

Short abstract
Elevated N-methyl-D-aspartate receptor activity contributes to central sensitization. Our laboratories and others recently reported that disrupting protein–protein interactions downstream of N-methyl-D-aspartate receptors suppresses pain. Specifically, disrupting binding between the enzyme neuronal nitric oxide synthase and either its upstream (postsynaptic density 95 kDa, PSD95) or downstream (e.g. nitric oxide synthase 1 adaptor protein, NOS1AP) protein partners suppressed inflammatory and/or neuropathic pain. However, the lack of a small-molecule neuronal nitric oxide synthase-NOS1AP inhibitor has hindered efforts to validate the therapeutic utility of disrupting the neuronal nitric oxide synthase-NOS1AP interface as an analgesic strategy. We, therefore, evaluated the ability of a putative small-molecule neuronal nitric oxide synthase-NOS1AP inhibitor ZLc002 to disrupt binding between neuronal nitric oxide synthase and NOS1AP using ex vivo, in vitro, and purified recombinant systems and asked whether ZLc002 would suppress inflammatory and neuropathic pain in vivo. In vitro, ZLc002 reduced co-immunoprecipitation of full-length NOS1AP and neuronal nitric oxide synthase in cultured neurons and in HEK293T cells co-expressing full-length neuronal nitric oxide synthase and NOS1AP. However, using a cell-free biochemical binding assay, ZLc002 failed to disrupt the in vitro binding between His-neuronal nitric oxide synthase1-299 and glutathione S-transferase-NOS1AP400-506, protein sequences containing the required binding domains for this protein–protein interaction, suggesting an indirect mode of action in intact cells. ZLc002 (4–10 mg/kg i.p.) suppressed formalin-evoked inflammatory pain in rats and reduced Fos protein-like immunoreactivity in the lumbar spinal dorsal horn. ZLc002 also suppressed mechanical and cold allodynia in a mouse model of paclitaxel-induced neuropathic pain. Anti-allodynic efficacy was sustained for at least four days of once daily repeated dosing. ZLc002 also synergized with paclitaxel when administered in combination to reduce breast (4T1) or ovarian (HeyA8) tumor cell line viability but did not alter tumor cell viability without paclitaxel. Our results verify that ZLc002 disrupts neuronal nitric oxide synthase-NOS1AP interaction in intact cells and demonstrate, for the first time, that systemic administration of a putative small-molecule inhibitor of neuronal nitric oxide synthase-NOS1AP suppresses inflammatory and neuropathic pain.
Keywords: N-methyl-D-aspartate, central sensitization, neuronal nitric oxide synthase, NOS1AP, postsynaptic density 95 kDa (PSD95)
Introduction
Elevated N-methyl-D-aspartate receptor (NMDAR) activity is one of the key mechanisms contributing to central sensitization.1,2 However, NMDAR antagonists have limited therapeutic applications due to unwanted side effects (e.g. motor impairment, memory deficits, cognitive dysfunction, dissociation from reality, and abuse liability).3–5 Strategies that counteract or prevent aberrant elevated NMDAR activity without altering the basal activity of NMDAR would, therefore, be advantageous. Our laboratories and others have previously demonstrated that disrupting protein–protein interactions downstream of NMDAR (i.e. NR2B-PSD95, PSD95-nNOS, and nNOS–NOS1AP) produces antinociceptive efficacy without unwanted side effects associated with NMDAR antagonists.6–10 We recently proposed that the nNOS–NOS1AP protein–protein interface was a previously unrecognized target for analgesic drug development.11 We showed that TAT-GESV, a peptide inhibitor of the nNOS–NOS1AP interface, disrupted binding between nNOS and NOS1AP in vitro, suppressed glutamate-induced cell death in cultured cortical neurons, and produced antinociceptive efficacy in mechanistically distinct models of neuropathic pain.11 TAT-GESV, administered intrathecally (i.t.), suppressed mechanical and cold allodynia induced by either toxic challenge with the chemotherapeutic agent paclitaxel or traumatic nerve injury produced by a partial sciatic nerve ligation.11 These effects were not observed following the administration of a control peptide (e.g. TAT-GESVΔ1, which lacks the terminal valine residue of TAT-GESV) which also failed to disrupt nNOS–NOS1AP interactions.11 However, peptides are not ideal therapeutics due to limited bioavailability and poor pharmacokinetics.12,13 Moreover, the impact of nNOS–NOS1AP disruption on inflammatory pain is unknown. Systemically active small-molecule nNOS–NOS1AP inhibitors are needed to better evaluate the therapeutic potential of disrupting the nNOS–NOS1AP interface.
A putative small-molecule inhibitor of nNOS–NOS1AP, ZLc002 (Figure 1) was recently shown to exhibit anxiolytic-like efficacy in a mouse model of chronic mild stress without altering appetite, general activity, or locomotor activity or interfering with the resting potential of neurons.14 ZLc002 also inhibited co-immunoprecipitation of NOS1AP with nNOS in hippocampal cells.14 However, despite the promising preclinical therapeutic and side effect profile of ZLc002, it is unclear whether these effects occur through direct disruption of the nNOS–NOS1AP complex. This evaluation is important because the ability of ZLc002 to uncouple NOS1AP from nNOS in hippocampal cells ex vivo could occur through either direct or indirect mechanisms. Moreover, whether ZLc002 produces antinociceptive efficacy or suppresses neurochemical markers of pain-evoked neuronal activation is unknown.
Figure 1.
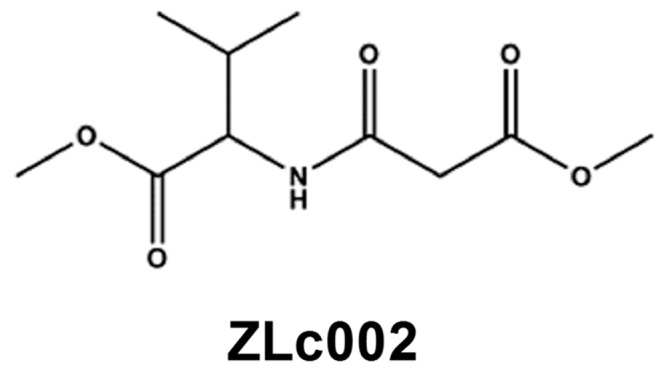
Chemical structure of ZLc002, putative small-molecule inhibitor of nNOS–NOS1AP interaction.
We evaluated the ability of ZLc002 to suppress inflammatory and neuropathic pain in vivo and further characterized its mechanism of action in vitro. We verified that ZLc002 disrupts nNOS–NOS1AP interaction in primary cultures of cortical neurons. We documented that, in a cell-free binding assay, ZLc002 did not directly disrupt binding between purified nNOS and NOS1AP containing the known interacting sites.15,16 We also established that, in intact HEK293T cells transfected with the full-length tagged proteins, ZLc002 disrupted the co-immunoprecipitation of nNOS with NOS1AP. We demonstrated that ZLc002 suppressed formalin-evoked pain behavior and neuronal activation in lumbar spinal dorsal horn. We established that ZLc002 suppressed the maintenance of chemotherapy-induced neuropathic pain induced by paclitaxel, which is used clinically to treat breast, ovarian, and lung cancers but produces dose-limiting toxic neuropathy in humans.17 Finally, we showed that ZLc002 acted synergistically with paclitaxel to enhance ovarian and breast cancer tumor cell line cytotoxicity in vitro.
