

Abstract
Mutations in FARS2, the gene encoding the mitochondrial phenylalanine‐tRNA synthetase (mtPheRS), have been linked to a range of phenotypes including epileptic encephalopathy, developmental delay, and motor dysfunction. We report a 9‐year‐old boy with novel compound heterozygous variants of FARS2, presenting with a pure spastic paraplegia syndrome associated with bilateral signal abnormalities in the dentate nuclei. Exome sequencing identified a paternal nonsense variant (Q216X) lacking the catalytic core and anticodon‐binding regions, and a maternal missense variant (P136H) possessing partial enzymatic activity. This case confirms and expands the phenotype related to FARS2 mutations with regards to clinical presentation and neuroimaging findings.
Introduction
Mitochondrial aminoacyl‐tRNA synthetases (mt‐aaRSs) play an integral role in mitochondrial protein synthesis and translation by charging transfer RNAs (tRNA) with their associated amino acids. To date, most mt‐aaRSs mutations involve a wide spectrum of central nervous system disorders with a broad range of phenotypes.1 For example, mutations in GARS cause peripheral neuropathy, MARS2 mutations have been associated with spastic ataxia with leukoencephalopathy, and DARS2 variants have been implicated in leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation.1, 2, 3, 4
FARS2 is a nuclear gene encoding the mitochondrial phenylalanyl‐tRNA synthetase (mtPheRS; MIM#611592), which transfers phenylalanine to its specific mitochondrial tRNAPhe. mtPheRS contains four major domains: a mitochondrial localization signal (residues 1–37) in the N‐terminal region (residues 1–83), a catalytic domain (residues 84–325), a linker region (residues 326–358), and an anticodon‐binding domain (359–451).5 Similar to many mt‐aaRSs, FARS2 has been linked with a severe mitochondrial syndrome associated with an infantile‐onset epileptic encephalopathy and/or fatal Alpers‐like syndrome.5, 6, 7, 8, 9 More moderate phenotypes consisting of cognitive disability, motor delays, and epilepsy have also been reported.10, 11 In its mildest form, FARS2 presented with a pure spastic paraplegia syndrome in a consanguineous Chinese family with homozygous mutations (spastic paraplegia type 77; SPG77 MIM#617046).12
We report a 9‐year‐old boy with progressive lower extremity spasticity and normal cognition associated with bilateral signal abnormalities in the dentate nuclei. Exome sequencing revealed novel compound heterozygous FARS2 mutations: a paternal nonsense variant upstream of the catalytic core and anticodon‐binding region (c.646 C>T; p.Q216X) and a maternal missense variant (c.407 C>A; p.P136H) associated with decreased enzyme activity. This combination of alleles with null and partial activity resulted in this unique case of SPG77.
Material and Methods
A detailed description of materials and methods used can be found in the Data S1.
Results
Subject
The subject was a 9‐year‐old boy, born to healthy nonconsanguineous parents. His parents were of northern European and Ashkenazi Jewish ancestry and were without neurological issues. Family history was negative. His early development was normal. He started walking at 14 months and was speaking in sentences by age 2. At approximately 2.5 years of age, the patient was noted to develop toe‐walking with a tendency to trip and skip. His ambulation has progressively worsened, but his development has otherwise been normal.
Examination showed spasticity in his lower extremities with preserved bulk and mild weakness. Deep tendon reflexes were brisk in both legs. He had bilateral extensor plantar responses and unsustained ankle clonus. He had a spastic gait with toe‐walking. Sensation and coordination were normal. Diagnostic studies included neuroimaging at 9 years with nonspecific signal abnormalities in the dentate nuclei consistent with edema (hypointense abnormalities on T1‐weighted images and hyperintense abnormalities on T2‐weighted images; Fig. 1); otherwise, serum lactate, plasma amino acids (including alanine), urine organic acids, and acylcarnitine profile were normal.
Figure 1.
