

Abstract
Objective
Neuronal Ceroid Lipofuscinoses (NCL) are fatal inherited neurodegenerative diseases with established neuronal cell death and increased ceramide levels in brain, hence, a need for disease‐modifying drug candidates, with potential to enhance growth, reduce apoptosis and lower ceramide in neuronal precursor PC12 cells and human NCL cell lines using enhanced flupirtine aromatic carbamate derivatives in vitro.
Methods
Aromatic carbamate derivatives were tested by establishing growth curves under pro‐apoptotic conditions and activity evaluated by trypan blue and JC‐1 staining, as well as a drop in pro‐apoptotic ceramide in neuronal precursor PC12 cells following siRNA knockdown of the CLN3 gene, and CLN1‐/CLN2‐/CLN3‐/CLN6‐/CLN8 patient‐derived lymphoblasts. Ceramide levels were determined in CLN1‐/CLN2‐/CLN3‐/CLN6‐/CLN8 patient‐derived lymphoblasts before and after treatment. Expression of BCL‐2, ceramide synthesis enzymes (CERS2/CERS6/SMPD1/DEGS2) and Caspases 3/8/9 levels were compared in treated versus untreated CLN3‐deficient PC12 cells by qRT‐PCR.
Results
Retigabine, the benzyl‐derivatized carbamate and an allyl carbamate derivative were neuroprotective in CLN3‐defective PC12 cells and rescued CLN1‐/CLN2‐/CLN3‐/CLN6‐/CLN8 patient‐derived lymphoblasts from diminished growth and accelerated apoptosis. All drugs decreased ceramide in CLN1‐/CLN2‐/CLN3‐/CLN6‐/CLN8 patient‐derived lymphoblasts. Increased BCL‐2 and decreased ceramide synthesis enzyme expression were established in CLN3‐derived PC12 cells treated with the benzyl and allyl carbamate derivatives. They down‐regulated Caspase 3/Caspase 8 expression. Caspase 9 expression was reduced by the benzyl‐derivatized carbamate.
Interpretation
These findings establish that compounds analogous to flupirtine demonstrate anti‐apoptotic activity with potential for treatment of NCL disease and use of ceramide as a marker for these diseases.
Introduction
The NCLs comprise 13 neurodegenerative diseases and cause blindness/neuro‐cognitive decline/spasticity/seizures and early death. Dysregulation of apoptosis,1 autophagy, inflammation, and galactosylceramide transport are documented.2, 3, 4
Autosomal recessive inheritance predominates with some dominant CLN4 cases.5 CLN3 disease is the juvenile form. Infantile CLN1 disease and late infantile CLN2 disease are caused by defective palmitoyl protein thioesterase 1 (PPT1) and tripeptidyl peptidase I (TPP1), respectively. Elevated ceramide and BCL‐2 are documented in CLN2/CLN3 disease brain.6 Ceramide impacts cell differentiation and growth inducing cell cycle arrest and apoptosis, all key mechanisms in neurodegenerative disorders,1, 3, 6 particularly CLN2/CLN3 human disease.1, 7, 8, 9 CLN3 protein downregulates ceramide.4
BCL‐2 prevents cytochrome c release maintaining the electrical gradient across the mitochondrial membrane.10, 11, 12 Cytochrome c bound to apoptosis protease activating factor (APAF1) activates caspases 8/9/caspase 3 causing cell death.13
Flupirtine, a nonopioid analgesic, upregulates BCL‐2, protects postmitotic neurons from death by increasing glutathione, activating G protein inwardly rectifying potassium channels, delaying calcium loss within the intermitochondrial membrane space and also possesses muscle‐relaxant/anticonvulsant properties beneficial in NCL.14 Flupirtine protects neuronal/photoreceptor cells/normal/CLN1/CLN2/CLN3 and CLN6‐deficient lymphoblasts from etoposide‐induced apoptosis.14
Intrathecal injection of TPP1 slows down CLN2 disease in mice15, 16 and humans. Flupirtine and aromatic carbamate derivatives17 antiapoptotic properties provide the basis for use as potential therapies in NCL disease.
Methods
Tissue culture
Immortalized lymphoblasts from normal controls/patients with defects in CLN1/2/3/6/8 genes were used. Use of patient cells is approved by an American University of Beirut Medical Center (AUBMC) University Institutional Review Board protocol. Lymphoblasts are grown at 37°C in a humidified atmosphere, 5% CO2/95% air in RPMI 1640 medium (Lonza) supplemented with 10% heat‐inactivated fetal bovine serum (FBS) (Sigma), 1X Gentamycin (Sigma), and 1% penicillin/streptomycin (Lonza). PC12 neuronal precursor cells are maintained at 37°C in a humidified atmosphere, 5% CO2/95% air in DMEM medium (Lonza) supplemented with 10% heat‐inactivated horse serum (Sigma), 5% heat‐inactivated FBS (Sigma), 1% Sodium pyruvate (Lonza), and 1% penicillin/streptomycin (Lonza). Medium is changed every 2 days, and cells split weekly.
siRNA CLN3 knockdown in PC12 cells
PC‐12 cells are transfected with siRNA for CLN3 knock‐down (BLOCK‐iT RNA™ Designer, Invitrogen)/scrambled control (HiPerfect Transfection, Qiagen). CLN3 knockdown is validated by quantitative real‐time PCR (qRT‐PCR) at 24/48/72 h, and normalized to β‐actin. Expression levels are calculated using the ΔΔCT method. Primer sequences (Tm = 60○C) include: CLN3 forward, 5′AGACCCTCATCCCTCCCGT3′; reverse, 5′GAATCCGAAAAGCGCCGCC3′; β‐Actin forward, 5′ACACTGTGCCCATCTACGAG3′; reverse, 5′ATTTCCCTCTCAGCTGTGGT3′.
Treatment with flupirtine/aromatic carbamate derivatives
A total of 24 h after seeding, cells are treated with 20 μmol/L flupirtine dissolved in 0.004% ethanol (Vehicle 1); 20 or 50 μmol/L retigabine dissolved in 0.1% DMSO (Vehicle 3); 20 μmol/L methyl carbamate analog (compound 3) dissolved in 0.004% ethanol (Vehicle 1); 20 or 50 μmol/L isobutyl carbamate derivative (compound 4) dissolved in 0.004% ethanol (Vehicle 1); 3, 20 or 50 μmol/L benzyl‐derivatized carbamate (compound 5) dissolved in 0.07% DMSO (Vehicle 2); 3, 20 or 50 μmol/L allyl carbamate derivative (compound 6) dissolved in 0.07% DMSO (Vehicle 2); 20 or 50 μmol/L tert‐butyl carbamate derivative (compound 7) dissolved in 0.1% DMSO (Vehicle 3); 20 or 50 μmol/L carbamate derivative with substitution of chlorine at the para position (compound 8) dissolved in 0.004% ethanol (Vehicle 1); 20 or 50 μmol/L 1‐chloroethyl analog (compound 9) dissolved in 0.004% ethanol (Vehicle 1); 0.1, 1 or 10 μmol/L 4‐trifluoromethyl derivative (compound 10) dissolved in 0.004% ethanol (Vehicle 1); or with the corresponding vehicle. After 4 h, cells are washed, centrifuged (only for lymphoblasts) and fresh media added.
Cell growth/Viability by trypan blue dye exclusion
1.5 × 105 PC12 cells are seeded/well and treated with 10 μg/mL etoposide (Sigma) for 18 h or transfected with CLN3 or scrambled siRNA for 24 h. 1.5 × 105 normal/patient‐derived lymphoblast cells (CLN1/2/3/6/8) are seeded/well. 24 h later, cells are washed, centrifuged and media containing compound is added. 4 h later, cells are washed, centrifuged and fresh media is added. 24 h later, cells are stained with trypan blue dye (0.4%) and white (viable) and blue (dead) cells counted at 24/48/72 h in triplicate using a light microscope and a hemocytometer.
Propidium Iodide (PI) staining
1 × 105 PC12 cells, treated with 10 μg/mL etoposide (Sigma)/vehicle for 18 h, are grown on coverslips, then washed, centrifuged and media containing compound added. 4 h later, cells are washed, centrifuged and media added. 24 h later, cells are stained with PI (5μg/mL) for 5 min. Three fields of vision are chosen randomly. Total number of cells/field of vision are counted. Number of PI‐positive red apoptotic cells is determined under fluorescence, and percentage of PI‐positive cells/total cells/field of vision calculated.