Materials and methods
Drugs and chemicals
Peptides were purchased from GeneCust (Dudelange, Luxembourg) or GenicBio (Shanghai, China) with at least 95% of purity: L-TAT-GESV (GRKKRRQRRRYAGQWGESV); L-TAT-GESVΔ1 (GRKKRRQRRRYAGQWGES): lacking the last C-terminal valine residue. Peptides were dissolved in phosphate-buffered saline (PBS) for AlphaScreen. ZLc002 was synthesized by RTI international (Research Triangle Park, NC) for Anagin Inc. (Indianapolis, IN) and provided to the investigators. MK-801 and ZLc002 were dissolved in dimethyl sulfoxide (DMSO) (Sigma Aldrich, St. Louis, MO, USA) at 20 mM for AlphaScreen biochemical binding assays and in a vehicle composed of 3% DMSO, 1:1:18 of emulphor (Alkamuls EL 620L; Solvay), 95% ethanol (Sigma Aldrich), 0.9% NaCl (Aquilite System; Hospira, Inc, Lake Forest, IL) for in vivo administration. All drugs were delivered via intraperitoneal (i.p.) injection in a volume of 5 ml/kg for in vivo studies performed in mice and in a volume of 1 ml/kg for in vivo studies performed in rats. In the rat formalin study, ZLc002 and MK-801 were dissolved in a vehicle containing 20% DMSO (Sigma Aldrich), and the remaining 80% consisting of 95% ethanol, emulphor, and 0.9% saline at a ratio of 1:1:8 (final ratio of 5: 2: 2: 16). MK-801 and formaldehyde (37% in H2O) were purchased from Sigma Aldrich. Formalin was diluted to 2.5% in saline from formaldehyde stock (100% formalin) and administered via local intraplantar (i.pl.) injection in a volume of 50 µl.
Protein purification
Purification of glutathione S-transferase (GST), His-tagged nNOS, and NOS1AP was performed as previously described.9,18 nNOS1-299 containing the core PSD-95, Dlg (discs large homolog), and ZO-1 (zona occludens-1) (PDZ) domain that binds NOS1AP and the β-finger that binds to PDZ2 of PSD95, but lacking the catalytic domain, was expressed as an N-terminal His-tagged fusion protein. GST-NOS1AP400-506, containing the internal ExF motif and the C-terminal tail that is recognized by the core PDZ domain of nNOS, was expressed as an N-terminal GST-tagged fusion protein.
AlphaScreen assay
AlphaScreen assays were set up and performed as previously described.9 Briefly, binding between nNOS and NOS1AP was set up using His-nNOS1-299 and GST-NOS1AP400-506 proteins. AlphaScreen nickel chelate acceptor beads (PerkinElmer, Waltham, MA) and AlphaScreen glutathione donor beads (PerkinElmer) were sequentially added and incubated at room temperature for 1 h with each addition. The reaction was carried out in a 40 μl final volume using 96-well ½ area plates in 1X PBS containing bovine serum albumin (1 mg/mL) and Tween-20 (0.1%). An EnSpire® Multimode Plate Reader (PerkinElmer, Waltham, MA) equipped with AlphaScreen optical detection module was used to read plates. Titration was performed to determine 50% binding between His-nNOS1-299 and GST-NOS1AP400-506 (0–100 nM each). To test the disruption of the protein–protein binding by inhibitors, the reaction was carried out using concentrations of His-nNOS1-299 and GST-NOS1AP400-506 which lead to 50% of maximum binding. Inhibitors or vehicle (PBS or DMSO) were added to the protein pairs at the beginning of the experiments. All the peptides used in this experiment were dissolved in PBS. ZLc002 was dissolved in DMSO. Peptides and ZLc002 were prepared as 20 mM stocks, and subsequent dilutions were made from this stock for use in each assay. The concentrations of peptides and ZLc002 ranging from 0 to 100 µM were used to determine IC50. Each data point represents the mean % AlphaScreen signal count derived from at least four determinations (i.e. duplicate determinations obtained in two independent assays performed on separate days).
Cell culture and transfection
HEK293T cells were cultured in Dulbecco’s minimal essential medium (with 10% fetal bovine serum, 19.4 mM supplementary glucose, 2 mM glutamine, 50 μg/ml streptomycin sulfate, and 50 U/ml penicillin) at 37°C under a 5% CO2 humidified atmosphere. The cells were transfected by the calcium phosphate method as previously described19 with full-length plasmid DNA pEGFP-nNOSα (human sequence fused to enhanced green fluorescent protein) and full-length pLuc-NOS1AP (human, transcript variant 1, fused to firefly luciferase) or pLuc-PSD95-PDZ2 (encoding aa 159–249 of human Dlg4 transcript variant4, fused to firefly luciferase) as indicated, or empty luciferase vector pLuc-C1 as negative control. These constructs have been previously described in the studies by Li et al.15,18
Inhibitor treatment and co-immunoprecipitation assay using HEK293T cells
Twenty-two hours after transfection, HEK293T cells were treated in 0.5 mM probenecid-supplemented conditioned cell culture medium with or without 10 μM ZLc002 for 90 min. The probenecid was added because cell lines, in contrast to neurons, have a high rate of extrusion of esters like calcium dyes20 and, therefore, most likely ZLc002 also. Cells were then lysed in low stringency buffer21 supplemented with protease inhibitors, 1 mM DTT, and 0.5% Igepal CA-630 and precleared at 4°C by centrifugation at 20,000g. Cell lysates were precipitated by agarose-coupled single-chain anti-green fluorescent protein (GFP) camelid antibody-based protein (“GFP-Trap,” Chromotek) for 1 h at 4°C. The co-precipitated luciferase-fused proteins were measured using a tube luminometer and % co-immunoprecipitation determined by normalization to lysate levels of luciferase-fusion. Equal immunoprecipitation of the EGFP-nNOS in all samples was determined by Western blotting with anti-GFP antibody (mouse monoclonal clone JL8, RRID:AB_2313808, used at 0.1 μg/ml; Clontech).
Cortical neuronal culture
Cortical neuron cultures were prepared from P0 rats of either sex (mixed) as described previously.22 The isolation of cells and tissues from animals was performed in accordance with the corresponding local, national, and European Union regulations. The neurons were cultured in Neurobasal-A/B-27 medium (Thermofisher), and one third of medium was changed every three days.
Inhibitor treatment and co-immunoprecipitation assay using cortical neurons
At eight days in vitro, neuronal medium was replaced by minimum essential medium (MEM cat. # 11700077, Thermofisher) and neurons were then treated with or without 10 µM ZLc002 for 90 min. Following pretreatments for 90 min, neurons were stimulated with 50 µM NMDA for 10 min, followed by immediate lysis in low stringency buffer (LSB), supplemented with protease inhibitors, 1 mM DTT, and 0.5% Igepal CA-630 and precleared at 4°C by centrifugation at 20,000g.21 Immunoprecipitating (IP) antibody, nNOS (mouse monoclonal clone A-11, RRID: AB_626757, Santa Cruz Biotechnology, 2.5 μg/ml) was added to the lysate. Samples were rotated for 2 h at 4°C, after which 5 μl of protein-A resin (GenScript) was added, and rotation continued for 1 h. The resin was then washed three times with the LSB, and protein was eluted from drained resin by boiling at 95° for 10 min in SDS-PAGE sample loading buffer and analyzed by Western blotting.18,23 Immunoprecipitated nNOS and co-immunoprecipitated NOS1AP in all samples were determined by Western blotting with anti-nNOS antibody (A-11) and anti-NOS1AP antibody (rabbit polyclonal IgG, R-300, RRID: AB_2251417, Santa Cruz Biotechnology), respectively.