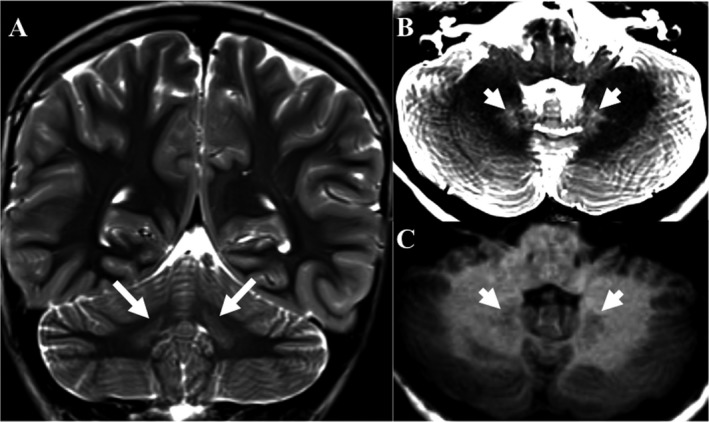
Magnetic resonance imaging of the brain (A) T2‐weighted coronal and axial (B) images demonstrates abnormal signal hyperintensities in the bilateral dentate nuclei (arrows and arrowheads, respectively) (C) T1‐weighted axial images demonstrated abnormal signal hypointensities. Together these findings are consistent with localized edema of the dentate nuclei.
Genetic findings
Exome sequencing identified two novel variants. One paternal nonsense variant, (c.646 C>T; p.Q216X) and a maternal missense variant, (c.407 C>A; p.P136H) (Fig. 2). Neither of these were observed in ~6500 individuals in the NHLBI Exome Sequencing Project. The P136H variant is a nonconservative amino acid substitution at a residue that is conserved among vertebrates (Fig. 2). The new residue differs in size, polarity, and charge. The Q216X variant truncates mtPheRS near its midpoint, omitting the enzymatic catalytic core and anticodon‐binding domain (Fig. 2).
Figure 2.
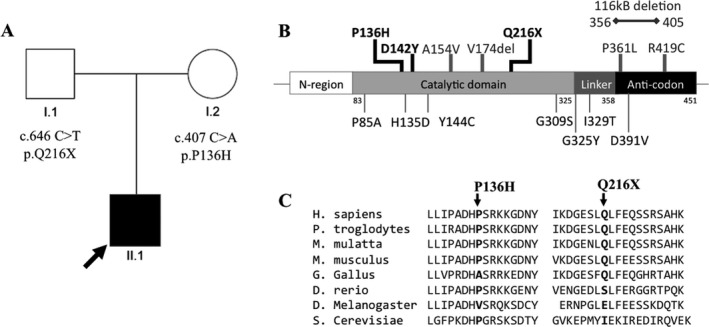
Pedigree and FARS2 mutations. (A) Family Pedigree. The proband is indicated by an arrow and the affected individual is in black. (B) FARS2 gene schematic and location of mutations. Our subject's mutations (P136H and Q216X) and previously reported cases with pure spastic paraplegia are in black. Previously reported moderate cases are in gray and above the schematic, previously reported severe cases are in gray and below the schematic. (C) Protein homology at mutated residues across species. The P136 and Q216 residues are conserved across most vertebrate species.
Biochemical evaluation
Both the wild‐type‐ (WT) and P136H‐mtPheRS recombinant proteins were overexpressed and purified; however, the Q216X variant was unable to be isolated despite numerous attempts with nickel affinity and anion exchange columns. We were unable to isolate the Q216X protein in the context of the c.646 C>T point mutation, nor in our attempt to specifically only express the first 216 amino acids of mtPheRS. This was presumably due to the instability or protease degradation of the truncated peptide and indicates that the truncated protein has no residual activity as a result of this instability.
Aminoacylation activity of the WT and P136H mtPheRS was evaluated (Fig. 3). The P136H mutant had a 50% reduction in activity compared to the WT enzyme (Fig. 3). This was further shown to equate to a 10‐fold decrease in the k cat/K M for P136H compared to the WT mtPheRS (Fig. 3). A large decrease in k cat/K M indicates a potentially physiologically significant reduction in the efficiency with which tRNAPhe can be aminoacylated by phenylalanine. Thermostability assays were completed with active site titrations after 1‐h incubation at room temperature and 37°C. These studies revealed that mtPheRS‐P136H's thermal stability was similar to WT (Fig. S1).