JC‐1 Staining
JC‐1 stain (5,50–60‐tetrachloro‐1,1,3,3‐tetraethylbenzimidazolylcarbocyanine iodide; Molecular Probes, Eugene, OR) assesses decrease in mitochondrial membrane potential after treatment with compounds. This dye forms J‐aggregates with cytochrome c‐APAF‐1 complex. Onset of apoptosis is visualized with shift in emission from red (595 nm) to green (535 nm). 5x104 normal/patient lymphoblasts (CLN1/2/3/6/8) are grown/well in triplicate. Then, cells are washed, centrifuged, and media containing compound added. 4 h later, cells are washed, centrifuged and media added. 24 h later, cells are incubated with 200 μmol/L of JC‐1 stain at 37°C for 30 min. Samples are analyzed by a fluorescent plate reader. In healthy cells, JC‐1 forms J‐aggregates which display strong red fluorescent intensity with excitation/emission at 560 nm and 595 nm, respectively. In apoptotic cells, JC‐1 exists as monomers exhibiting strong green fluorescence with excitation and emission at 485 nm and 53 nm. Ratio of fluorescence of J‐aggregates (red) to monomers (green) is used as an indicator of cell death.
Ceramide levels
Cell Homogenization
Cell pellets are washed with ice‐cold PBS, suspended in methanol/chloroform (2:1) and stored at −80°C for 48–36 h.
Lipid Extraction
A total of 700 μL of distilled water are added to the homogenate, then 1 mL chloroform and 1 mL of distilled water, and the sample centrifuged at 956 ×g for 10 min at 4°C. The lower phase is lyophilized. Lipids are resuspended in 1 mL chloroform.
Ceramide Assay (Diacyl glycerol kinase (DGK) assay)
Ceramide standards (Non‐hydroxy Fatty Acid Ceramide from Sigma‐Aldrich) and samples were dried under vacuum. β‐octylglucoside (BOG) (Sigma‐Aldrich) and 1,2‐dioleoyl‐sn‐glycero‐3‐phospho‐(1′‐rac‐glycerol) (DOPG) (Sigma‐Aldrich) were added as micelles mixture to samples/standards and sonicated for 30 min. Reaction mixtures were added to the ATP mix containing 1.3 μCi ATP, [γ‐32P] (IZOTOP) per sample and incubated at room temperature (RT) for 45 min. The reaction was stopped using methanol/chloroform/distilled water prior to lipid extraction. Lower phase is dried under vaccum, resuspended in chloroform/methanol (9:1) and run on a TLC plate (SILICYCLE) using chloroform/acetone/methanol/acetic acid/water as running buffer (50:20:15:10:5). Plates were dried, x‐ray film overlaid and kept at −80°C overnight to visualize ceramide bands. Lanes of the TLC plate are scraped into scintillation vials, and counts/minute detected using a liquid scintillation counter. Results are expressed as pmols of ceramide/nmol of total phospholipids.
Phosphate determination
After lyophilization, 150 μL of 70% perchloric acid (Fluka) were added to disodium hydrogen phosphate (Na2HPO4) (MERCK) standards. Tubes were capped with methanol‐soaked glass balls, placed at 180°C for 1 h, cooled at RT, and distilled water/2.5% ammonium molybdate (Riedel‐de Haën)/10% ascorbic acid (Biochemical) added. Mixtures were incubated for 15 min at 50°C, and concentration determined spectrophotometerically (820 nm wavelength).
RNA isolation
Using RIBOZOL™ (AMRESCO), total RNA is isolated from cells, according to manufacturer protocols and stored at −80°C. RNA quality is assessed by analyzing A260/A280 and A260/A230 ratios with a ND‐1000 spectrometer (Nanodrop Technologies).
qRT‐PCR
Total RNA is reverse‐transcribed using RevertAid Reverse Transcriptase (Thermo Scientific) with 1000 ng of input RNA and random primers (Thermo Scientific). qRT‐PCR reactions are performed using specific primers (TIB MOLBIOL) and the iQ™ SYBR® Green Supermix (BioRad) as fluorescent detection dye, in CFX96™ Real‐Time PCR (BioRad), in 12.5 μL in duplicate. To characterize generated amplicons and to control contamination by unspecific by‐products, melt curve analysis is performed. Results are normalized to β‐actin and expression levels calculated using ΔΔCt method. Primer sequences (Tm = 60°C): SMPD1 forward, 5′CTATGAAGCGATGGCCAAG3′; reverse, 5′TGGGGAAAGAGCATAGAACC3′; CerS2 forward, 5′GCTGGAGATTCACATTTTACC3′; reverse, 5′CGAAGACGATGAAGATGTTGT3′; CerS6 forward, 5′TTTAGGGCACAGTTCTTTGG3′; reverse, 5′ACAGGGGGAGGATGAGATAC3′; DEGS2 forward, 5′GACTTCGAGTGGGTCTACAC3′; reverse, 5′GGTCCACGTGGTACTTCTTG3′; BCL‐2 forward, 5′TGTGTGTGGAGAGCGTCAAC3′; reverse, 5′TGAGCAGAGTCTTCAGAGAC3′; Caspase 3 forward, 5′TGGTTCATCCAGTCGCTTTG3′; reverse, 5′CATTCTGTTGCCACCTTTCG3′; Caspase 8 forward, 5′CTGCTGGGGATGGCCACTGTG3′; reverse, 5′TCGCCTCGAGGACATCGCTCTC3′; Caspase 9 forward, 5′CGAACTAACAGGCAAGCAGC3′; reverse, 5′ACCTCACCAAATCCTCCAGAAC3′; β‐Actin forward, 5′ACACTGTGCCCATCTACGAG3′; reverse, 5′ATTTCCCTCTCAGCTGTGGT3′.
Statistical analysis
Continuous data were expressed as means ± SEM, and compared by two‐tailed Student's t‐test and one‐way ANOVA followed by posthoc tests.
GraphPad Prism 6 (GraphPad Software, Inc., California, USA) was used. All tests were two‐sided and P < 0.05 was considered as statistically significant.
Results
Flupirtine/aromatic carbamate derivatives positively modulate growth and protect PC12 cells from etoposide‐induced apoptosis
Flupirtine and flupirtine analogues (Fig. 1A) enhanced growth and protected neuronal precursor PC12 cells from apoptosis induced by etoposide. The degree of protection was variable depending on the carbamate used. Treatment with 20 μmol/L flupirtine exhibited slightly enhanced growth at 24 h compared to vehicle‐treated PC12 cells, when cells were pretreated with etoposide (Fig. 1B). The benzyl‐derivatized carbamate (compound 5) and the allyl carbamate derivative (compound 6) were significantly protective after addition of etoposide to PC12 cells, based on trypan blue dye exclusion (data not shown). Treatment with benzyl‐derivatized carbamate (compound 5) at 50 μmol/L resulted in significantly increased cell growth compared with vehicle‐treated PC12 cells after etoposide (Fig. 1C, asterisk symbol). Similarly, treatment with allyl carbamate derivative (compound 6) at 20 μmol/L caused significantly increased cell growth compared with vehicle‐treated PC12 cells treated with etoposide (Fig. 1D, asterisk symbol). Treatment with 20 μmol/L retigabine conferred significant protection against etoposide‐induced apoptosis compared with vehicle‐treated PC12 cells, at 24 h (Fig. 1E, asterisk symbol). This protection was confirmed by PI staining whereby flupirtine/compound 5/6 and retigabine significantly decreased, in equal manner, total number of dead cells compared to vehicle‐treated PC12 cells after etoposide treatment (Fig. 1F, † symbol).
Figure 1.