Tumor cell viability assay
4T1 mouse breast cancer cells were a gift from Dr Harikrishna Nashatri (IUPUI) and were maintained in RPMI-1640 supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin. HeyA8 human ovarian cancer cells were a gift from Dr Kenneth Nephew (IU Bloomington) and were maintained in DMEM supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin. All the cells were kept in a 37°C incubator equipped with 5% CO2. Tumor cell viability was measured with the 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay according to the manufacturer’s instructions (Roche, Indianapolis, IN), as described in our previously published work.24 Briefly, cells were seeded at a density of 3000 cells/well in a 96-well plate and cultured overnight. The following day, 4T1 and HeyA8 cells were treated with an increasing concentration of ZLc002 (0 − 50 µM), paclitaxel (0 − 500 nM), or the combination of both and incubated for a further 72 h. At the end of the incubation, 10 μL MTT solution (5 mg/mL) was added to each of the wells. After 4 h of incubation, 100 μL solubilization solution was added. The solubilized crystals were measured at optical density 570 nm. The effect of the drugs on cells was expressed as a percentage of viability compared to untreated cells. Data were derived from multiple experiments (n = 7 for 4T1 cell line and n = 3 for HeyA8), all performed on separate days; all data sets were normalized and subjected to nonlinear regression analysis to generate IC50. The combination response (additivity, synergy, or antagonism) was analyzed using Combenefit (Cancer Research UK Cambridge Institute; Cambridge, UK), a software tool that enables the visualization, analysis, and quantification of drug combination effects.25 The data from the combination treatments were processed using three synergy reference models: the Bliss independence model, the Loewe additivity model, and the highest single agent (HSA) model using Combenefit (‘Combination Benefit’).25
Subjects
Adult C57BL/6J male mice, weighing 23–33 g (Jackson Laboratory, Bar Harbor, ME), were used in the studies of chemotherapy-induced peripheral neuropathic pain produced by paclitaxel. Adult male Sprague Dawley rats, weighing 285–446 g (Envigo, Indianapolis, IN, USA), were used in the formalin study. The positive control (MK-801) and vehicle-treated groups that appear in the rat formalin study described here (vehicle and MK801 data points in Figure 5 only6) were published previously by our group as comparators in a separate evaluation of structurally distinct PSD95-nNOS protein–protein interaction disruptors (i.e. IC87201 and ZL006) and are included here with permission from the publisher.6 The vehicle- and MK-801-treated groups were tested, perfused, and processed concurrently, under blinded conditions, with the ZLc002-treated groups described for the first time in the present report, in accordance with our obligations to comply with the guidelines from the Association for Assessment and Accreditation of Laboratory Animal Care to reduce unnecessary animal use.6 Tissue from all subjects was processed concurrently for Fos immunohistochemistry. Animals were housed in a temperature-controlled facility (73 ± 2°F), 45% humidity under a 12-h light/dark cycle with standard rodent chow and water ad libitum. All experimental procedures were approved by the Bloomington Institutional Animal Care and Use Committee of Indiana University and followed guidelines of the International Association for the Study of Pain.
Figure 5.

ZLc002 reduces formalin-evoked pain behavior and Fos-like immunoreactivity in spinal dorsal horn. (a) ZLc002 (4 and 10 mg/kg, i.p.) reduced composite pain scores (CPS) from 30 to 50 min postformalin relative to vehicle. MK-801 (0.1 mg/kg) reduced CPS from 25 to 50 min postformalin relative to vehicle. (b) ZLc002 (4 and 10 mg/kg i.p.) (p < 0.001) and MK-801 (0.1 mg/kg i.p.) reduced formalin-evoked AUC of pain behavior scores relative to vehicle in phase 2 but not in phase 1 of formalin-evoked pain behavior. Data are mean ± S.E.M. (n = 5–6 per group) ***p < 0.001; **p < 0.01; *p < 0.05 vs. vehicle; ###p < 0.001 vs. all other groups, (two-way ANOVA followed by Bonferroni’s multiple comparison test). MK-801 and vehicle groups were published previously but run and processed concurrently with ZLc002-treated groups shown here (see methods and Carey et al.6). CPS: composite pain score; AUC: area under the curve.
Formalin test
Rats received a single i.p. injection of ZLc002 (4 or 10 mg/kg), MK-801 (0.1 mg/kg) or vehicle 30 min before i.pl. formalin injection. Animals were placed on an elevated clear glass table in Plexiglass observation chambers immediately following i.p. injection and were allowed to habituate to the testing apparatus for 30 min. Next, rats received a unilateral i.pl. injection of 2.5% formalin (50 µl) into the superficial plantar surface of the hind paw. Behavior was videotaped for 60 min immediately following i.pl. formalin and nociceptive behaviors were quantified by a single experimenter (LMC) blinded to the experimental conditions. Composite pain scores (CPS) were calculated for every 5 min time bin as described in our previous work.6,26 No pain behavior was scored as 0, lifting of the paw was scored as 1, and shaking/biting/flinching was scored as 2. The area under the curve (AUC) for formalin-evoked pain was calculated for the early phase of pain behavior (phase 1, 0–10 min) and the late phase (phase 2, 10–60 min) for each subject.
Tissue preparation for immunohistochemistry
Immunohistochemical experiments were conducted on tissues from the same subjects that are used to evaluate the impact of ZLc002 (4 or 10 mg/kg i.p.), MK-801 (0.1 mg/kg i.p.) and vehicle on formalin-evoked pain behavior. Immediately after concluding behavioral procedures, rats were anesthetized with 25% urethane and immediately perfused transcardially with 0.1% heparinized 0.1 M PBS followed by 4% paraformaldehyde (i.e. 1 h post i.pl. formalin). Lumbar spinal cord tissue was dissected and kept in the same fixative for 24 h and then cryoprotected in 30% sucrose for three days prior to sectioning.
Immunohistochemistry
Immunohistochemical experiments were conducted as described previously.6,27–30 Briefly, transverse sections (30 μm) of the L4-L5 lumbar spinal cord were cut on a cryostat and kept in an antifreeze solution (50% sucrose in ethylene glycol and 0.1 M PBS) prior to staining. Every fourth section was processed for immunostaining to avoid counting the same cell twice in adjacent sections. Sections were washed in 0.1 M PBS, then endogenous peroxidases were quenched in 0.3% H2O2 for 30 min. Tissue was then incubated for 1 h in a blocking solution consisting of 5% normal goat serum and 0.3% Triton X-100 in 0.1 M PBS. Tissue was incubated with rabbit polyclonal Fos protein antibody (1:1500, Santa Cruz Biotechnology, Dallas, TX, USA) for 24 h at 4°C. Fos protein expression was visualized using the avidin-biotin peroxidase method using diaminobenzidine as the chromagen. Three sections per animal which displayed the greatest numbers of Fos-like immunoreactive (FLI) cells, based upon qualitative evaluation, were quantified by an experimenter blinded to treatment conditions. Images were obtained using a Retiga 1300 digital camera mounted on a Leica DMLB microscope. The number of FLI cells were counted manually using ImageJ software in spinal subdivisions as first described by Presley et al.31 The spinal subdivisions subjected to quantification were the superficial dorsal horn (laminae I and II), the nucleus proprius (lamina III and IV), the neck region of the dorsal horn (laminae V and VI), and the ventral horn (laminae VII-X). Statistical analyses were conducted on the number of FLI cells per subdivision averaged across the three sections quantified per animal to generate a single mean for each subdivision per animal. FLI cells were largely absent in rats receiving an i.pl. injection of saline in lieu of formalin (data not shown and Carey et al.6).