Figure 3.
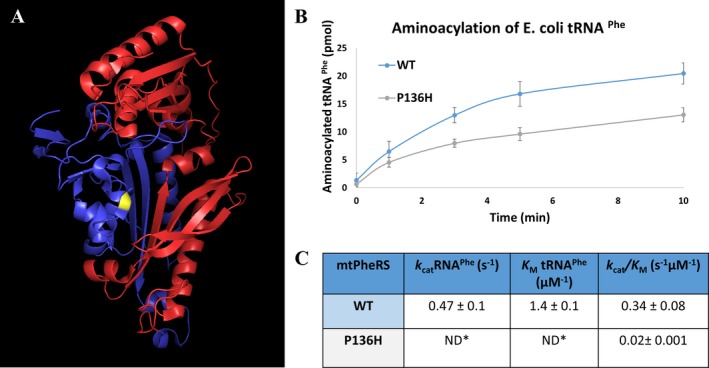
Biochemical analysis of variants. (A) Crystal structure of mtPheRS. Yellow residue denotes the P136H residue. Red region denotes the portion of mtPheRS removed in Q216X truncation. Blue region is the portion of mtPheRS present in Q216X variant15 (B) Aminoacylation curve of generation of Phe‐tRNAPhe. Blue line is WT mtPheRS and gray line is P136H mtPheRS. (C) Steady‐state kinetic parameters tRNAPhe. *Individual kinetic parameters could not be obtained due to the high K M compared to practical tRNA concentrations ([S] ≪ [E]). Therefore, the k cat/K M was directly estimated from V = k cat/K M([E][S]).
Discussion
Pure spastic paraplegia consists of lower extremity spasticity and dysfunction, without other neurological or cognitive issues. Our subject possessed novel compound heterozygous mutations in FARS2 (Q216X and P136H) resulting in a pure spastic paraplegia syndrome that was also associated with bilateral signal abnormalities of the dentate nuclei. His phenotype was similar to a previously reported consanguineous Chinese family with pure spastic paraplegia due to homozygous mutations within the catalytic domain of mtPheRS (D142Y), although their neuroimaging was normal.12 To date, these subjects represent the mildest cases of FARS2‐related disease. Overall, approximately 21 subjects with FARS2‐related disease have been reported, with most cases (12 of 21; see Table S1) presenting with a severe phenotype consisting of epileptic encephalopathy and diffuse cortical dysfunction (lower extremity spasticity, motor delays, and cognitive disability). Many of these severe cases were also associated with lactic acidosis or liver disease.6, 7, 8 Several moderate phenotypes have also been reported with diffuse cortical dysfunction, but had more functional capabilities.10, 11, 12 Examples included two siblings with compound heterozygous mutations (R419C & microdeletion) presenting with intellectual disability, truncal hypotonia, appendicular hypertonicity, and neonatal epilepsy).11 Additional moderate subjects included two unrelated children with diffuse cortical dysfunction and lactic acidosis.10 One child (P361L & V174del) had additional findings of bradykinesia and dystonia, whereas the other (P361L & A154V) also developed epilepsy. Both had neuroimaging abnormalities in the frontopontine pyramidal tracts, thalamus, and cerebellar atrophy.10 In contrast to the subjects with pure spastic paraplegia, these individuals could be classified as complex spastic paraplegia due to their combination of spasticity and other significant neurological issues.
