

Abstract
This case study examined the impact of a respiratory strength training program targeting inspiratory and expiratory musculature in an individual with C9orf72 amyotrophic lateral sclerosis (ALS). The individual tolerated 24 months of respiratory training completed at home, 50 repetitions per day, and 5 days per week. Significant increases in maximum inspiratory pressure (from 71 to 134 centimeters of water), maximum expiratory pressure (from 108 to 197 centimeters of water) and peak cough flow (from 331 to 655 Liters per minute) were noted and forced vital capacity remained unchanged. A moderate intensity respiratory strength training program applied early in the disease progression improved function in this C9orf72 ALS individual.
Introduction
Amyotrophic lateral sclerosis (ALS) is a terminal neurodegenerative disease characterized by the rapid and progressive deterioration of upper and lower motor neurons resulting in progressive muscle paralysis. The disease typically presents focally in either the limb or bulbar muscles, but gradually spreads leading to impairments in mobility, speech, swallowing, and respiration.1 Respiratory failure represents the leading terminal event in ALS.2 Involvement of inspiratory muscles leads to impaired ventilation while expiratory muscle weakness reduces subglottic air pressure generation, airway clearance physiologic ability (cough production) and increases the occurrence of pulmonary infection.2 Pulmonary infection occurs as a result of bulbar muscle impairment and the inability to adequately protect the airway during swallowing, leading to aspiration pneumonia3 and increased risk of malnutrition, which negatively impacts survival in ALS.4
Current management strategies for bulbar and respiratory impairment in ALS include: assistive ventilation, mechanical insufflation‐exsufflation, assistive communication devices, compensatory swallowing strategies, diet modification, and feeding tube placement.1 These therapeutic strategies are palliative in nature and are typically implemented to compensate for the loss of function, energy conservation, pain management, and to optimize quality of life. The role of active interventions such as exercise are controversial in this patient population, however, ALS animal models5 and human clinical trials6 suggest mild intensity exercise programs applied early in disease progression might serve a beneficial role in maintaining function and improving overall survival.
Expiratory muscle strength training (EMST) is an exercise technique noted to improve subglottic air pressure generation, airway clearance abilities, and swallowing in several neurologic patient populations.7 We recently applied EMST in ALS patients to determine its feasibility, safety and impact on bulbar and respiratory outcomes and reported that a 5‐week program of EMST led to significant improvements in maximum expiratory pressure generation, and, in one individual, improved airway protection.8 We also reported a case study of an individual with ALS who underwent 8‐weeks of EMST with a noted 102% improvement in subglottic air pressure generation and improved cough production.9
Given the occurrence of both inspiratory and expiratory muscle impairment in ALS, we wanted to extend our previous work to determine the effects of a combined respiratory strength program that includes both inspiratory (IMST) and expiratory muscles to target the vital functions of both ventilation (inspiratory) and airway clearance (expiratory). Here we present a case study of an ALS individual who underwent combined respiratory strength training over 24‐months.
Methods
Participant
A.C. is a 58‐year‐old male who presented to our laboratory 2 months post diagnosis with bulbar‐onset ALS (El‐Escorial Revised Criteria) and a confirmed chromosome 9 open reading frame 72 (C9orf72) ALS gene mutation (see Table 1 for demographics). He reported slurred speech 1.5 years prior to diagnosis; therefore, he was a slowly progressing ALS patient. This study received approval from our university's Institutional Review Board and the participant signed an informed consent (IRB201501172).
Table 1.
Summary of physiologic data for A.C. over the 24‐month treatment period
| Outcome | Baseline | 3‐Month | 6‐Month | 9‐Month | 12‐Month | 18‐Month | 24‐Month | Change (raw, %) |
|---|---|---|---|---|---|---|---|---|
| MIP (cmH20) | 71 | 129 | 125 | 113 | 140 | 133 | 134 | +63 (+88.7%) |
| MEP (cmH20) | 108 | 144 | 139 | 148 | 186 | 186 | 197 | +89 (+82.4%) |
| FVC (%Predicted) | 104% | 102% | 112% | 109% | 114% | 101% | 104% | 0% (±0%) |
| PEF (L/min) | 331 | 618 | 570 | 474 | 544 | 642 | 655 | +324 (+97.8%) |
|
ALSFRS‐R ALSFRS‐R (b) ALSFRS‐R (r) |
46 10 12 |
45 10 12 |
44 9 12 |
44 9 12 |
−2 (−4.35%) −1 (−10%) 0 (±0%) |
MEP, maximum expiratory pressure; MIP, maximum inspiratory pressure; FVC, forced vital capacity; PEF, peak expiratory flow; ALSFRS‐R, amyotrophic lateral sclerosis functional rating scale revised total score; ALSFRS‐R (b), amyotrophic lateral sclerosis functional rating scale revised bulbar subscale score; ALSFRS‐R (r), amyotrophic functional rating scale revised respiratory subscale score.