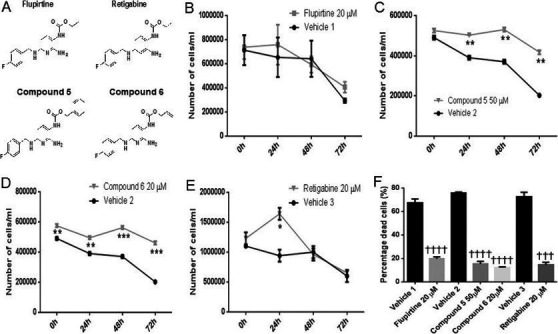
Treatment with flupirtine, retigabine, the benzyl (compound 5) or allyl (compound 6) carbamate derivatives enhances cell growth and decreases apoptosis in etoposide‐treated PC12 neuronal precursor cells. (A) Chemical structure of flupirtine and aromatic carbamate derivatives. Growth curve of PC12 neuronal precursor cells treated with etoposide and in response to treatment with (B) 20 μmol/L flupirtine, (C) 50 μmol/L compound 5, (D) 20 μmol/L compound 6, or (E) 20 μmol/L retigabine. Live cells are counted in triplicate at time‐points 0, 24, 48, and 72 h. (F) Propidium Iodide staining of PC12 neuronal precursor cells treated with etoposide in response to treatment with 20 μmol/L flupirtine, 50 μmol/L compound 5, 20 μmol/L compound 6, or 20 μmol/L retigabine. These drugs increase growth and decrease the number of PI‐positive apoptotic cells compared to vehicle‐treated PC12 neuronal precursor cells (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001). Flupirtine is compared to vehicle 1, benzyl (compound 5), and allyl (compound 6) carbamate derivatives are compared to vehicle 2, and retigabine is compared to vehicle 3. Results in panels B to E represent the mean ± SEM of three‐independent experiments analyzed by multiple t‐tests with Holm–Sidak correction to determine statistical significance, with alpha=0.05 (asterisk symbol). Results in panel F represent the mean ± SEM of three‐independent experiments analyzed by one‐way ANOVA (alpha = 0.05) followed by Sidak's posthoc multiple comparisons († symbol) ††† P < 0.001 and †††† P < 0.0001.
Flupirtine/aromatic carbamate derivatives rescue PC12 cells from CLN3‐knockdown apoptosis
PC12 cells demonstrated significant growth inhibition when cells were transfected with siRNA against CLN3 versus scrambled siRNA, at 24 h showing maximum CLN3 knockdown of 75% (P < 0.001) (Fig. 2A). Treatment with 20 μmol/L flupirtine, 50 μmol/L compound 5, 20 μmol/L compound 6, and 20 μmol/L retigabine resulted in significant increase in growth in siCLN3‐transfected PC12 cells compared to vehicle‐treated siCLN3‐transfected PC12 cells (Fig. 2B, asterisk symbol). Cell growth was significantly higher after treatment with compounds 5/6 compared to flupirtine and retigabine treatment (Fig. 2B, † symbol).
Figure 2.
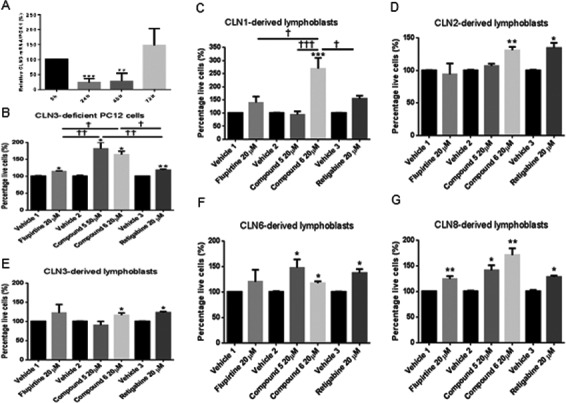
Treatment with flupirtine, retigabine, the benzyl (compound 5) or allyl (compound 6) carbamate derivatives corrects growth and decreases apoptosis in CLN3‐deficient PC12 neuronal precursor cells and in CLN1/CLN2/CLN3/CLN6/CLN8 patient‐derived lymphoblast cells. (A) Maximum siCLN3 knock‐down in PC12 cells is determined by qRT‐PCR. Relative CLN3 mRNA/PGK1 (%) of PC12 transfected cells with siCLN3 at 24, 48, and 72 h compared to scrambled siRNA. (B) Growth curve of PC12 neuronal precursor cells transfected with siCLN3 for 24 h and (C) CLN1/(D) CLN2/(E) CLN3/(F) CLN6/(G) CLN8‐derived lymphoblast cell lines treated with 20 μmol/L flupirtine, 50 μmol/L compound 5, 20 μmol/L compound 6, or 20 μmol/L retigabine. Live cells are counted in triplicate at 24 h. (A) CLN3‐deficient PC12 cells treated with flupirtine (*P < 0.05), compound 5 (*P < 0.05), compound 6 (*P < 0.05), and retigabine (**P < 0.01) exhibit an increase in number of live cells compared to vehicle‐treated cells. (C) Treatment with compound 6 and retigabine significantly increases cell growth compared to vehicle‐treated CLN1‐derived lymphoblast cell lines (*P < 0.05). (D) Treatment with compound 6 (**P < 0.01) and retigabine (*P < 0.05) significantly increases cell growth compared with vehicle‐treated CLN2‐derived lymphoblast cells. (E) Treatment with compound 6 and retigabine significantly increases the percentage of live cells compared to vehicle‐treated CLN3‐derived lymphoblast cells (*P < 0.05). (F) Treatment with compounds 5 and 6 and retigabine significantly increases number of live cells compared to vehicle‐treated CLN6‐derived lymphoblast cells (*P < 0.05). (G) Treatment with flupirtine (**P < 0.01), compound 5 (*P < 0.05), compound 6 (**P < 0.01) and retigabine (*P < 0.05) significantly increases the percentage of live cells compared to vehicle‐treated CLN8‐derived lymphoblast cells. Flupirtine is compared to vehicle 1, the benzyl (compound 5) and allyl (compound 6) carbamate derivatives are compared to vehicle 2, and retigabine is compared to vehicle 3. Results represent the mean ± SEM of three‐independent experiments analyzed by Student's t‐test when comparing each compound to its vehicle (asterisk symbol) and by one‐way ANOVA (alpha = 0.05) followed by Tukey's posthoc multiple comparisons when comparing all compounds among each other († symbol) † P < 0.05, †† P < 0.01, and ††† P < 0.001.
Flupirtine/aromatic carbamate derivatives block apoptosis selectively in CLN1/2/3/6/8‐derived lymphoblasts
Treatment of CLN1/2/3/6/8‐derived lymphoblasts with flupirtine, retigabine, and aromatic carbamate derivatives enhances cell growth (Fig. 2C–G, asterisk symbol). Degree of protection was compound‐specific and disease cell line‐dependent. Treatment with 20 μmol/L flupirtine significantly enhanced growth in CLN8‐derived lymphoblasts compared to vehicle‐treated cells. Treatment with benzyl‐derivatized carbamate (compound 5) at 50 μmol/L improved growth significantly compared to growth of vehicle‐treated CLN6/8‐derived patient lymphoblasts. Treatment with allyl carbamate derivative (compound 6) at 20 μmol/L exhibited significant increase in growth compared to vehicle‐treated CLN1/2/3/6/8‐derived patient lymphoblasts. Similarly, treatment with 20 μmol/L retigabine conferred significant protection against apoptosis compared to corresponding vehicle‐treated patient cell lines. In CLN1/CLN8‐derived lymphoblasts, cell growth was significantly higher after treatment with compound 6 compared to other compounds (Fig. 2C and G, † symbol).
This protection was confirmed by JC‐1 staining of compound versus vehicle‐treated CLN1/2/3/6/8‐derived lymphoblasts (Fig. 3A–E, asterisk symbol). Treatment of patient lymphoblasts with 20 μmol/L flupirtine resulted in a significant reduction in number of apoptotic cells with J‐aggregates compared with vehicle‐treated cells, suggesting that flupirtine rescues CLN1‐ (P < 0.05), CLN2‐ (P < 0.01), CLN3‐ (P < 0.05), CLN6‐ (P < 0.01), and CLN8‐derived (P < 0.01) patient lymphoblasts from apoptosis. Treatment of patient lymphoblasts with compound 5 (50 μmol/L) reduced the number of apoptotic cells with J‐aggregates compared to vehicle‐treated cells. This was significant in CLN2‐ (P < 0.05)/CLN8‐derived (P < 0.05) patient lymphoblasts. Treatment of patient lymphoblasts with compound 6 (20 μmol/L) significantly reduced the number of apoptotic cells with J‐aggregates compared to vehicle‐treated cells in CLN1‐ (P < 0.05), CLN2‐ (P < 0.01), CLN3‐ (P < 0.05), CLN6‐ (P < 0.05), and CLN8‐derived (P < 0.05) patient lymphoblasts. Treatment of patient lymphoblasts with retigabine (20 μmol/L) resulted in significant reduction in the number of apoptotic cells with J‐aggregates compared to vehicle‐treated CLN1‐ (P < 0.05), CLN2‐ (P < 0.05), CLN3‐ (P < 0.05), CLN6‐ (P < 0.05), and CLN8‐derived (P < 0.01) patient lymphoblasts. In CLN1‐derived lymphoblasts, rescue from apoptosis was significantly higher after flupirtine compared to other compounds (Fig. 3A, † symbol). In CLN2‐derived lymphoblasts, rescue from apoptosis was significantly higher after treatment with compound 6 compared to others (Fig. 3B, † symbol).