Paclitaxel-induced neuropathic pain
Paclitaxel (Tecoland Corporation, Irvine, CA) was dissolved in a vehicle consisting of a 1:1:4 ratio of cremophor EL (Sigma Aldrich), ethanol (Sigma Aldrich) and saline (Aquilite System; Hospira, Inc, Lake Forest, IL). Mice were injected with either the cremophor-vehicle or paclitaxel (4 mg/kg, i.p.) on days 0, 2, 4, and 6 following initiation of paclitaxel dosing (16 mg/kg i.p. cumulative dose). Responsiveness to mechanical and cold stimulation was assessed before initiation of paclitaxel or cremophor-vehicle dosing (i.e. baseline, day 0) and during development and maintenance phases of paclitaxel-induced hypersensitivity on days 4, 7, 11, and 15 as previously described.
Time course of anti-allodynic effects of ZLc002 in the paclitaxel model of neuropathic pain
Paclitaxel-treated mice were randomly divided into two groups and injected systemically with either vehicle or ZLc002 (10 mg/kg, i.p.) on day 16 following initiation of paclitaxel dosing. Responsiveness to mechanical and cold stimulation was assessed starting at 30 min after drug injection and reevaluated at 60, 90, and 150 min postinjection.
Effects of repeated systemic dosing with ZLc002 in the paclitaxel model of neuropathic pain
The same mice used in the time course evaluation were repeatedly injected with vehicle or ZLc002 (10 mg/kg, i.p.) once daily for another seven consecutive days. Repeated dosing was initiated on day 16 following initiation of paclitaxel dosing. Responsiveness to mechanical stimulation was assessed at 30 min posttreatment on days 1, 4, and 8 of chronic injection.
Assessment of mechanical allodynia
Withdrawal thresholds (g) to mechanical stimulation were measured in duplicate for each paw using an electronic von Frey anesthesiometer supplied with a 90-g semi-flexible probe (IITC Life Science, Woodland Hills, CA) as described previously.32,33
Assessment of cold allodynia
Cold allodynia was assessed by applying one drop (∼5–6 µl) of acetone (Sigma Aldrich) to the plantar surface of the hind paw. Time spent reacting to acetone stimulation was measured in triplicate for each paw.33,34
Statistical analysis
Data were analyzed using GraphPad Prism for Windows 5 (Graphpad Software, San Diego, CA USA). IC50 values in AlphaScreen were calculated by nonlinear regression analysis using the equation of a sigmoid concentration–response curve using GraphPad Prism. Co-immunoprecipitation data were analyzed by one-way analysis of variance (ANOVA) followed by post hoc Bonferroni test. In vivo data were analyzed by two-way repeated measures ANOVA and one-way ANOVA, as appropriate. Post hoc comparisons were performed using Bonferroni’s post hoc tests or, in the case of comparisons to control, Bonferroni’s multiple comparison test. P < 0.05 was considered statistically significant.
Results
ZLc002 reduced co-immunoprecipitation of NOS1AP with nNOS immunoprecipitated from primary cultured cortical neurons
We verified that ZLc002 can disrupt nNOS–NOS1AP interactions in primary neuronal cultures as previously suggested14 using methodology published previously by our group.18 Our results confirm that NMDA (50 µM) challenge elevated nNOS–NOS1AP interaction relative to control conditions in primary cortical neurons as determined with co-immunoprecipitation (Figure 2). Moreover, pretreatment with ZLc002 (10 µM) reduced NMDA-induced nNOS–NOS1AP interaction (F2,11 = 21.26, p < 0.001, Figure 2). NMDA-treated cells displayed higher levels of co-immunoprecipitated nNOS–NOS1AP than either control cells not treated with NMDA (p < 0.001; Bonferroni’s post hoc test) or NMDA-treated cells pretreated with ZLc002 (p < 0.01; Bonferroni’s post hoc test) (Figure 2).
Figure 2.

ZLc002 reduces co-immunoprecipitation of NOS1AP with nNOS in primary cortical neurons challenged with NMDA (50 µM). (a) Immunoblots show that nNOS–NOS1AP association was increased after exposure of cells to NMDA and this effect was reduced by ZLc002 (10 µM) pretreatment. (b) Quantification of mean ± S.E.M from n = 4 experiments (from two separate primary neuronal cultures) as shown in (a). **p < 0.01; ***p < 0.001 vs. NMDA-treated controls (one-way ANOVA followed by Bonferroni’s post hoc test). NMDA: N-methyl-D-aspartate; nNOS: neuronal nitric oxide synthase.
ZLc002 failed to disrupt nNOS–NOS1AP protein–protein interactions in the AlphaScreen in vitro binding assay
To investigate whether ZLc002 disrupts the binding between nNOS and NOS1AP through a direct mechanism, we set up AlphaScreen assays for cell-free measurement of direct binding between purified His-nNOS1-299 (containing the extended PDZ domain of nNOS) and GST-NOS1AP400-506 (containing the ExF motif and C-terminal tail). ZLc002 failed to disrupt this interaction event at the highest concentration tested (100 μM) in this reductionist Alphascreen binding assay, whereas the consensus peptide inhibitor of nNOS–NOS1AP, TAT-GESV, reliably disrupted the interaction with an IC50 of 4.9 μM (Figure 3) under analogous conditions. Moreover, consistent with our previous findings, the inactive peptide TAT-GESVΔ1, had no effect on binding between nNOS and NOS1AP (Figure 3). These results are consistent with ZLc002 acting in cells as a prodrug as previously suggested14 and therefore showing no activity in a cell-free assay.
Figure 3.

ZLc002 failed to disrupt the interaction between His-nNOS1-299 and GST-NOS1AP400-506 in AlphaScreen. The peptide nNOS–NOS1AP disruptor TAT-GESV disrupted this interaction with an IC50 of 4.9 μM, whereas the inactive peptide TAT-GESVΔ1 failed to do so (n = 4 replicates derived from two separate assays performed on separate days). Data are mean ± S.E.M.
ZLc002 reduced co-immunoprecipitation of full-length NOS1AP but not of PSD95-PDZ2 from HEK293T cells co-expressing full-length nNOS
Although ZLc002 disrupts NMDA-evoked interaction of nNOS with NOS1AP (Figure 2), this effect need not be direct and could, instead, result from an indirect effect on NMDA receptor signaling. For example, nNOS inhibitors also reduce NMDA-evoked nNOS–NOS1AP interaction in neurons.18 To determine whether ZLc002 directly disrupts the targeted protein–protein interaction14 in intact cells as intended, we evaluated the effect of ZLc002 exposure on the co-immunoprecipitation of NOS1AP preassembled with over-expressed nNOS in HEK293T cells, cells that would not be expected to express endogenous nNOS, PSD95, or NMDAR subunits. Full-length GFP-tagged nNOS was immunoprecipitated from transfected 293T cells co-expressing full-length NOS1AP (tagged with luciferase tag for quantification, see methods) with or without a 90-min preexposure to 10 µM ZLc002. Immunoblotting demonstrated comparable immunoprecipitation of GFP-nNOS in all samples in each replicate (n = 3), and quantification of co-immunoprecipitated NOS1AP showed that ZLc002 reduced the nNOS–NOS1AP interaction by ∼40% (F2,8 = 495.5, p < 0.0001; Figure 4(a)). To evaluate the selectivity of this inhibition for the nNOS interaction with NOS1AP, the experiments were repeated using nNOS and PSD95-PDZ2. While higher levels of binding of nNOS to PSD95-PDZ2 were observed in control relative to empty vector samples (F2,8 = 859.4, p < 0.0001; p < 0.0001; Figure 4(b)), ZLc002 had no effect on co-immunoprecipitation of PSD95-PDZ2 with nNOS. These observations suggest that ZLc002 is selective for a specific function of the nNOS-PDZ domain, i.e. the recruitment of NOS1AP.