Assessments
Pulmonary function
Maximum inspiratory pressure (MIP) and maximum expiratory pressure (MEP) were measured in accordance with the American Thoracic Society (ATS) guidelines. The inspiratory or expiratory pressure valve was fitted to the manometer (Micro RPM Pressure Meter, MDSpiro/Micro Direct, Lewiston, ME), a bacterial filter was attached to the valve and a rubber‐flanged mouthpiece (MTH6400, MDSpiro/Micro Direct, Lewiston, ME) was utilized, allowing positioning in the mouth without labial leakage. During testing, the participant was seated comfortably with a nose clip in place. Three trials were completed for both MIP and MEP's and the highest value obtained used for trainer calibration and data analysis. Forced vital capacity (FVC) was assessed in a seated position wearing nose clips and using a Micro I handheld spirometer (Carefusion, Yorba Linda, CA) in accordance with ATS guidelines. Raw FVC (L) and the percent predicted in accordance for age, height, gender, and race10 were recorded.
Voluntary cough
Voluntary cough strength was measured using a digital peak flow meter (Microlife PF 100; Microlife, Clearwater, FL). The patient was in the seated position, with nose clips in place, and instructed to “cough hard like something is stuck in your throat”. Three trials were completed and the highest value for peak cough flow (L/min) was recorded.
Global disease progression
To index A.C.'s global disease progression throughout the 2‐year period, the validated Amyotrophic Lateral Sclerosis Functional Rating Scale Revised11 (ALSFRS‐R) survey was administered every 6 months.
Respiratory training program
Immediately following his initial evaluation, A.C. commenced an at‐home inspiratory and expiratory muscle strength training program using calibrated hand‐held resistance trainers. Inspiratory breath sets were completed using a Phillips Threshold Inspiratory Muscle Trainer (Respironics, Murraysville, PA) and expiratory repetitions were completed using the Expiratory Muscle Strength Trainer (EMST 150, Gainesville, FL). Respiratory trainers were calibrated at 30% of A.C.'s inspiratory and expiratory pressure generating capacities to reflect a moderate training load. A.C. was instructed to complete 25 inspiratory and 25 expiratory repetitions, 5 days per week, resulting in 50 daily repetitions and 250 weekly repetitions.
Data analysis
Descriptive statistics were utilized to report A.C.'s performance across the physiologic domains over the training period.
Results
Patient compliance and tolerability
A.C. self‐reported completing 100% of trainings without any reports of fatigue, discomfort or adverse effects.
Maximum inspiratory and expiratory pressure
Following 24 months of respiratory training, MIPs increased by 89% from 71 cmH2O to 134 cmH2O. MEPs increased by 82% from 108 cmH2O to 197 cmH2O.
Pulmonary function
No change in the raw FVC value was noted over the 24‐month period (5.02 L vs. 4.95 L), and the associated percent predicted value remained stable at 104% predicted.
Voluntary cough production
A.C.'s baseline PEF was 331 L/min and increased by 98% to 655 L/min after 24‐months of respiratory training.
Global disease progression
Total ALSFRS‐R score for A.C. dropped by two points (from 46 to 44) over the treatment period. The respiratory subscale score remained stable at 12, and the bulbar subscale score declined from 10 to 9. A.C.'s response for the handwriting question declined from a score of 4 to 3.
Discussion
To our knowledge, this case report represents the first documentation of combined respiratory training in ALS to target both ventilatory (inspiratory training) and airway clearance (expiratory training) physiologic functions. In this individual with confirmed C9orf72 ALS, combined respiratory strength training was safe, feasible, and lead to significant increases in MIP's, MEP's and peak cough flow. Pulmonary function (FVC) was maintained and global disease progression was noted to be slower than established rates of ALS decline.12
A.C.'s MIP improved by 89% over baseline (71–134 cmH2O) following respiratory training (see Figure 1). A previous study investigating IMST in ALS reported non‐significant improvements in MIPs following 3‐months of training.13 The current treatment paradigm extended beyond this previous investigation and demonstrates the benefit of continued inspiratory exercise despite advancing disease duration. A.C. increased and maintained MIP values, suggesting improvement to inspiratory pressure physiologic reserve that may have clinically significant implications. Importantly, progressive inspiratory muscle weakness has been identified as the main contributor to inadequate ventilation in patients with neuromuscular disease.3 Further, reduced MIP and MEP values are predictive indices of tracheostomy free survival in ALS with MIPs also serving as a primary indicator of diaphragm muscle weakness.14 In addition to prolonging the vital function of ventilation, improved MIP may facilitate more positive outcomes regarding survival time and preservation of diaphragm muscle strength.