Figure 3.
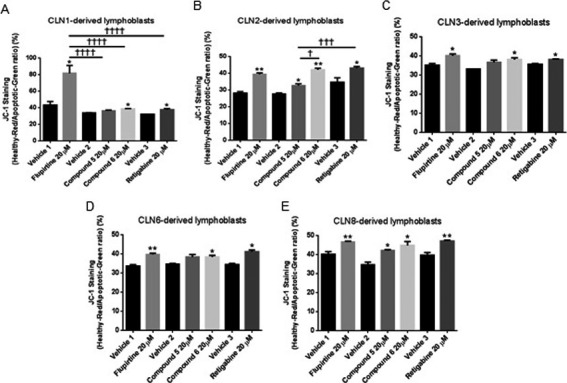
Treatment with flupirtine, retigabine, benzyl (compound 5), or allyl (compound 6) carbamate derivatives decreases apoptosis in CLN1/CLN2/CLN3/CLN6/CLN8 patient‐derived lymphoblast cells. JC‐1 staining of (A) CLN1/(B) CLN2/(C) CLN3/(D) CLN6/(E) CLN8‐derived lymphoblast cell lines treated with 20 μmol/L flupirtine, 50 μmol/L compound 5, 20 μmol/L compound 6, or 20 μmol/L retigabine. (A) Treatment with flupirtine, compound 6 and retigabine significantly decreases the number of apoptotic cells compared to apoptotic cells in corresponding vehicle‐treated CLN1‐derived lymphoblast cell lines (*P < 0.05). (B) Treatment with flupirtine (**P < 0.01), compound 5 (*P < 0.05), compound 6 (**P < 0.01), and retigabine (*P < 0.05) significantly decreases the number of apoptotic cells compared to corresponding vehicle‐treated CLN2‐derived lymphoblast cell lines. (C) Treatment with flupirtine, compound 6 and retigabine significantly decreases the number of apoptotic cells compared to corresponding vehicle‐treated CLN3‐derived lymphoblast cell lines (*P < 0.05). (D) Treatment with flupirtine (**P < 0.01), compound 6 (*P < 0.05) and retigabine (*P < 0.05) significantly decreases the number of apoptotic cells compared with apoptotic cells in corresponding vehicle‐treated CLN6‐derived lymphoblast cell lines. (E) Treatment with flupirtine (**P < 0.01), compound 5 (*P < 0.05), compound 6 (*P < 0.05) and retigabine (**P < 0.01) significantly decreases the number of apoptotic cells compared to corresponding vehicle‐treated CLN8‐derived lymphoblast cell lines. Flupirtine is compared to vehicle 1, the benzyl (compound 5) and allyl (compound 6) carbamate derivatives are compared to vehicle 2, and retigabine is compared to vehicle 3. Results represent the mean ± SEM of three‐independent experiments analyzed by Student's t‐test when comparing each compound to its vehicle (asterisk symbol) and by one‐way ANOVA (alpha = 0.05) followed by Tukey's posthoc multiple comparisons when comparing all compounds among each other († symbol) † P < 0.05, ††† P < 0.001, and †††† P < 0.0001.
Flupirtine/aromatic carbamate derivatives impact ceramide signaling pathways in PC12 cells and CLN1/2/3/6/8‐derived lymphoblasts
Ceramide levels are elevated in CLN3 disease patient sera (El‐Sitt et al., Elevated ceramide levels in CLN3 patients and Cln3 Δex7/8 mice and developmental comparison between wild type and Cln3Δex7/8 mouse sera and brain, submitted to Pediatric Research), cells, and brain.3 The level of ceramide in CLN1/2/3/6/8‐derived lymphoblast cell lines is significantly higher than level in normal lymphoblasts (Fig. 4A, asterisk symbol). Level of ceramide in CLN6/CLN8‐derived lymphoblasts is significantly higher than level in CLN1‐derived lymphoblasts (Fig. 4A, † symbol).
Figure 4.
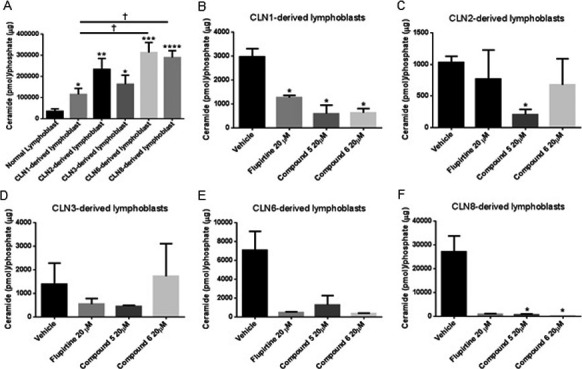
Ceramide levels in normal and CLN1/CLN2/CLN3/CLN6/CLN8 patient‐derived lymphoblast cells treated with flupirtine, benzyl (compound 5), or allyl (compound 6) carbamate derivatives. (A) Measurement of ceramide levels in CLN1‐, CLN2‐, CLN3‐, CLN6‐ and CLN8‐derived lymphoblast cells compared to normal lymphoblasts. Results represent the mean ± SEM of five independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by Student's t‐test as compared to normal lymphoblasts. Ceramide levels of (B) CLN1/(C) CLN2/(D) CLN3/(E) CLN6/(F) CLN8‐derived lymphoblast cells treated with 20 μmol/L flupirtine, 50 μmol/L compound 5, or 20 μmol/L compound 6. Treatment with drugs decreases ceramide levels compared to levels in corresponding vehicle‐treated CLN1/CLN2/CLN3/CLN6/CLN8 patient‐derived lymphoblast cells. Results in panel A represent the mean ± SEM of three‐independent experiments analyzed by Student's t‐test when comparing each CLN‐deficient lymphoblast cell line to the normal lymphoblasts (asterisk symbol) and by one‐way ANOVA (alpha = 0.05) followed by Tukey's posthoc multiple comparisons when comparing all cell lines among each other († symbol). Results in panels B to F represent the mean ± SEM of three‐independent experiments analyzed by Student's t‐test when comparing each compound to the vehicle (asterisk symbol) and by one‐way ANOVA (alpha = 0.05) followed by Tukey's posthoc multiple comparisons when comparing all compounds among each other († symbol) † P < 0.05.
Flupirtine significantly decreases level of ceramide in CLN1‐derived lymphoblasts versus ceramide in vehicle‐treated cells. Similarly, flupirtine decreases ceramide levels in CLN2‐ and CLN3‐lymphoblasts versus ceramide in vehicle‐treated cells. This decrease was not statistically significant. Although in CLN6‐ (P‐value = 0.08) and CLN8‐lymphoblasts (P‐value = 0.06), the decrease in ceramide levels trended toward significance, but was not statistically significant. Benzyl‐derivatized carbamate (compound 5) significantly decreases ceramide in CLN1‐, CLN2‐, and CLN8‐derived lymphoblasts compared to ceramide in vehicle‐treated cells. In CLN3‐ (P‐value = 0.55) and CLN6‐derived lymphoblasts (P‐value = 0.12), decrease in ceramide by compound 5 trended toward significance, but was not statistically significant. Treatment with allyl carbamate derivative (compound 6) significantly decreases ceramide in CLN1‐ and CLN8‐lymphoblasts versus ceramide in vehicle‐treated cells. In CLN2‐ and CLN3‐derived lymphoblasts, decrease in ceramide by compound 6 was not significant. Although in CLN6‐derived lymphoblasts, the decrease in ceramide levels trended toward significance, but was not statistically significant (P‐value = 0.08) (Fig. 4B–F, asterisk symbol).
Flupirtine/aromatic carbamate derivatives regulate expression of Bcl‐2, caspases 3/8/9
CLN3 knockdown of 75% (P < 0.001) was determined at 24 h (Fig. 2A). Transfection of PC12 cells with siRNA against CLN3 for 24 h causes a significant decrease in expression of BCL‐2 compared to scrambled siRNA‐treated cells (Fig. 5A, asterisk symbol). Treatment with 20 μmol/L flupirtine, 50 μmol/L compound 5, or 20 μmol/L compound 6 significantly upregulated, in equal manner, BCL‐2 expression versus vehicle‐treated siCLN3‐transfected cells. Blocking CLN3 expression significantly increased Caspases 3/8/9 expression versus scrambled siRNA‐treated cells (Fig. 5B–D, asterisk symbol). Flupirtine/compounds 5/6 significantly downregulated Caspases 3/8 expression versus vehicle‐treated siCLN3‐transfected cells (Fig. 5B and C, asterisk symbol). Caspase 9 expression was significantly reduced in flupirtine‐/compound 5‐treated cells versus vehicle‐treated siCLN3‐transfected cells (Fig. 5D, asterisk symbol). Caspase 3/9 downregulation was significantly higher after treatment with flupirtine and compound 5 versus compound 6 (Fig. 5B and D, † symbol). Caspase 8 downregulation was significantly higher after treatment with compounds 5/6 compared to flupirtine treatment (Fig. 5C, † symbol).
