomal dominant Alzheimer's disease that aβ 1–42 levels plateau many years before the onset of dementia.24, 36, 37It must be noted that of the 12 subjects showing clinical progression, 10 progressed to MCI and only two progressed to dementia, and so it might be that the rate of amyloid decreases plateaus in the MCI stage.38, 39 Another possibility is that in late onset AD the pathophysiological cascade may take less time to unfold than in early onset AD, as the aging brain might be more vulnerable for pathophysiological changes.
Subjects with fast decreasing aβ 1–42 showed amyloid aggregation on PET about a year later. The observation that amyloid in CSF may become abnormal before PET is in line with previous reports.12, 24, 32, 40, 41 An implication of these results is that for the earliest disease stages aβ 1–42 CSF might be more useful than amyloid PET to serve as an outcome measure for trials testing drugs that target aβ 1–42 production. However, it must be noted that although the concordance between CSF and PET measures for abnormal amyloid is often high,42 it is also possible for individuals to have an abnormal amyloid PET scan and normal amyloid CSF.42, 43 In some cases, CSF amyloid may decrease at a later point in time,13 indicating that not all individuals seem to follow the same temporal trajectories of CSF and PET changes. Such discrepancies may reflect differences between these modalities in terms of sensitivity and/or specificity to detect certain pathological aspects of amyloid plaques among individuals. Brain glucose metabolism only decreased in subjects with fast decreasing aβ 1–42. Tau levels increased in subjects showing intermediate and fast decreasing aβ 1–42 levels, suggesting that the process of amyloid aggregation and tau increase might be coupled together in time, which has also been observed in autosomal dominant AD.24 Potentially, decreasing aβ 1–42 levels reflect the presence of toxic aβ 1–42 oligomers that can trigger tau pathology.21, 44 However, no continuous correlation between tau levels and continuous aβ 1–42 slopes was observed, and so alternatively these processes might be independent from each other. We further observed that hippocampal volumes reduced for all subjects, independently of the rates of decreasing aβ 1–42. This suggests that reductions in hippocampal volumes over time can in part reflect normal aging processes, or neurodegenerative processes unrelated to Alzheimer's disease. As such hippocampal atrophy, although closely linked to the disease and to memory loss, may not necessarily be specific for Alzheimer's disease.33, 45 At this point, research schemes for disease staging allow use CSF tau levels, glucose metabolism or hippocampal atrophy to indicate neuronal injury.46 The present observations suggest that these markers reflect different aspects of neuronal injury, and so these markers should not be used interchangeably.
The majority of subjects that showed decreasing aβ 1–42 levels did not carry an APOE ε4 allele, which was unexpected as APOE ε4 is the major genetic risk factor for Alzheimer's disease. The relatively large proportion of subjects lacking this allele might be explained by their average age of 75 years, which is an age when about 22% in noncarriers are expected to have abnormal amyloid versus 60% APOE ε4 carriers.27 Consequently, it is unclear to what extent our findings can be generalized to APOE ε4 allele carriers. One study reported higher aβ 1‐40 levels with older age in cognitively normal noncarriers, but observed no association with age in carriers.12 Another study reported that higher levels of aβ 1–‐40 correlated with higher plaque burden in carriers, although of weaker strength than in noncarriers.10 Although, our results highlight the importance of repeated CSF sampling over a long period of time to improve our understanding of the pathophysiological mechanisms leading to Alzheimer's disease, more longitudinal studies are required to study this question in younger participants, when the proportion of APOE ε4 carriers with normal amyloid is expected to be larger.12, 27
A limitation of the present study is that even though this sample of individuals with repeated CSF and detailed follow‐up information over a period spanning 10 years is currently one of the largest samples available, due to the average age and the present inclusion criterion of normal amyloid the proportion of individuals who showed clinical decline was small. Another potential limitation is that there were no repeated measurements available for APP markers, and so it remains unclear whether those markers change over time. Furthermore, amyloid aggregation on PET was analyzed using SUVr values, which might not be the most optimal approach to reliably detect (small) changes over time.47 In addition, the slow and intermediate tertiles of decreasing aβ 1–42 showed annual changes that were similar to measuring variability. Since those groups showed no or little decline in cognition and other biomarkers, it is conceivable amyloid levels will remain largely normal in those individuals. However, subjects in the fast tertile of decreasing aβ 1–42 showed consistent, subsequent clinical and cognitive decline, as well as decline in other biomarkers, suggesting that these individuals are in the earliest stage of Alzheimer's disease.
In conclusion, we observed in cognitively normal subjects with initial normal CSF biomarkers that higher levels of APP processing seem to be at the start of a pre‐amyloid stage of Alzheimer's disease, during which aβ 1–42 concentrations show rapid decreases over time. Trials that target amyloid production are currently on going. Although these results require replication in larger samples, an implication is that CSF aβ 1–42 might have use as an outcome marker in trials targeting amyloid beta when levels are still normal, and that such therapeutic strategies might be of benefit for this population.
Author Contributions
Study concept and design: B.M.T., P.J.V.; Analysis of the data: B.M.T.; Drafting the manuscript: B.M.T.; Critical revision of the manuscript: L.V, M.D.Z, A.C.H., W.M.F, C.T., P.S., P.J.V.