Figure 4.

ZLc002 reduces co-immunoprecipitation of NOS1AP with nNOS but not PSD95-PDZ2 in HEK293T cells co-expressing the full-length proteins. ZLc002 (10 µM) treatment disrupts co-immunoprecipitation with full-length EGFP-nNOS of (a) full-length pLuc-NOS1AP but not of (b) pLuc-PSD95-PDZ2 from HEK293T cell lysates. Data are mean ± S.E.M. (n = 3) ***p < 0.001 (one-way ANOVA followed by Bonferroni’s post hoc test). Immunoblots under each bar chart demonstrate equal levels of nNOS among compared samples. GFP: green fluorescent protein.
ZLc002 reduced formalin-evoked nociceptive behavior and Fos-like immunoreactivity in the spinal dorsal horn
The i.pl. injection of formalin increased CPS in a biphasic manner (F12,18 = 19.22, p < 0.0001; Figure 5(a)). ZLc002, administered 30 min prior to i.pl. formalin injection, reduced formalin-evoked CPS (F3,18 = 9.964, p < 0.001, Figure 5(a)), and the interaction between time and drug treatment was significant (F38,18 = 4.187, p < 0.0001, Figure 5(a)). Post hoc analyses revealed that both the high (10 mg/kg i.p.) and low dose of ZLc002 (4 mg/kg i.p.) reduced formalin-evoked CPS from 30–50 min following formalin (i.pl.) injection relative to vehicle (p < 0.05 for each comparison; Bonferroni’s multiple comparison test). MK-801 similarly reduced formalin-evoked CPS from 25 to 50 min postformalin relative to vehicle (p < 0.05 for each comparison, Bonferroni’s multiple comparison test (Figure 5(a)).
The i.pl. formalin increased the AUC of formalin-induced pain behavior in a phase-dependent manner (F1,18 = 40.49, p < 0.0001; Figure 5(b)). ZLc002 treatment decreased the AUC (F3,18 = 40.49, p < 0.0001; Figure 5(b)), and the interaction between phase and treatment was significant (F3,18 = 8.205, p < 0.01; Figure 5(b)). None of the treatment groups altered phase 1 of formalin-evoked pain behavior (p > 0.5; Figure 5(b)). The NMDAR antagonist MK-801 (0.1 mg/kg i.p.) (p < 0.001), used here as a positive control, and both the high (10 mg/kg i.p.) (p < 0.001) and low (4 mg/kg) (p < 0.001) dose of ZLc002, all reduced the AUC of phase 2 of formalin-evoked pain behavior relative to vehicle treatment (Figure 5(b)).
ZLc002 reduced formalin-evoked Fos protein-like immunoreactivity (F5,24 = 85.86, p < 0.0001; Figure 5(c) and (d)) in a lamina-dependent manner (F3,24 = 21.43, p < 0.0001; Figure 6(a) and (b)), and the interaction between drug treatment and spinal cord laminar expression of Fos-protein like immunoreactivity was significant (F15,24 = 8.7, p < 0.0001; Figure 6(a)). ZLc002, at doses of both 4 and 10 mg/kg i.p., reduced formalin-evoked Fos-like immunoreactivity in the superficial dorsal horn (p < 0.001; Bonferroni’s post hoc test) and the neck region of the dorsal horn (p < 0.001; Bonferroni’s post hoc test) but not in the nucleus proprius or the ventral horn (p > 0.05 for each comparison; Bonferroni’s post hoc test) relative to vehicle treatment. By contrast, MK-801 (0.1 mg/kg i.p.) reduced formalin-evoked Fos-like immunoreactivity in laminae I-IV (p < 0.001) (Figure 6(a) and (b)) and in the ventral horn (p < 0.01) relative to vehicle (Figure 6(a) and (b)). Effects of ZLc002 (4 and 10 mg//kg i.p.) did not differ from each other in any spinal cord region (P > 0.05). Example photomicrographs depicting the impact of vehicle, ZLc002, and MK-801 on formalin-evoked Fos protein expression are shown in Figure 6(c) and (f).
Figure 6.

(a) ZLc002 (4 and 10 mg/kg i.p.) reduced formalin-evoked Fos-like immunoreactivity in laminae I-II (p < 0.001) and laminae V-VI (p < 0.001) relative to vehicle. MK-801 reduced formalin-evoked Fos-like immunoreactivity in laminae I-II (p < 0.001), laminae III-IV (p < 0.001), V-VI (p < 0.001), and the ventral horn (p < 0.01) relative to rats treated with vehicle. (b) Schematic adapted from the rat brain atlas of Paxinos and Watson35 showing spinal cord laminae quantified for Fos-like immunoreactivity. Data are mean ± S.E.M. (n = 5–6 per group) ***p < 0.001; **p < 0.01; *p < 0.05 vs. vehicle (two-way ANOVA followed by Bonferroni’s multiple comparison test). MK-801 and vehicle groups were published previously but run and processed concurrently with ZLc002-treated groups shown here (see methods and Carey et al.6). Example photomicrographs taken at 10x magnification showing formalin-evoked Fos-like immunoreactivity in lumbar dorsal horn of rats treated with vehicle (c), ZLc002 (4 mg/kg i.p.) (d), ZLc002 (10 mg/kg i.p.) (e), and MK-801 (0.1 mg/kg) (f). Scale bar is equal to 100 µm.
ZLc002 attenuates mechanical and cold allodynia evoked by paclitaxel in mice
Paclitaxel treatment decreased mechanical paw withdrawal thresholds, mechanical paw withdrawal thresholds differed across test days, and the interaction between treatment and test day was significant (F1,22 = 33.7, p < 0.0001 (treatment); F4,88 = 20.81, p < 0.0001 (day); F4,88 = 17.49, p < 0.0001 (interaction); Figure 7(a)). Similarly, paclitaxel increased the duration of time spent responding to cold, cold responsiveness differed across test days, and the interaction between treatment and test day was significant (F1,22 = 43.491, p < 0.0001 (treatment); F4,88 = 34.02, p < 0.0001 (day); F4,88 = 13.31, p < 0.0001 (interaction); Figure 7(b)). Mechanical and cold hypersensitivity was present on day 7, was maintained throughout the observation interval, and remained ongoing on day 15, prior to initiation of pharmacological manipulations (p < 0.0001, Bonferroni’s post hoc test for both mechanical and cold assessment), relative to day 0 prepaclitaxel baseline response (Figure 7(a) and (b)).
Figure 7.

The putative nNOS–NOS1AP disruptor ZLc002 suppresses mechanical and cold allodynia induced by paclitaxel treatment. Paclitaxel (a) lowers mechanical paw withdrawal thresholds consistent with the development of mechanical allodynia and (b) increases cold response time, consistent with the development of cold allodynia. ZLc002 (10 mg/kg i.p.) increases (c) mechanical paw withdrawal thresholds and (d) lowers duration of response to cold in paclitaxel-treated mice. ZLc002 (10 mg/kg i.p.) does not alter responsiveness to (e) mechanical or (f) cold stimulation in control mice receiving the cremophor vehicle in lieu of paclitaxel. Data are mean ± S.E.M. *p < 0.05; **p < 0.01; ***p < 0.001 (two-way repeated measures ANOVA followed by Bonferroni’s post hoc test); #paired sample t-test baseline vs. postpaclitaxel predrug response. BL: baseline; PTX: paclitaxel.