Figure 5.
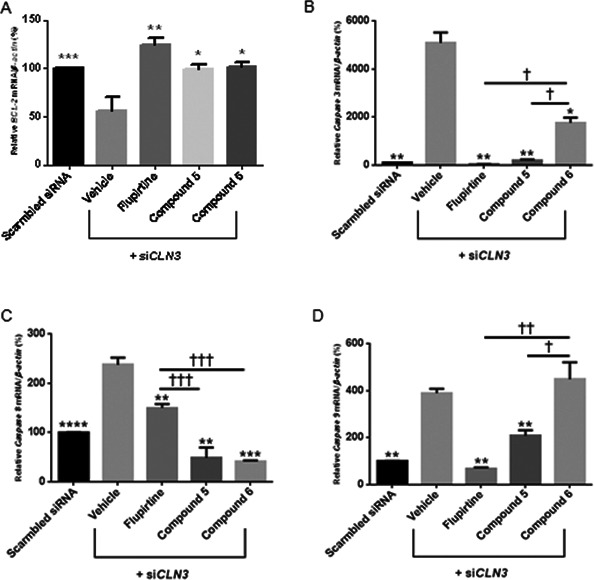
Expression of BCL‐2, Caspases 3, 8 and 9 in CLN3‐deficient PC12 neuronal precursor cells. Maximum siCLN3 knock‐down in PC12 cells shown in Fig 2A. PC12 neuronal precursor cells are transfected with siCLN3 for 24 h and treated with 20 μmol/L flupirtine, 50 μmol/L compound 5, or 20 μmol/L compound 6. Total RNA is extracted and quantitative real‐time PCR experiments performed using specific primers for (A) BCL‐2, (B) Caspase 3, (C) Caspase 8 and (D) Caspase 9. Values are means of the fold change normalized to β‐Actin mRNA expression, with standard errors of the mean (SEM) represented by vertical bars. Transfection of neuronal precursor PC12 cells with siCLN3 decreases BCL‐2 (***P < 0.001) and increases Caspase 3 (**P < 0.01), Caspase 8 (****P < 0.0001) and Caspase 9 (**P < 0.01) expression compared to transfection with scrambled siRNA. Treatment with flupirtine, compounds 5 and 6 increases BCL‐2 levels and decreases levels of Caspases 3, 8 and 9. Results represent the mean ± SEM of three‐independent experiments analyzed by Student's t‐test when comparing all bars to the vehicle bar (asterisk symbol) and by one‐way ANOVA (alpha = 0.05) followed by Tukey's posthoc multiple comparisons when comparing all compounds among each other († symbol) † P < 0.05, †† P < 0.01 and ††† P < 0.001.
Flupirtine/aromatic carbamate derivatives regulate expression of ceramide synthesis enzymes
To evaluate the molecular effects of flupirtine and aromatic carbamate derivatives on ceramide synthetic pathways, expression levels of ceramide synthesis enzymes, including ceramide synthase 2 (CERS2), ceramide synthase 6 (CERS6), acidic sphingomyelinase (SMPD1), and delta(4)‐desaturase sphingolipid 2 (DEGS2) were analyzed. Maximum CLN3 knockdown of 75% (P < 0.001) was determined at 24 h (Fig. 2A). Blocking CLN3 expression by siRNA for 24 h increases pro‐apoptotic ceramide9, 18 and significantly upregulates expression of ceramide synthesis enzymes CERS2, CERS6, SMPD1, and DEGS2 compared to expression in scrambled siRNA controls. Addition of flupirtine, compound 5/6 attenuated, in equal manner CERS2/CERS6, SMPD1/DEGS2 expression versus expression in vehicle‐treated siCLN3‐transfected cells (Figs. 6A–D, asterisk symbol).
Figure 6.
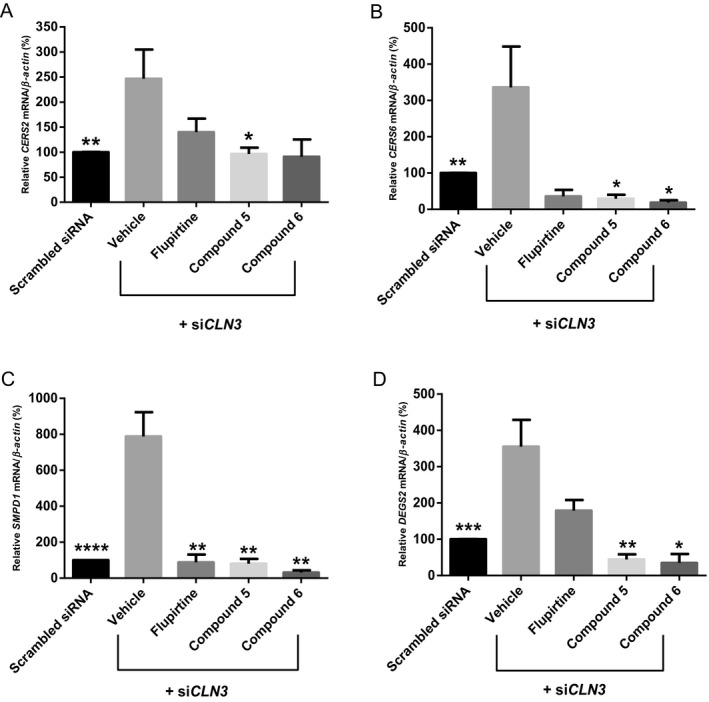
Expression of ceramide synthesis enzymes (CERS2, CERS6, SMPD1 and DEGS2) in CLN3‐deficient PC12 neuronal precursor cells. Maximum siCLN3 knock‐down in PC12 cells shown in Fig 2A. PC12 neuronal precursor cells are transfected with siCLN3 for 24 h and treated with 20 μmol/L flupirtine, 50 μmol/L compound 5, or 20 μmol/L compound 6. Total RNA is extracted and quantitative real‐time PCR performed using specific primers for (A) CERS2, (B) CERS6, (C) SMPD1 and (D) DEGS2 cDNA. Values are means of the fold change normalized to β‐Actin mRNA expression, with standard errors of the mean (SEM) represented by vertical bars. Transfection of neuronal precursor PC12 cells with siCLN3 increases CERS2 (**P < 0.01), CERS6 (**P < 0.01), SMPD1 (****P < 0.0001) and DEGS2 (***P < 0.001) expression levels as compared to scrambled siRNA. Treatment with flupirtine, compounds 5 and 6 decreases the levels of CERS2,CERS6,SMPD1 and DEGS2. Results represent the mean ± SEM of three‐independent experiments analyzed by Student's t‐test when comparing all bars to the vehicle bar (asterisk symbol) and by one‐way ANOVA (alpha = 0.05) followed by Tukey's posthoc multiple comparisons when comparing all compounds among each other († symbol).