Conflict of Interest
Nothing to report.
Acknowledgments
This work has been supported by ZonMW Memorabel grant programme #73305056 (BMT) and #733050824 (BMT and PJV).
ADNI acknowledgements: Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (http://www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Funding Information
This work has been supported by ZonMW Memorabel grant programme #73305056 (BMT) and #733050824 (BMT and PJV). Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada.
Funding Statement
This work was funded by ZonMw grants 73305056 and 733050824; Alzheimer's Disease Neuroimaging Initiative grant ; National Institutes of Health grant U01 AG024904; DOD ADNI grant W81XWH‐12‐2‐0012; National Institute on Aging grant ; National Institute of Biomedical Imaging and Bioengineering grant ; The Canadian Institutes of Health Research grant .
References
- 1. Toledo JB, Xie SX, Trojanowski JQ, Shaw LM. Longitudinal change in CSF Tau and Aβ biomarkers for up to 48 months in ADNI. Acta Neuropathol 2013;126:659–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Landau SM, Lu M, Joshi AD, et al. Comparing positron emission tomography imaging and cerebrospinal fluid measurements of β‐amyloid. Ann Neurol 2013;74:826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ower AK, Hadjichrysanthou C, Gras L, et al. Temporal association patterns and dynamics of amyloid‐ß and tau in Alzheimer's disease. Eur J Epidemiol 2017;1–10. 10.1007/s1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mattsson N, Insel PS, Donohue M, et al. Predicting reduction of cerebrospinal fluid β‐amyloid 42 in cognitively healthy controls. JAMA Neurol 2015;72:554–560. [DOI] [PubMed] [Google Scholar]
- 5. Gomar JJ, Conejero‐Goldberg C, Davies P, et al. Anti‐correlated cerebrospinal fluid biomarker trajectories in preclinical Alzheimer's disease. J Alzheimers Dis 2016;51:1085–1097. [DOI] [PubMed] [Google Scholar]
- 6. Bertens D, Tijms BM, Vermunt L, et al. The effect of diagnostic criteria on outcome measures in preclinical and prodromal Alzheimer's disease: implications for trial design. Alzheimer's Dement 2017;3:513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vassar R, Bennett BD, Babu‐Khan S, et al. β‐Secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999;286:735–741. [DOI] [PubMed] [Google Scholar]
- 8. Portelius E, Price E, Brinkmalm G, et al. A novel pathway for amyloid precursor protein processing. Neurobiol Aging 2011;32:1090–1098. [DOI] [PubMed] [Google Scholar]
- 9. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. 2002;297:353–356. [DOI] [PubMed] [Google Scholar]
- 10. Mattsson N, Insel PS, Palmqvist S, et al. Increased amyloidogenic APP processing in APOE ɛ4‐negative individuals with cerebral β‐amyloidosis. Nat Commun 2016;7:10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fagan AM, Mintun MA, Shah AR, et al. Cerebrospinal fluid tau and ptau 181increase with cortical amyloid deposition in cognitively normal individuals: implications for future clinical trials of Alzheimer's disease. EMBO Mol Med 2009;1:371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sutphen CL, Jasielec MS, Shah AR, et al. Longitudinal cerebrospinal fluid biomarker changes in preclinical Alzheimer Disease during middle age. JAMA Neurol 2015;72:1029–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shaw LM, Vanderstichele H, Knapik‐Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 2009;65:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wu G, Sankaranarayanan S, Hsieh SHK, et al. Decrease in brain soluble amyloid precursor protein β (sAPPβ) in Alzheimer's disease cortex. J Neurosci Res 2011;89:822–832. [DOI] [PubMed] [Google Scholar]
- 15. Wu G, Sankaranarayanan S, Tugusheva K, et al. Decrease in age‐adjusted cerebrospinal fluid β‐secretase activity in Alzheimer's subjects. Clin Biochem 2008;41:986–996. [DOI] [PubMed] [Google Scholar]
- 16. Korecka M, Waligórska T, Figurski M, et al. Qualification of a surrogate matrix‐based absolute quantification method for amyloid‐β₄₂ in human cerebrospinal fluid using 2D UPLC‐tandem mass spectrometry. J Alzheimers Dis 2014;41:441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. FreeSurfer Fischl B. NeuroImage 2012;62:774–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reuter M, Schmansky NJ, Rosas HD, Fischl B. Within‐subject template estimation for unbiased longitudinal image analysis. NeuroImage 2012;61:1402–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Landau SM, Breault C, Joshi AD, et al. Amyloid‐ imaging with pittsburgh compound b and florbetapir: comparing radiotracers and quantification methods. J Nucl Med 2013;54:70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Landau SM, Harvey D, Madison CM, et al. Associations between cognitive, functional, and FDG‐PET measures of decline in AD and MCI. Neurobiol Aging 2011;32:1207–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. 1992;256:184–185. [DOI] [PubMed] [Google Scholar]
- 22. Gomez‐Isla T, Growdon WB, McNamara MJ, et al. The impact of different presenilin 1 andpresenilin 2 mutations on amyloid deposition, neurofibrillary changes and neuronal loss in the familial Alzheimer's disease brain. Brain 1999;122:1709–1719. [DOI] [PubMed] [Google Scholar]