Paclitaxel lowered mechanical paw withdrawal thresholds (t11 = 9, p < 0.0001; Figure 7(c)) and increased duration of time spent responding to cold (t11 = 8.28, p < 0.0001; Figure 7(d)) relative to prepaclitaxel baseline responses. In paclitaxel-treated mice, ZLc002 (10 mg/kg, i.p.) increased postinjection mechanical paw withdrawal thresholds, mechanical paw withdrawal thresholds differed across postinjection times, and the interaction between drug treatment and injection time was significant (F1,10 = 14.81, p = 0.0032 (drug); F2,20 = 9.05, p = 0.0016 (time); F2,20 = 4.20, p = 0.030 (interaction); two-way repeated measures ANOVA; Figure 7(c)). In paclitaxel-treated mice, ZLc002 elevated mechanical paw withdrawal thresholds relative to vehicle treatment from 30 min (p < 0.01, Bonferroni’s post hoc test; Figure 7(c)) to 90 min postinjection (p < 0.05, Bonferroni’s post hoc test; Figure 7(c)). In paclitaxel-treated mice, ZLc002 (10 mg/kg, i.p.) decreased postinjection cold responsiveness, cold responsiveness did not differ reliably across postinjection times, and the interaction between drug treatment and injection time was not significant (F1,10 = 5.46, p = 0.0415 (drug); F2,20 = 1.99, p = 0.1632 (time); F2,20 = 0.16, p = 0.8556 (interaction); two-way repeated measures ANOVA; Figure 7(d)). Thus, ZLc002 attenuated paclitaxel-induced cold responsiveness throughout the observation interval (Figure 7(d)).
In mice that received the cremophor-based vehicle in lieu of paclitaxel, ZLc002 treatment did not reliably alter postinjection mechanical or cold responsiveness, behavioral responsiveness was stable across postinjection times, and the interaction between drug treatment and time was not significant (Mechanical: F1,10 = 3.30, p = 0.0992 (drug); F2,20 = 0.84, p = 0.4469 (time); F2,20 = 0.16, p = 0.8565 (interaction); Cold: F1,10 = 2.40, p = 0.1522 (drug); F2,20 = 1.51, p = 0.2441 (time); F2,20 = 2.15, p = 0.1430 (interaction); two-way repeated measures ANOVA; Figure 7(e) to (f)).
Effects of repeated dosing with nNOS–NOS1AP disruptor in a mouse model of paclitaxel-induced neuropathic pain
In paclitaxel-treated mice, once daily dosing with ZLc002 (10 mg/kg i.p. x 8 days) increased mechanical paw withdrawal thresholds relative to the vehicle-treated group across the observation interval (F1,10 = 26.59, p = 0.0004 (drug)) (Figure 8(a)). Mechanical paw withdrawal thresholds also differed across injection days, and the interaction between treatment and injection day was significant (F2,20 = 4.13, p = 0.0316 (day); F2,20 = 8.25, p = 0.0024 (interaction)) (Figure 8(a)). ZLc002 increased mechanical paw withdrawal thresholds relative to vehicle in paclitaxel-treated mice on day 1 (p < 0.01; Bonferroni’s post hoc test) and day 4 (p < 0.001; Bonferroni’s post hoc test) but not on day 8 of chronic dosing (p > 0.05 for each comparison; Bonferroni’s post hoc test).
Figure 8.

Effects of repeated sytemic dosing with nNOS–NOS1AP disruptor ZLc002 on paclitaxel-induced mechanical and cold allodynia. In paclitaxel-treated mice, ZLc002 (10 mg/kg i.p. × 8 days) increased mechanical paw withdrawal threshold (a) and reduced cold response durations (b) relative to vehicle. ZLc002 reduced paclitaxel-induced allodynia on days 1 and 4 but not on day 8 of repeated dosing. Data are mean ± S.E.M. *p < 0.05; **p < 0.01; ***p < 0.05 vs. corresponding vehicle condition (two-way repeated measures ANOVA, followed by Bonferroni’s post hoc test). #paired sample t-test baseline vs. postpaclitaxel predrug response. PTX: paclitaxel; BL: baseline.
Once daily dosing with ZLc002 (10 mg/kg i.p. × 8 days) also lowered paclitaxel-induced cold hypersensitivity relative to the vehicle-treated group across the observation interval (F1,10 = 12.5, p = 0.0054 (drug)), and these effects were also time dependent (F2,20 = 8.20, p = 0.0025 (interaction)) (Figure 8( b)). Cold responsiveness did not differ across injection days (F2,20 = 0.15, p = 0.8579 (day)) (Figure 8(a)). ZLc002 lowered cold response times relative to vehicle in paclitaxel-treated mice on day 1 (p < 0.05; Bonferroni’s post hoc test) and day 4 (p < 0.001; Bonferroni’s post hoc test) but not on day 8 (p > 0.05 for each comparison; Bonferroni’s post hoc test) of chronic dosing (Figure 8(a) and (b)).
Impact of ZLc002 in the presence and absence of paclitaxel on breast and ovarian tumor cell line viability
The impact of ZLc002 and paclitaxel over a wide range of molar ratios (i.e. dose–response matrix between eight concentrations of ZLc002 and eight concentrations of paclitaxel) on 4T1 and HeyA8 tumor cell line cytotoxicity is shown in Figures 9 and 10, respectively. ZLc002 alone had no effect on the viability of either 4T1 (Figure 9(a)) or HeyA8 (Figure 10(a)) tumor cells, which was markedly inhibited by paclitaxel in each case (Figures 9(b) and 10(b)). Nonetheless, quantification of the drug combination responses indicates that the combination between ZLc002 and paclitaxel is synergistic using the Bliss model (4T1: Figure 9(e); HeyA8: Figure 10(e)), Loewe additivity model (4T1: Figure 9(f); HeyA8: Figure 10(f)), and HSA model (4T1: Figure 9(g); HeyA8: Figure 10(g)). The synergy maps showed that ZLc002 and paclitaxel have synergistic effects (blue areas in the model graph) on inhibiting tumor cell proliferation at a wide range of drug combination ratios in both 4T1 (Figure 9(e) to (g)) and HeyA8 (Figure 10(e) to (g)) cells.
Figure 9.

ZLc002 synergizes with paclitaxel in 4T1 cells to reduce breast cancer tumor cell line viability. Dose–response matrix for the effect of (a) ZLc002 and (b) paclitaxel in 4T1 cells. Abs EC50, absolute EC50 > 50 µM, reflects little or no inhibition of 4T1 tumor cell viability by ZLc002; Abs EC50, absolute EC50 = 33.5 nM, reflects efficacy of paclitaxel in reducing 4T1 tumor cell viability by 50% of maximum. (c–d) Single-agent and combination responses determined by an MTT viability assay in 4T1 cells. The landscape of the combination responses for ZLc002 and paclitaxel based on the (e) Bliss model, (f) Loewe model, and (g) highest single agent (HSA) model. Each model supports synergism of the combination of ZLc002 with paclitaxel in reducing tumor cell line viability. (n = 7 experiments). HSA: highest single agent.
Figure 10.

ZLc002 synergizes with paclitaxel in HeyA8 cells to reduce ovarian tumor cell line viability. Dose–response matrix delineating the effect of (a) ZLc002 and (b) paclitaxel in HeyA8 cells. Rel EC95, Relative EC95 = 0 µM, reflects absence of inhibition of HeyA8 tumor cell viability by ZLc002; Abs EC50, Absolute EC50 = 10.2 nM, reflects efficacy of paclitaxel in reducing HeyA8 tumor cell viability by 50% of maximum. (c–d) Single-agent and combination responses determined by an MTT viability assay in 4T1 cells. The landscape of the combination responses for ZLc002 and paclitaxel based on the (E) Bliss model, (F) Loewe model, and (G) highest single agent (HSA) model. Each model supports synergism of the combination of ZLc002 with paclitaxel in reducing tumor cell line viability. (n = 3 experiments). HSA: highest single agent.