Discussion
The NCLs are fatal disorders. Intrathecal TPP1 is effective in slowing disease in CLN2 disease, but is expensive and medical coverage available only in few countries. Gene and/or protein replacement are not options for NCLs due to defective membrane proteins (CLN3/CLN6/CLN8), hence the need for alternate novel therapies accessible to all.19 Flupirtine is neuroprotective in human postmitotic neurons and photoreceptors.14 The significant protection flupirtine imparts to CLN1/2/3/8‐derived lymphoblasts, as well as CLN2/3‐deficient postmitotic human‐derived neurons provides the basis for use in some NCLs.14 Flupirtine is a derivative of triaminopyridine in the form of ethyl‐N‐[2‐amino‐6‐(4‐fluorophenylmethylamino) pyridin‐3‐yl]carbamate (Fig. 1A). The carbamate group is cleavable under strong basic and acid conditions.20 Retigabine, a phenyl bioisostere of flupirtine, is approved for use as an anticonvulsant and analgesic drug. Flupirtine penetrates the blood‐brain barrier. Retigabine exerts neuroprotective properties in vitro in rat organotypic hippocampal slice cultures exposed to oxygen, glucose deprivation, and serum withdrawal.21 Efficacy of flupirtine on cognitive function in Creutzfeldt‐Jakob disease patients is reported.22 Flupirtine and retigabine may be promising drugs for treatment of Alzheimer's disease.23 Also, retigabine is in trials for treatment of amyotrophic lateral sclerosis (Clinicaltrial.gov ID: NCT02450552). A case report on one case treated with flupirtine in NCL disease is published.24
Retigabine, benzyl‐derivatized carbamate (compound 5) and allyl carbamate derivative (compound 6) possess neuroprotective activity in etoposide‐induced PC12 cells due to blunting of CLN3 expression (Fig. 1A). Etoposide increases apoptosis in neuronal precursor PC12 cells, and flupirtine affords statistically significant protection to cells. Blocking CLN3 expression in cells provides a good in vitro model for CLN3 disease. In patient‐derived lymphoblasts, the neuroprotective effect of flupirtine/retigabine and novel aromatic carbamates was drug‐dose, and disease‐specific. Patient lymphoblasts provide a peripheral tissue for testing of NCL variants. Prevention of apoptosis in lymphoblasts constitutes a surrogate marker for testing therapeutic agents with potential neuroprotective activity in the brain. The concentrations of the compounds used in this study were based on the concentration of flupirtine (20 μmol/L) already in use in the literature proven to be neuroprotective in vitro in several cell models.14 An important question arises whether drug concentrations with which we observed neuroprotective effects in vitro correlate with concentrations of flupirtine used in humans.25 In fact, plasma concentrations of flupirtine required for analgesic activity in humans correlates with in vitro concentrations of 2.5–6.5 μmol/L.
Ceramide is involved in apoptosis and cell death. Elevated ceramide levels are documented in CLN2/3 disease patients and in Cln3‐knockout mouse brains, contributing to accelerated apoptosis.6, 26 Wild‐type CLN3p downregulates ceramide and has significant anti‐apoptotic activity (Fig. 7).9 Ceramide accumulates in CLN3‐deficient cells with CLN3 transfection restoring levels to normal.4, 6, 9, 27 Baseline levels of ceramide in CLN1/2/3/6/8‐derived lymphoblast cells were significantly higher than in normal lymphoblasts. Treatment with flupirtine, benzyl‐derivatized carbamate (compound 5) or allyl carbamate derivative (compound 6) decreased ceramide in a compound and NCL disease‐specific manner. The actions of these drugs could be acting upstream of ceramide generation on one or more of the de novo ceramide pathway enzymes such as ceramide synthases 2 and 6 (CERS2 and 6), dihydroceramide desaturase 2 (DEGS2), or acid sphingomyelinase phosphodiesterase 1 (SMPD1) (Fig. 7).
Figure 7.
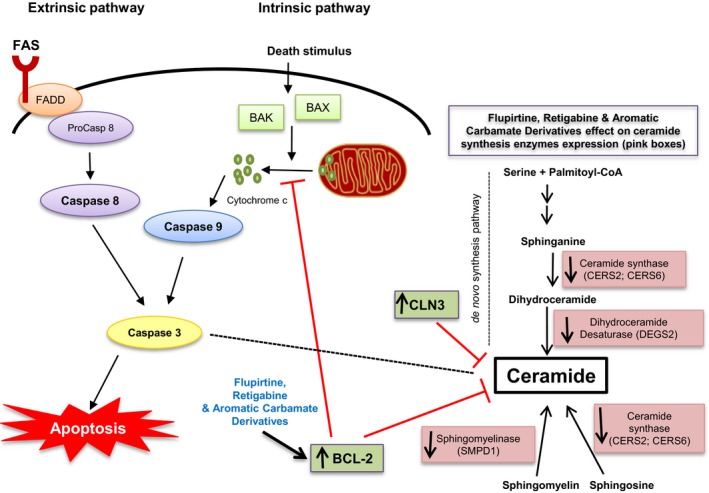
Schematic representation of the effects of CLN3, flupirtine, retigabine, and benzyl (compound 5) or allyl (compound 6) carbamate derivatives on ceramide synthesis and intrinsic/extrinsic apoptotic pathways. Schematic representation of sphingolipid synthetic pathways and apoptotic events. The sites of action of the various sphingolipid synthesis enzymes and the location of the CLN3 action are indicated. Enzymes downregulated are shown in pink. Apoptosis extrinsic and intrinsic pathways are indicated. Each requires specific triggering signals to begin an energy‐dependent cascade of molecular events. Each pathway activates its own initiator caspase (caspase 8 or 9), which in turn activates executioner caspase 3. The execution pathway results in cell shrinkage, chromatin condensation, formation of cytoplasmic blebs and apoptotic bodies and ultimate phagocytosis of apoptotic bodies by adjacent parenchymal cells, neoplastic cells, or macrophages. Arrows indicate activation. Red T bar indicates inhibition. CLN3: Ceroid Lipofuscinosis, Neuronal 3; CERS2: Ceramide Synthase 2; CERS6: Ceramide Synthase 6; DEGS2: Delta 4‐Desaturase, Sphingolipid 2; SMPD1: Sphingomyelin Phosphodiesterase 1. FADD: FAS‐associated death domain; BAK: Bcl‐2 antagonist/killer‐1; BAX: BCL‐2‐associated X protein; BCL‐2: B‐cell lymphoma 2.
Flupirtine imparts its anti‐apoptotic effect by upregulating BCL‐2.14 The BCL‐2 family encodes proteins that regulate apoptosis including pro‐apoptotic BAX and BAK, anti‐apoptotic BCL‐2 and Bcl‐xL, and others. BAX and BCL‐2 are present in the mitochondrial membrane and maintain balance between cell survival and death by regulating cytochrome c release and activation of the caspase cascade and apoptosis (Fig. 7).28, 29 Blocking CLN3 expression decreased expression of BCL‐2 mRNA. Flupirtine/carbamate derivatives 5 and 6 reversed BCL‐2 downregulation.
Apoptosis and autophagy are interdependent contributors to cell death, with caspases initiating the process that is well‐documented in CLN3 patient cells.3 Inhibition of BCL‐2 expression, release of cytochrome c, and activation of caspases 3/8/9 result in an apoptotic cascade. Caspase‐dependent extrinsic apoptosis is launched via the death receptor pathway, which activates initiator caspase 8, then effector caspase 3. The intrinsic pathway is activated from mitochondria by cell signals via initiator caspase 9, then effector caspase 330 (Fig. 7). Caspase 3 increased expression is a marker for apoptosis.31 Involvement of caspase 8 is documented in apoptotic pathways in neurodegenerative diseases.3, 32 Caspase 8 upregulation is indispensable for the signal transduction cascade via the extrinsic apoptotic pathway (Fig. 7).30 Caspase 9 is activated by release of cytochrome c from damaged mitochondria through interaction with APAF‐1 followed by activation of caspase 3.33 Caspase 3 then cleaves poly (ADP‐ribose) polymerase‐1 (PARP‐1) involved in DNA damage inducing programmed cell death (Fig. 7).34 Elevated Caspases 3/8/9 expression is consistent with increase in number of J‐aggregates by JC‐1 staining, following blocking of CLN3 expression. Flupirtine effectively downregulated expression of Caspases 3/8/9, and decreased the apoptotic cell count. Similarly, benzyl‐derivatized carbamate (compound 5) significantly downregulated expression of Caspases 3/8/9. Allyl carbamate derivative (compound 6) downregulated expression of Caspases 3/8, keeping Caspase 9 expression intact. These findings imply that flupirtine and compound 5 act via the extrinsic/intrinsic pathways, whereas compound 6 exerts its effect only via the extrinsic pathway.
BCL‐2 protein (BCL‐2p) inhibits ceramide channel formation in the outer mitochondrial membrane35 in vitro. Ghafourifar et al. demonstrate that anti‐apoptosis by BCL‐2p is exerted downstream of ceramide since pre‐incubation with BCL‐2p prevents ceramide‐induced cytochrome c release in mitochondria.36 Other groups report that BCL‐2 overexpression rescues from ceramide‐induced apoptosis37, 38 or ceramidase inhibitor‐induced apoptosis.39 While BCL‐2p acts downstream of ceramide, Bcl‐xL targets the ceramide pathway inhibiting ceramide generation, acting upstream of ceramide.40 Other studies demonstrate that ceramide accumulation after DNA damage can be reduced by BCL‐2 or Bcl‐xL overexpression, suggesting an upstream action of both proteins.41, 42, 43 This highlights importance of the BCL‐2 family in initiation and regulation of the ceramide pathway (Fig. 7). Puranam et al. established that transfection of CLN3‐derived cells with CLN3 cDNA corrects apoptosis and lowers ceramide back to normal.9 CLN3p, which resides in the membrane, fine‐tune regulation of the apoptotic pathway by attenuating ceramide generation. Also, ceramide accumulation occurs upstream of caspase activation44 induces caspase‐dependent apoptosis, unlike BCL‐2p, which acts downstream of ceramide, but upstream of caspase 3. CLN3 protein modulates the generation of ceramide which occurs upstream of BCL‐2p and caspase activation in CLN3‐derived cells.