Figure 1.
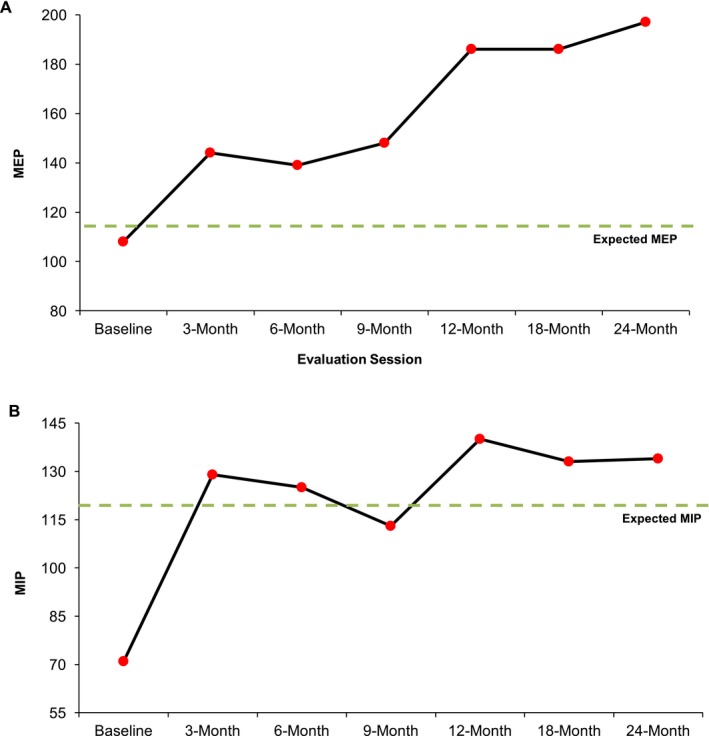
(A) Maximum expiratory pressure (MEP cmH2O) and (B) Maximum inspiratory pressure (MIP cmH2O) measurements for the combined respiratory training participant over the 24‐month training period. At baseline, A.C.'s MEP was 108 cmH2O representing a 6 cmH2O reduction as compared to established normative data based on his age and gender. His baseline MIP (71 cmH2O) was reduced by 47 cmH2O. After 24 months of training, his MEP and MIP improved by 89% and 82%, respectively, and are presently above expected values for his age and gender.
A.C.'s MEP improved 82% from baseline (108–197 cmH2O; see Figure 1). A case study in a Parkinson's disease patient reported similar findings of improved MEP following 20 weeks of EMST.15 Expiratory muscle weakness and inability to generate subglottic air pressure contributes to ineffective cough production and, thus, an increasingly diminished ability to protect the airway during swallowing.3 Indeed reduced voluntary cough strength has been associated with unsafe swallowing in ALS.16 Increased subglottic air pressure generation capacity likely contributed to the noted increases in peak cough flow that more than doubled (from 331 to 655 L/min). This finding is consistent with other reports of improvements in voluntary cough measures following a program of EMST.17 Since A.C. presented with bulbar‐onset ALS, maintenance of adequate airway protection and the ability to expel and clear secretions or tracheal aspirate is of great clinical significance. This is particularly relevant in this patient whose baseline peak cough flow was very close to the threshold needed for an effective cough (270 L/min).18
A.C. demonstrated relative maintenance of pulmonary function and global disease progression compared to established ALS rates of decline for FVC (3.5% per month19) and ALSFRS‐R scores (0.8–0.9 points per month12). It is unclear if disease maintenance is related to the experimental behavioral intervention or the potential he represents a slow progressing patient. Future studies in other ALS individuals will help to elucidate the disease modifying potential of this active intervention.
Limitations
Given that this is a case study in a slow progressing patient, results should be interpreted with caution. Furthermore, previous research has examined detraining effects following EMST.20 A.C. continuously trained during the study, therefore, it remains unclear how a detraining period might affect the observed improvements in function.
Conclusions
The results observed provide emerging evidence that a combined respiratory intervention applied early in the disease is feasible, safe, and contributes to improved function. We are currently conducting a large randomized sham‐controlled clinical trial to investigate the impact of the same respiratory strength training program that will serve to validate these findings.