Discussion
Our studies support a role for disruption of nNOS–NOS1AP protein–protein interactions downstream of NMDARs as a therapeutic strategy for suppressing inflammatory and neuropathic pain. We verified that the putative small-molecule nNOS–NOS1AP inhibitor ZLc002 disrupts the NMDA-induced interaction between full-length nNOS and NOS1AP proteins in primary cortical neurons. We also showed that ZLc002 selectively disrupted the preestablished interaction in HEK293T cells transfected with full-length nNOS and NOS1AP proteins without altering the interactions between full-length nNOS and PSD95-PDZ2. These observations are consistent with a direct action on the nNOS–NOS1AP protein–protein interaction itself rather than on the upstream, NMDA-evoked mechanism of interaction. Our studies suggest that the nNOS–NOS1AP interface is a previously unrecognized target for inflammatory pain. We revealed, for the first time, that a small-molecule nNOS–NOS1AP disruptor exhibits anti-allodynic efficacy in models of inflammatory and neuropathic pain. Importantly, ZLc002, administered systemically, suppressed both formalin-evoked pain behavior as well as inflammation-evoked neuronal activation in lumbar spinal dorsal horn of the same subjects, similar to the NMDAR antagonist MK-801. Moreover, ZLc002 suppressed both mechanical and cold allodynia in a mouse model of neuropathic pain induced by paclitaxel treatment and enhanced the ability of paclitaxel to reduce tumor cell viability in vitro.
ZLc002 was previously shown to disrupt nNOS–NOS1AP interactions, as judged by the observation of reduced co-immunoprecipitation of NOS1AP with nNOS measured in hippocampal cells.14 However, such actions in a complex cellular system could occur through direct or indirect mechanisms. Our studies verify and extend these published observations by showing that ZLc002 reduces the co-immunoprecipitation of NOS1AP but not PSD95-PDZ2 with nNOS in HEK293T cells. Although both of these interactions involve the extended PDZ domain at the first 130 N-terminal amino acids of nNOS, the stable NOS1AP interaction requires the docking of a PDZ ligand motif into the nNOS-PDZ pocket,15 whereas PSD95-PDZ2 interacts with a distinct β-finger extension of the core nNOS-PDZ domain.36 The two sites of interaction are, therefore, spatially very close to one another but are distinct,15,16,18 making this comparison a good evaluation of selectivity. Importantly, because endogenous nNOS, PSD95, or NMDAR subunits can be expected to be absent in HEK293T cells and ZLc002 disrupts the constitutive nNOS–NOS1AP interaction in this system, ZLc002 is unlikely to reduce co-immunoprecipitation of nNOS and NOS1AP in cultured neurons by acting through NMDA-evoked mechanisms involving extraneous targets that are present in neurons but not measured herein.
To determine whether disruption of nNOS–NOS1AP interactions induced by ZLc002 resulted from a direct mechanism, we used a cell-free AlphaScreen biochemical binding assay employing purified nNOS and NOS1AP fragments containing the interacting interface.15,16,18 Intriguingly, we failed to detect the disruption by ZLc002 of His-nNOS1-299-GST-NOS1AP400-506 binding with the fragments that are critical for nNOS–NOS1AP interactions15 in this reductionist assay. By contrast, the peptide nNOS–NOS1AP disruptor TAT-GESV disrupted these interactions in the same AlphaScreen assay, whereas a putative inactive peptide TAT-GESVΔ1, lacking the terminal valine residue, failed to do so, consistent with the known critical role of the peptide ligand terminal valine in target recognition.37 Therefore, the potency of ZLc002 for disrupting binding of nNOS–NOS1AP could not be determined using AlphaScreen, which uses a cell-free system consisting of only a single pair of purified protein fragments. It is plausible that ZLc002 disrupts the interactions through a mechanism distinct from the direct disruption at this interacting interface (e.g. allosteric mechanisms and/or via an active metabolite of ZLc002 that is produced in cells).
ZLc002 has recently been hypothesized to act as a pro-drug.14 This observation could account for our observation that ZLc002 disrupted the co-immunoprecipitation of NOS1AP with nNOS from primary cortical neurons expressing enzymes such as esterases that would, presumably, be available to transform ZLc002 into other bioactive mediators, but it failed to disrupt the interaction between nNOS and NOS1AP in our cell-free AlphaScreen assay even though the peptide nNOS–NOS1AP disruptor TAT-GESV was able to potently disrupt this interaction. Support for this hypothesis is derived from the fact that ZLc002 also disrupted co-immunoprecipitation between full-length nNOS and NOS1AP in transfected HEK293T cells in the immunoprecipitation assay. In our study, ZLc002 disrupted nNOS–NOS1AP interactions in HEK293T cells transfected with full-length nNOS and full-length NOS1AP but not between full-length nNOS and PDZ2 of PSD95. This finding is consistent with previous work showing that ZLc002 inhibits the co-immunoprecipitation of NOS1AP with nNOS but not of PSD95 with nNOS.14 More work is needed to determine the precise location at which ZLc002 binds within the complex, if indeed it binds at all, and whether the currently identified two nNOS interacting sites on NOS1AP may affect ZLc002’s binding differentially from TAT-GESV, therefore, giving us different results in different protein–protein disruption assays. Our observations, nonetheless, suggest that ZLc002 produces a functional disruption of nNOS–NOS1AP interactions in intact cells.
ZLc002 exhibits anxiolytic efficacy in mice and disrupts co-immunoprecipitation of nNOS and NOS1AP in ZLc002-treated hippocampal cells without changing PSD95 expression or its association with NMDARs.14 Increases in association of nNOS and NOS1AP also accompany anxiogenic-like behaviors.14 However, prior to the present report, whether this small molecule disrupts nNOS–NOS1AP binding through a direct mechanism or suppresses pathological pain was unknown.14 Our studies demonstrate, for the first time, that disruption of nNOS–NOS1AP protein–protein interactions suppresses inflammatory nociception. ZLc002, administered systemically, produced antinociceptive efficacy in the formalin test and reduced the number of formalin-evoked Fos-like immunoreactive cells in the lumbar spinal dorsal horn. ZLc002 selectively suppressed phase 2, but not phase 1, of formalin-induced pain behavior, similar to the NMDAR antagonist MK-801. These observations are consistent with the role of NMDARs in contributing to central nervous system sensitization and, specifically, phase 2 of formalin-induced pain behavior.2,6,38–40 Moreover, ZLc002 selectively suppressed the number of formalin-evoked Fos protein-like immunoreactive cells, a marker of neuronal activation, in dorsal horn regions implicated in nociceptive processing but did not reliably alter Fos protein expression in ventral horn regions typically associated with motor function. Notably, ZLc002 suppressed formalin-evoked Fos protein expression in the superficial dorsal horn (lamina I, II) and neck region (lamina V, VI) of the dorsal horn but not in the nucleus proprius (lamina III, IV) or ventral horn. The ZLc002-induced suppression of Fos protein expression was observed in the same subjects that exhibited ZLc002-induced antinociception in the formalin test. These observations are consistent with the hypothesis that the suppression of inflammation-evoked Fos protein expression induced by antinociceptive doses of ZLc002 reflects a suppression of nociceptive processing. Moreover, the pattern of changes in both pain behavior and Fos protein expression was similar to those observed previously by our group with PSD95-nNOS inhibitors IC87201 and ZL006.6
We previously demonstrated that the nNOS–NOS1AP inhibitor TAT-GESV, but not the inactive peptide TAT-GESVΔ1, reversed established neuropathic pain due to paclitaxel treatment.11 Anti-allodynic efficacy of TAT-GESV (i.t.) was preserved following repeated intrathecal injection of the peptide inhibitor.11 Here, we show that ZLc002, administered systemically, reduces the maintenance of paclitaxel-evoked mechanical and cold allodynia. The anti-allodynic effects of ZLc002 were preserved for at least four days of repeated dosing, although loss of anti-allodynic efficacy was observed by day 8 of repeated dosing. More work is necessary to determine whether differences in the route of administration contributed to loss of efficacy (i.e. tolerance) observed with repeated systemic, but not intrathecal, administration. A compensatory mechanism in NMDAR-PSD95-nNOS–NOS1AP-mediated nociceptive signaling could be engaged following repeated systemic dosing of the small molecule but not following repeated intrathecal dosing with the peptide nNOS–NOS1AP disruptor. Tolerance was not observed with repeated intrathecal dosing of either TAT-GESV or MK-801 in the same paclitaxel model of neuropathic pain in our previous work.9,11 More work is necessary to establish the site of action of systemically administered small-molecule nNOS–NOS1AP disruptors and determine whether mechanisms of anti-allodynic efficacy and tolerance could differ at spinal and supraspinal levels.