Ceramide formation or breakdown in the cell has at least 5 known origins: (1) sphingomyelin hydrolysis via neutral/acid sphingomyelinase (SMPD); (2) breakdown to sphingosine by ceramidase; (3) de novo synthesis of ceramide by ceramide synthases (CerS); (4) generation of cerebrosides via cerebroside synthase; and (5) formation of ceramide phosphate by ceramide kinase.9, 45 Upregulation of ceramide de novo synthesis enzymes (CERS2, CERS6, and DEGS2) and ceramide hydrolytic enzyme (SMPD1) was observed in CLN3‐derived PC12 cells, in line with the increase in ceramide levels. Downregulation of CERS2/CERS6/SMPD1/DEGS2 in neuronal precursor PC12 cells treated with flupirtine/compounds 5 or 6 reflects the impact of these aromatic carbamate derivatives on ceramide synthesis. They are specifically involved in de novo ceramide synthesis (CERS2/CERS6/DEGS2), and synthesis of ceramide from sphingosine (CERS2/CERS6), and degradation of sphingomyelin (SMPD1) to ceramide (Fig. 7). Alterations in these enzymes may affect the pathophysiology of various forms of NCL disease and shed light on the mechanism of action of these novel carbamates.
Dihydroceramide synthases (CERS2/CERS6) acylate sphinganine and sphingosine to form dihydroceramide and ceramide, respectively. Higher levels of dihydroceramide synthases may directly/indirectly lead to increased ceramide in neuronal tissue accelerating neurodegeneration. DEGS2 is responsible for conversion of dihydroceramides generated via de novo synthesis to ceramide.46 Blocking DEGS2 leads to decreased cell proliferation and cell cycle arrest.47 The fourth enzyme, SMPD1, or acid sphingomyelinase (SMase) catalyzes hydrolysis of sphingomyelin to ceramide and phosphocholine. Several SMases have been identified48 and are upregulated in tumor tissue, but correlation with neurodegeneration is not established yet.49 The significant downregulation in ceramide pathway enzyme expression after treatment with flupirtine/compounds 5/6 are consistent with the observed reduction in ceramide levels. Ceramide participation has proven central to many neurodegenerative diseases. Neurodegenerative disorders are marked by extensive neuronal apoptosis and gliosis. Regulatory mechanisms of cell death are not fully elucidated. Understanding cell death mechanisms is enhanced by exploring impact of compounds on the ceramide synthetic pathway. The clarification of flupirtine and other aromatic carbamate derivatives’ impact on this pathway add to an emerging field of therapeutically significant drugs impacting ceramide regulation potentially effective in treating neurodegenerative disorders.50, 51
There were differences noted in effects of flupirtine, the benzyl or the allyl carbamate derivative on cell growth, apoptosis, ceramide levels, and mRNA expression levels of caspases or sphingolipid metabolism enzymes. In fact, the differences documented in cell models for NCL disease variants could suggest that differences in magnitude and/or mechanisms may be at play in the neuroprotective effects observed for flupirtine or the different aromatic carbamate derivatives that are NCL disease variant specific.
In conclusion, allyl carbamate derivative (compound 6) possesses greater anti‐apoptotic activity than flupirtine in neuronal precursor PC12 cells and in CLN1/2/3/6/8 patient lymphoblasts. These experiments serve as preclinical assays for potential use of these aromatic carbamate derivatives to treat CLN3 disease and these CLN1/2/6/8 diseases in animal models and humans. Alteration in BCL‐2/ceramide synthesis enzyme expression levels support that flupirtine analogues impact the ceramide synthesis pathway. Future experiments in Cln3 Δex7/8 knock‐in mice/CLN2/Cln6 nclf/CLN8‐derived mouse models will shed further light on the role of aromatic carbamate derivatives of flupirtine in treatment of NCL disease variants.
Conflict of Interest
J.M., F.S, KA.M., S.E., and J.A. have nothing to declare. N.K., P.T., RM.B. have a provisional patent application detailing the aromatic carbamates described herein has been filed: ‘Functionalized Pyridine Carbamates with Enhanced Neuroprotective Activity’ U. S. Patent Appl. US 62/532624, 2017. RM.B. has an Application for Method of Treating Batten Disease. Inventor: Rose‐Mary Boustany. Duke (File No. 5405‐240 PR). US Patent and Trademark No.10/148,859 (U.S.National Phase); Use Patent issued 11/23/2004 US Patent # 6 821 995, expired 11/23/2014.
Acknowledgments
We would like to thank the OpenMinds fund for their financial support of this work.
Funding Information
We would like to thank the OpenMinds fund for their financial support of this work.
Funding Statement
This work was funded by OpenMinds grant .
References
- 1. Lane SC, Jolly RD, Schmechel DE, et al. Apoptosis as the mechanism of neurodegeneration in Batten's disease. J Neurochem 1996;67:677–683. [DOI] [PubMed] [Google Scholar]
- 2. Lockhart EM, Warner DS, Pearlstein RD, et al. Allopregnanolone attenuates N‐methyl‐D‐aspartate‐induced excitotoxicity and apoptosis in the human NT2 cell line in culture. Neurosci Lett 2002;328:33–36. [DOI] [PubMed] [Google Scholar]
- 3. Persaud‐Sawin DA, Boustany RM. Cell death pathways in juvenile Batten disease. Apoptosis 2005;10:973–985. [DOI] [PubMed] [Google Scholar]
- 4. Persaud‐Sawin DA, McNamara JO 2nd, Rylova S, et al. A galactosylceramide binding domain is involved in trafficking of CLN3 from Golgi to rafts via recycling endosomes. Pediatr Res 2004;56:449–463. [DOI] [PubMed] [Google Scholar]
- 5. Haltia M, Goebel HH. The neuronal ceroid‐lipofuscinoses: a historical introduction. Biochem Biophys Acta 2013;1832:1795–1800. [DOI] [PubMed] [Google Scholar]
- 6. Puranam K, Qian WH, Nikbakht K, et al. Upregulation of Bcl‐2 and elevation of ceramide in Batten disease. Neuropediatrics 1997;28:37–41. [DOI] [PubMed] [Google Scholar]
- 7. Cho S, Dawson PE, Dawson G. Role of palmitoyl‐protein thioesterase in cell death: implications for infantile neuronal ceroid lipofuscinosis. Eur J Paediatr Neurol 2001;5 Suppl A:53–55. [DOI] [PubMed] [Google Scholar]
- 8. Haddad SE, Khoury M, Daoud M, et al. CLN5 and CLN8 protein association with ceramide synthase: biochemical and proteomic approaches. Electrophoresis 2012;33:3798–3809. [DOI] [PubMed] [Google Scholar]
- 9. Puranam KL, Guo WX, Qian WH, et al. CLN3 defines a novel antiapoptotic pathway operative in neurodegeneration and mediated by ceramide. Mol Genet Metab 1999;66:294–308. [DOI] [PubMed] [Google Scholar]
- 10. Schendel SL, Xie Z, Montal MO, et al. Channel formation by antiapoptotic protein Bcl‐2. Proc Natl Acad Sci USA 1997;94:5113–5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tsujimoto Y, Shimizu S. VDAC regulation by the Bcl‐2 family of proteins. Cell Death Differ 2000;7:1174–1181. [DOI] [PubMed] [Google Scholar]
- 12. Waterhouse NJ, Green DR. Mitochondria and apoptosis: HQ or high‐security prison? J Clin Immunol 1999;19:378–387. [DOI] [PubMed] [Google Scholar]
- 13. Bratton SB, Walker G, Srinivasula SM, et al. Recruitment, activation and retention of caspases‐9 and ‐3 by Apaf‐1 apoptosome and associated XIAP complexes. EMBO J 2001;20:998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dhar S, Bitting RL, Rylova SN, et al. Flupirtine blocks apoptosis in batten patient lymphoblasts and in human postmitotic CLN3‐ and CLN2‐deficient neurons. Ann Neurol 2002;51:448–466. [DOI] [PubMed] [Google Scholar]