The variable severity of the FARS2 phenotypes may be related to the amount of residual mtPheRS activity each individual possesses. This residual activity may play a role in both neuro‐development, as well as the long‐term maintenance of the nervous system. Previous work revealed mitochondrial dysfunction can disrupt migration of cortical interneurons and lead to epileptic encephalopathy and/or intellectual disability.13 One could hypothesize that individuals with reduced mtPheRS activity leading to severe FARS2 phenotypes (associated with larger energy deficits) could possess these migrational anomalies, as well as being vulnerable to developing later‐onset injury in cells with higher energy requirements (e.g., cerebellar Purkinje or cortical pyramidal cells), leading to their complex spastic paraplegia. Alternatively, individuals with higher residual mtPheRS activity may generate enough energy for normal interneuron migration, resulting in their phenotype solely being a function of later‐onset injury to cells with higher energy requirements. Our subject had residual mtPheRS activity as a result of a partially active substitution mutation (P136H) combined with an unstable null truncation variant (Q216X). This may be why his phenotype is currently limited to a pure spastic paraplegia syndrome.
Although our subject presented with the same phenotype as the consanguineous Chinese family, his neuroimaging revealed bilateral signal abnormalities of the dentate nuclei. Of note, our subject was not the only FARS2‐related patient with changes in this location. Similar abnormalities were also seen in a child with a more severe clinical phenotype and additional neuroimaging features that included a thin corpus callosum, white matter changes, and diffuse cerebral cortical atrophy.9 Interestingly, another case with a similar presentation to our subject involved SARS2, a gene encoding a mt‐aaRS for serine.14 In that report, homozygous splice site mutations led to partial activity of the mitochondrial seryl‐tRNA synthetase (mtSerRS) resulting in spastic paraplegia and mild intellectual disability. Interestingly, this child also presented with increased T2 hyperintensities in the dentate nuclei with later development of cerebellar atrophy. Both our subject and this child were without clinical cerebellar findings. It may be that in both subjects’ dentate changes are not contributing to their spasticity; nevertheless, cerebellar dysfunction is frequently associated with spastic paraplegia and in this context, may be playing a role. It could also be that with time, both subjects develop cerebellar symptoms. The identification and long‐term follow‐up of more FARS2 or other mt‐aaRS patients with pure spastic paraplegia will be required to better understand these associations.
Aminoacyl‐tRNA synthetases have been associated with a range of symptoms from early‐onset mitochondrial phenotypes to milder forms with later‐onset neurological findings. The more common phenotype of FARS2 disorders is a severe disorder with epileptic encephalopathy and severe neurological abnormalities affecting the function of deep gray nuclei and the cerebral cortex. We report a subject presenting with pure spastic paraplegia without familial consanguinity. This case further underscores the idea that the phenotype related to FARS2 and other mt‐aaRSs may have a wide spectrum of disease severity. The subject reported here will require continued monitoring to determine if his course has plateaued or may progress; however, in the context of reported FARS2‐related disease, it may be that pure spastic paraplegia is the mildest presentation.
Conflict of Interest
None declared.
Author Contributions
S.K.S., J.M.G., M.A., N.S., and T. M. P. evaluated subject and collected clinical data. R.S., M.I., and T. M. P. conceived and designed the experiments. R.S., M.I., and T. M. P. performed the experiments. S.K.S., R.S., M.I., J.M.G., M.A., N.S., and T. M. P. analyzed the data. S.K.S., R.S., M.I., and T. M. P. wrote the manuscript.
Supporting information
Data S1.
Table S1. Currently Identified FARS2 Subjects and Data.
Figure S1. Thermostability of mtPheRS mutant.
Acknowledgments
We are grateful to Kelli Dejohn, Carmela Brito, Joanne Baez, and Barrington Burnett for clinical and technical assistance, and critical analysis. pET21c‐mtPheRS was a generous gift from L. Spremulli, University of North Carolina, Chapel Hill, NC, USA. We especially thank the family of our patient for the loving care for their child and cooperation with our work. MI was funded by the National Science Foundation (MCB 1715840) and RS by an Ohio State University Center for RNA Biology fellowship. TMP was funded by the Cedars‐Sinai institutional funding program and the Cedars‐Sinai Diana and Steve Marienhoff Fashion Industries Guild Endowed Fellowship in Pediatric Neuromuscular Diseases.