Authorship Contributions
R. R., L. C. T.‐G., J. P. W., and E. K. P. were involved in the clinical treatment and data collection of the patient. R. R. and E. K. P. were involved in data analysis and drafting of the manuscript. EKP was involved in study conceptualization and data management. All authors reviewed the manuscript.
Conflict of Interest
The authors declare that they have no conflict of interest.
Funding Information
This study was funded by the ALS Association [clinical management grant 17‐CM‐323].
Funding Statement
This work was funded by ALS Association grant 17‐CM‐323.
References
- 1. Wijesekera LC, Leigh PN. Amyotrophic lateral sclerosis. Orphanet J Rare Dis 2009;4:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gruis KL, Lechtzin N. Respiratory therapies for amyotrophic lateral sclerosis: a primer. Muscle Nerve 2012;46:313–331. [DOI] [PubMed] [Google Scholar]
- 3. Epstein SK, Parsons PE, Morrison RS. Respiratory muscle weakness due to neuromuscular disease: clinical manifestations and evaluation. UpToDate [online serial]. Waltham, MA: UpToDate 2010.
- 4. Desport JC, Preux PM, Truong TC, et al. Nutritional status is a prognostic factor for survival in ALS patients. Neurology 1999;53:1059–1063. [DOI] [PubMed] [Google Scholar]
- 5. Carreras I, Yuruker S, Aytan N, et al. Moderate exercise delays the motor performance decline in a transgenic model of ALS. Brain Res 2010;1313:192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clawson LL, Cudkowicz M, Krivickas L, et al. A randomized controlled trial of resistance and endurance exercise in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 2018;19:250–258. [DOI] [PubMed] [Google Scholar]
- 7. Laciuga H, Rosenbek JC, Davenport PW, Sapienza CM. Functional outcomes associated with expiratory muscle strength training: narrative review. J Rehabil Res Dev 2014;51:535–546. [DOI] [PubMed] [Google Scholar]
- 8. Plowman EK, Watts SA, Tabor L, et al. Impact of expiratory strength training in amyotrophic lateral sclerosis. Muscle Nerve 2016;54:48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tabor LC, Rosado KM, Robison R, et al. Respiratory training in an individual with amyotrophic lateral sclerosis. Ann Clin Transl Neurol 2016;3:819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med 1999;159:179–187. [DOI] [PubMed] [Google Scholar]
- 11. Cedarbaum JM, Stambler N, Malta E, et al. The ALSFRS‐R: a revised ALS functional rating scale that incorporates assessments of respiratory function. J Neurol Sci 1999;169:13–21. [DOI] [PubMed] [Google Scholar]
- 12. McElhiney M, Rabkin JG, Goetz R, et al. Seeking a measure of clinically meaningful change in ALS. Amyotroph Lateral Scler Frontotemporal Degener 2014;15:398–405. [DOI] [PubMed] [Google Scholar]
- 13. Cheah BC, Boland RA, Brodaty NE, et al. INSPIRATIonAL–INSPIRAtory muscle training in amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2009;10:384–392. [DOI] [PubMed] [Google Scholar]
- 14. Heffernan C, Jenkinson C, Holmes T, et al. Management of respiration in MND/ALS patients: an evidence based review. Amyotroph Lateral Scler 2006;7:5–15. [DOI] [PubMed] [Google Scholar]
- 15. Saleem AF, Sapienza CM, Okun MS. Respiratory muscle strength training: treatment and response duration in a patient with early idiopathic Parkinson's disease. NeuroRehabilitation 2005;20:323–333. [PubMed] [Google Scholar]
- 16. Plowman EK, Watts SA, Robison R, et al. Voluntary cough airflow differentiates safe versus unsafe swallowing in amyotrophic lateral sclerosis. Dysphagia 2016;31:383–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sapienza CM, Wheeler K. Respiratory muscle strength training: functional outcomes versus plasticity. Semin Speech Lang 2006;27:236–244. [DOI] [PubMed] [Google Scholar]
- 18. Servera E, Sancho J, Zafra MJ. Cough and neuromuscular diseases. Noninvasive airway secretion management. Arch Bronconeumol 2003;39:418–427. [DOI] [PubMed] [Google Scholar]
- 19. Lechtzin N, Rothstein J, Clawson L, et al. Amyotrophic lateral sclerosis: evaluation and treatment of respiratory impairment. Amyotroph Lateral Scler Other Motor Neuron Disord 2002;3:5–13. [DOI] [PubMed] [Google Scholar]
- 20. Troche MS, Rosenbek JC, Okun MS, Sapienza CM. Detraining outcomes with expiratory muscle strength training in Parkinson disease. J Rehabil Res Dev 2014;51:305–310. [DOI] [PMC free article] [PubMed] [Google Scholar]