- 15. Xu S, Wang L, El‐Banna M, et al. Large‐volume intrathecal enzyme delivery increases survival of a mouse model of late infantile neuronal ceroid lipofuscinosis. Mol. Ther. 2011;19:1842–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wiseman JA, Meng Y, Nemtsova Y, et al. Chronic enzyme replacement to the brain of a late infantile neuronal ceroid lipofuscinosis mouse has differential effects on phenotypes of disease. Mol Ther Methods Clin Dev 2017;17:204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kinarivala N, Patel R, Boustany RM, et al. Discovery of aromatic carbamates that confer neuroprotective activity by enhancing autophagy and inducing the anti‐apoptotic protein B‐cell lymphoma 2 (Bcl‐2). J Med Chem 2017;60:9739–9756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Makoukji J, Raad M, Genadry K, et al. Association between CLN3 (Neuronal Ceroid Lipofuscinosis, CLN3 Type) gene expression and clinical characteristics of breast cancer patients. Front Oncol 2015;5:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kinarivala N, Trippier PC. Progress in the Development of Small Molecule Therapeutics for the Treatment of Neuronal Ceroid Lipofuscinoses (NCLs). Journal of medicinal chemistry. 2016;59:4415–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klawe C, Maschke M. Flupirtine: pharmacology and clinical applications of a nonopioid analgesic and potentially neuroprotective compound. Expert Opin Pharmacother 2009;10:1495–1500. [DOI] [PubMed] [Google Scholar]
- 21. Boscia F, Annunziato L, Taglialatela M. Retigabine and flupirtine exert neuroprotective actions in organotypic hippocampal cultures. Neuropharmacology 2006;51:283–294. [DOI] [PubMed] [Google Scholar]
- 22. Otto M, Cepek L, Ratzka P, et al. Efficacy of flupirtine on cognitive function in patients with CJD: A double‐blind study. Neurology 2004;62:714–718. [DOI] [PubMed] [Google Scholar]
- 23. Gongadze N, Antelava N, Kezeli T, et al. The mechanisms of neurodegenerative processes and current pharmacotherapy of Alzheimer's disease. Georgian Med News 2008;155:44–48. [PubMed] [Google Scholar]
- 24. Cialone J, Augustine EF, Newhouse N, et al. Parent‐reported benefits of flupirtine in juvenile neuronal ceroid lipofuscinosis (Batten disease; CLN3) are not supported by quantitative data. J Inherit Metab Dis 2011;34:1075–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zimmer G, Balakirev M, Zwicker K, et al. Effect of the triaminopyridine flupirtine on calcium uptake, membrane potential and ATP synthesis in rat heart mitochondria. Br J Pharmacol 1998;123:1154–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mencarelli C, Martinez‐Martinez P. Ceramide function in the brain: when a slight tilt is enough. Cell Mol Life Sci 2013;70:181–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rylova SN, Amalfitano A, Persaud‐Sawin DA, et al. The CLN3 gene is a novel molecular target for cancer drug discovery. Can Res 2002;62:801–808. [PubMed] [Google Scholar]
- 28. Chu R, Upreti M, Ding WX, et al. Regulation of Bax by c‐Jun NH2‐terminal kinase and Bcl‐xL in vinblastine‐induced apoptosis. Biochem Pharmacol 2009;78:241–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jeong HS, Choi HY, Choi TW, et al. Differential regulation of the antiapoptotic action of B‐cell lymphoma 2 (Bcl‐2) and B‐cell lymphoma extra long (Bcl‐xL) by c‐Jun N‐terminal protein kinase (JNK) 1‐involved pathway in neuroglioma cells. Biol Pharm Bull 2008;31:1686–1690. [DOI] [PubMed] [Google Scholar]
- 30. Liu L, Mu LM, Yan Y, et al. The use of functional epirubicin liposomes to induce programmed death in refractory breast cancer. Int J Nanomed 2017;12:4163–4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gown AM, Willingham MC. Improved detection of apoptotic cells in archival paraffin sections: immunohistochemistry using antibodies to cleaved caspase 3. J Histochem Cytochem 2002;50:449–454. [DOI] [PubMed] [Google Scholar]
- 32. Torriglia A, Chaudun E, Chany‐Fournier F, et al. Involvement of L‐DNase II in nuclear degeneration during chick retina development. Exp Eye Res 2001;72:443–453. [DOI] [PubMed] [Google Scholar]
- 33. Falcon C, Al‐Obaidi M, Di Stasi A. Exploiting cell death pathways for inducible cell elimination to modulate graft‐versus‐host‐disease. Biomedicines 2017;5:pii. E30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Los M, Mozoluk M, Ferrari D, et al. Activation and caspase‐mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol Biol Cell 2002;13:978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Siskind LJ, Feinstein L, Yu T, et al. Anti‐apoptotic Bcl‐2 family proteins disassemble ceramide channels. J Biol Chem 2008;283:6622–6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ghafourifar P, Klein SD, Schucht O, et al. Ceramide induces cytochrome c release from isolated mitochondria. Importance of mitochondrial redox state. J Biol Chem 1999;274:6080–6084. [DOI] [PubMed] [Google Scholar]
- 37. Zhang J, Alter N, Reed JC, et al. Bcl‐2 interrupts the ceramide‐mediated pathway of cell death. Proc Natl Acad Sci USA 1996;93:5325–5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Smyth MJ, Perry DK, Zhang J, et al. prICE: a downstream target for ceramide‐induced apoptosis and for the inhibitory action of Bcl‐2. Biochem J 1996;316(Pt 1):25–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Raisova M, Goltz G, Bektas M, et al. Bcl‐2 overexpression prevents apoptosis induced by ceramidase inhibitors in malignant melanoma and HaCaT keratinocytes. FEBS Lett 2002;516:47–52. [DOI] [PubMed] [Google Scholar]
- 40. El‐Assaad W, El‐Sabban M, Awaraji C, et al. Distinct sites of action of Bcl‐2 and Bcl‐xL in the ceramide pathway of apoptosis. Biochem J 1998;336(Pt 3):735–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. El‐Assaad W, Kozhaya L, Araysi S, et al. Ceramide and glutathione define two independently regulated pathways of cell death initiated by p53 in Molt‐4 leukaemia cells. Biochem J 2003;376(Pt 3):725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sawada M, Nakashima S, Banno Y, et al. Influence of Bax or Bcl‐2 overexpression on the ceramide‐dependent apoptotic pathway in glioma cells. Oncogene 2000;19:3508–3520. [DOI] [PubMed] [Google Scholar]
- 43. Tepper AD, de Vries E, van Blitterswijk WJ, Borst J. Ordering of ceramide formation, caspase activation, and mitochondrial changes during CD95‐ and DNA damage‐induced apoptosis. J Clin Investig 1999;103:971–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hernandez‐Corbacho MJ, Canals D, Adada MM, et al. Tumor necrosis factor‐alpha (TNFalpha)‐induced ceramide generation via ceramide synthases regulates loss of focal adhesion kinase (FAK) and programmed cell death. J Biol Chem 2015;290:25356–25373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Taha TA, Mullen TD, Obeid LM. A house divided: ceramide, sphingosine, and sphingosine‐1‐phosphate in programmed cell death. Biochem Biophys Acta 2006;1758:2027–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Michel C, vanEchten‐Deckert G , Rother J, et al., Jr. Characterization of ceramide synthesis. A dihydroceramide desaturase introduces the 4,5‐trans‐double bond of sphingosine at the level of dihydroceramide. The Journal of Biological Chemistry 1997;272:22432–22437. [DOI] [PubMed] [Google Scholar]
- 47. Kraveka JM, Li L, Szulc ZM, et al. Involvement of dihydroceramide desaturase in cell cycle progression in human neuroblastoma cells. J Biol Chem 2007;282:16718–16728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim WJ, Okimoto RA, Purton LE, et al. Mutations in the neutral sphingomyelinase gene SMPD3 implicate the ceramide pathway in human leukemias. Blood 2008;111:4716–4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Osawa Y, Suetsugu A, Matsushima‐Nishiwaki R, et al. Liver acid sphingomyelinase inhibits growth of metastatic colon cancer. J Clin Investig 2013;123:834–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jana A, Hogan EL, Pahan K. Ceramide and neurodegeneration: susceptibility of neurons and oligodendrocytes to cell damage and death. J Neurol Sci 2009;278:5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhao L, Spassieva SD, Jucius TJ, et al. A deficiency of ceramide biosynthesis causes cerebellar purkinje cell neurodegeneration and lipofuscin accumulation. PLoS Genet 2011;7:e1002063. [DOI] [PMC free article] [PubMed] [Google Scholar]