Funding Information
MI was funded by the National Science Foundation (MCB 1715840) and RS by an Ohio State University Center for RNA Biology fellowship. TMP was funded by the Cedars‐Sinai institutional funding program and the Cedars‐Sinai Diana and Steve Marienhoff Fashion Industries Guild Endowed Fellowship in Pediatric Neuromuscular Diseases.
Funding Statement
This work was funded by National Science Foundation grant MCB 1715840; Ohio State University Center grant ; Cedars‐Sinai institutional funding program grant ; Cedars‐Sinai Diana grant ; Steve Marienhoff Fashion Industries Guild Endowed Fellowship grant .
References
- 1. Musante L, Püttmann L, Kahrizi K, et al. Mutations of the aminoacyl‐tRNA‐synthetases SARS and WARS2 are implicated in the etiology of autosomal recessive intellectual disability. Hum Mutat 2017;38:621–636. [DOI] [PubMed] [Google Scholar]
- 2. Yao P, Fox PL. Aminoacyl‐tRNA synthetases in medicine and disease. EMBO Mol Med 2013;5:332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stranneheim H, Wedell A. Exome and genome sequencing: a revolution for the discovery and diagnosis of monogenic disorders. J Intern Med 2016;279:3–15. [DOI] [PubMed] [Google Scholar]
- 4. Datt M, Sharma A. Evolutionary and structural annotation of disease‐associated mutations in human aminoacyl‐tRNA synthetases. BMC Genom 2014;15:1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Almalki A, Alston CL, Parker A, et al. Mutation of the human mitochondrial phenylalanine‐tRNA synthetase causes infantile‐onset epilepsy and cytochrome c oxidase deficiency. Biochim Biophys Acta 2014;1842:56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Elo JM, Yadavalli SS, Euro L, et al. Mitochondrial phenylalanyl‐tRNA synthetase mutations underlie fatal infantile Alpers encephalopathy. Hum Mol Genet 2012;21:4521–4529. [DOI] [PubMed] [Google Scholar]
- 7. Cho JS, Kim SH, Kim HY, et al. FARS2 mutation and epilepsy: possible link with early‐onset epileptic encephalopathy. Epilepsy Res 2017;129:118–124. [DOI] [PubMed] [Google Scholar]
- 8. Walker MA, Mohler KP, Hopkins KW, et al. Novel compound heterozygous mutations expand the recognized phenotypes of FARS2 ‐linked disease. J Child Neurol 2016;31:1127–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Raviglione F, Conte G, Ghezzi D, et al. Clinical findings in a patient with FARS2 mutations and early‐infantile‐encephalopathy with epilepsy. Am J Med Genet Part A 2016;170:3004–3007. [DOI] [PubMed] [Google Scholar]
- 10. Vantroys E, Larson A, Friederich M, et al. New insights into the phenotype of FARS2 deficiency. Mol Genet Metab 2017;122:172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vernon HJ, McClellan R, Batista DA, Naidu S. Mutations in FARS2 and non‐fatal mitochondrial dysfunction in two siblings. Am J Med Genet Part A 2015;167:1147–1151. [DOI] [PubMed] [Google Scholar]
- 12. Yang Y, Liu W, Fang Z, et al. A newly identified missense mutation in FARS2 causes autosomal‐recessive spastic paraplegia. Hum Mutat 2016;37:165–169. [DOI] [PubMed] [Google Scholar]
- 13. Lin‐Hendel EG, McManus MJ, Wallace DC, et al. Differential mitochondrial requirements for radially and non‐radially migrating cortical neurons: implications for mitochondrial disorders. Cell Rep 2018;15:229–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Linnankivi T, Neupane N, Richter U, et al. Splicing defect in mitochondrial Seryl‐tRNA synthetase gene causes progressive spastic paresis instead of HUPRA syndrome. Hum Mutat 2016;37:884–888. [DOI] [PubMed] [Google Scholar]
- 15. Klipcan L, Levin I, Kessler N, et al. The tRNA‐induced conformational activation of human mitochondrial phenylalanyl‐tRNA synthetase. Cell 2008;16:1095–1104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.
Table S1. Currently Identified FARS2 Subjects and Data.
Figure S1. Thermostability of mtPheRS mutant.