- 23. Murayama O, Tomita T, Nihonmatsu N, et al. Enhancement of amyloid beta 42 secretion by 28 different presenilin 1 mutations of familial Alzheimer's disease. Neurosci Lett 1999;265:61–63. [DOI] [PubMed] [Google Scholar]
- 24. Fagan AM, Xiong C, Jasielec MS, et al. Longitudinal change in CSF biomarkers in autosomal‐dominant Alzheimer's disease. Sci Transl Med 2014;6:226ra30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peters F, Salihoglu H, Rodrigues E, et al. BACE1 inhibition more effectively suppresses initiation than progression of β‐amyloid pathology. Acta Neuropathol 2018;135:695–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. De Leon MJ, Pirraglia E, Osorio RS, et al. The nonlinear relationship between cerebrospinal fluid Aβ42 and tau in preclinical Alzheimer's disease. PLoS ONE 2018;13: e0191240–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jansen WJ, Ossenkoppele R, Knol DL, et al. Prevalence of cerebral amyloid pathology in persons without dementia. JAMA 2015;313:1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bero AW, Yan P, Roh JH, et al. Neuronal activity regulates the regional vulnerability to amyloid‐β deposition. Nat Neurosci 2011;14:750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cirrito JR, Yamada KA, Finn MB, et al. Synaptic activity regulates interstitial fluid amyloid‐β levels in vivo. Neuron 2005;48:913–922. [DOI] [PubMed] [Google Scholar]
- 30. Bero AW, Bauer AQ, Stewart FR, et al. Bidirectional relationship between functional connectivity and amyloid‐ deposition in mouse brain. J Neurosci 2012;32:4334–4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Selkoe DJ. Alzheimer's disease is a synaptic failure. Science 2002;298:789–791. [DOI] [PubMed] [Google Scholar]
- 32. Palmqvist S, Schöll M, Strandberg O, et al. Earliest accumulation of β‐amyloid occurs within the default‐mode network and concurrently affects brain connectivity. Nat Commun 2017;8:1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Donohue MC, Sperling RA, Petersen R, et al. Association between elevated brain amyloid and subsequent cognitive decline among cognitively normal persons. JAMA 2017;317:2305–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vos SJB, Gordon BA, Su Y, et al. NIA‐AA staging of preclinical Alzheimer disease: discordance and concordance of CSF and imaging biomarkers. Neurobiol Aging 2016;44:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tijms BM, Bertens D, Slot RE, et al. Low normal cerebrospinal fluid Aβ42 levels predict clinical progression in nondemented subjects. Ann Neurol 2017;81:749–753. [DOI] [PubMed] [Google Scholar]
- 36. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bateman RJ, Xiong C, Benzinger TLS, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med 2012;367:795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Buchhave P, Minthon L, Zetterberg H, et al. Cerebrospinal fluid levels of β‐amyloid 1–42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch Gen Psychiatry 2012;69:98–106. [DOI] [PubMed] [Google Scholar]
- 39. Bertens D, Knol DL, Scheltens P, et al. Temporal evolution of biomarkers and cognitive markers in the asymptomatic, MCI, and dementia stage of Alzheimer's disease. Alzheimers Dement 2015;11:511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Palmqvist S, Mattsson N, Hansson O. Alzheimer's disease neuroimaging initiative. Cerebrospinal fluid analysis detects cerebral amyloid‐β accumulation earlier than positron emission tomography. Brain 2016;139(Pt 4):1226–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vlassenko AG, McCue L, Jasielec MS, et al. Imaging and cerebrospinal fluid biomarkers in early preclinical alzheimer disease. Ann Neurol 2016;80:379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zwan M, van Harten A, Ossenkoppele R, et al. Concordance between cerebrospinal fluid biomarkers and [11C]PIB PET in a memory clinic cohort. J Alzheimers Dis 2014;41:801–807. [DOI] [PubMed] [Google Scholar]
- 43. Forsberg A, Almkvist O, Engler H, et al. High PIB retention in Alzheimers disease is an early event with complex relationship with csf biomarkers and functional parameters. Curr Alzheimer Res 2010;7:56–66. [DOI] [PubMed] [Google Scholar]
- 44. Oddo S, Caccamo A, Tran L, et al. Temporal profile of amyloid‐beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J Biol Chem 2006;281:1599–1604. [DOI] [PubMed] [Google Scholar]
- 45. ten Kate M, Barkhof F, Visser PJ, et al. Amyloid‐independent atrophy patterns predict time to progression to dementia in mild cognitive impairment. Alzheimer's Res Ther 2017;9:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jack CR, Bennett DA, Blennow K, et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016;87:539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. van Berckel BNM, Ossenkoppele R, Tolboom N, et al. Longitudinal amyloid imaging using 11C‐PiB: methodologic considerations. J Nucl Med 2013;54:1570–1576. [DOI] [PubMed] [Google Scholar]