The anti-allodynic effects of ZLc002 observed herein were selective for the pathological pain state; ZLc002 did not alter responsiveness to either mechanical or cold stimulation in control animals that received the cremophor-based vehicle in lieu of paclitaxel. Our findings reveal, for the first time, that a functional small-molecule inhibitor of nNOS–NOS1AP interactions produces anti-allodynic efficacy in rodent models of both neuropathic and inflammatory pain.
ZLc002 failed to impede, and in fact, was synergistic with paclitaxel in reducing tumor cell line viability, without itself producing tumor cell cytotoxicity. Similar synergistic effects of ZLc002 with paclitaxel were observed in both breast cancer (4T1) and ovarian (HeyA8) tumor cell lines. The same conclusions were obtained using Combenefit analysis applied to different classical synergy models (i.e. the Bliss model, Loewe model, and HSA model). The fact that identical conclusions were derived from separate experiments employing distinct tumor cell lines and three different synergy models further validate our findings, although synergism appeared more robust in the breast cancer 4T1 cell line. These observations raise the possibility that nNOS–NOS1AP disruption may be highly efficacious clinically for suppressing chemotherapy-induced neuropathic pain in breast cancer patients without impeding the anti-tumor efficacy of paclitaxel. More work is necessary to evaluate whether ZLc002 could enhance the anti-cancer effects of paclitaxel in vivo and whether such effects translate to different chemotherapeutic agents.
We recently reported that nNOS–NOS1AP interactions are involved in pro-nociceptive signaling using a peptide nNOS–NOS1AP disruptor.11 TAT-GESV, administered i.t., attenuated mechanical and cold allodynia in two mechanistically distinct neuropathic pain models (i.e. chemotherapy-induced peripheral neuropathy produced by paclitaxel and traumatic nerve injury induced by partial sciatic nerve ligation).11 TAT-GESV, administered i.t., also reduced paclitaxel-evoked phosphorylation of p53 in the lumbar spinal cord, consistent with a spinal site of anti-allodynic efficacy.11 Because p53 is a downstream substrate of proinflammatory p38 MAPK that is known to be activated upon nNOS–NOS1AP association, it was used as a surrogate marker of p38 activation.18 More work is necessary to determine whether p38MAPK could also be activated by pathological pain and disrupted by ZLc002 at its site of action.
Elevated NMDAR activity contributes to central sensitization.2 However, targeting NMDAR produces unwanted side effects, such as motor and memory impairment rendering NMDAR antagonism undesirable.3 Thus, it is noteworthy that ZLc002 does not produce motor impairment,14 consistent with similar observations made by our group using the peptide nNOS–NOS1AP disruptor TAT-GESV11 and nNOS-PSD95 inhibitors IC87201 and ZL006.6–9,11 Novel strategies disrupting protein–protein interactions downstream of NMDARs—including NR2B-PSD95, PSD95-nNOS, and nNOS–NOS1AP interactions—thus remain promising alternative approaches capable of suppressing elevated NMDAR activity-mediated pro-nociceptive signaling and allodynia without unwanted on-target side effects of NMDAR antagonists.6–9,11 Further studies are required to determine whether therapeutic strategies disrupting nNOS–NOS1AP interactions are superior to those targeting NR2B-PSD95 or PSD95-nNOS interactions in the quest to develop safe and effective anti-hyperalgesic and anti-allodynic agents for the treatment of pain.
In conclusion, our findings collectively suggest that the putative nNOS–NOS1AP small-molecule inhibitor, ZLc002, disrupts nNOS–NOS1AP interactions in primary cortical neurons and in HEK293T cells transfected with the full-length proteins but not in a cell-free Alphascreen biochemical binding assay. ZLc002 suppresses neuropathic and inflammatory pain as well as a neurochemical marker of inflammation-evoked neuronal activation in pain processing regions of the spinal dorsal horn. ZLc002 reduces paclitaxel-induced neuropathic pain in vivo and synergized with paclitaxel to reduce both breast and ovarian tumor cell line viability in vitro. ZLc002 did not alter mechanical or cold sensitivity in the absence of paclitaxel, suggesting that the nNOS–NOS1AP disruptor reversed the sensitized responses to cutaneous (mechanical and cold) stimulation in a manner that was selective for the pathological pain state. Future medicinal chemistry efforts are required to identify small-molecule nNOS–NOS1AP inhibitors that themselves disrupt nNOS–NOS1AP in a cell-free system and exhibit better drug-like properties (i.e. enhanced duration of action, lack of tolerance, and show improved drug-like properties).
Author contributions
WL conducted the paclitaxel studies and drafted the initial manuscript. LLL conducted the immunoprecipitation experiments and prepared the expression constructs. LMC conducted the formalin and immunohistochemical experiments. ZX conducted the AlphaScreen and tumor cell viability assays. WL, LMC, LLL, ZX, and AGH analyzed data. MJC and AGH designed the study. AGH and MJC oversaw the project and wrote the manuscript with WL, LMC, ZX, LLL, and YYL.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: YYL is partially employed at Anagin, Inc.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Supported by CA200417 and DA041229 and DA042584 (to AGH) (to AGH and MJC). LMC is supported by NIDA T32 training grant DA024628 and the Harlan Scholars Research Program.
References
- 1.Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain 2009; 10: 895–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woolf CJ, Thompson SW. The induction and maintenance of central sensitization is dependent on N-methyl-D-aspartic acid receptor activation; implications for the treatment of post-injury pain hypersensitivity states. Pain 1991; 44: 293–299. [DOI] [PubMed] [Google Scholar]
- 3.Zhou HY, Chen SR, Pan HL. Targeting N-methyl-D-aspartate receptors for treatment of neuropathic pain. Expert Rev Clin Pharmacol 2011; 4: 379–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Jr, Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 1994; 51: 199–214. [DOI] [PubMed] [Google Scholar]
- 5.Pal HR, Berry N, Kumar R, Ray R. Ketamine dependence. Anaesth Intensive Care 2002; 30: 382–384. [DOI] [PubMed] [Google Scholar]
- 6.Carey LM, Lee WH, Gutierrez T, Kulkarni PM, Thakur GA, Lai YY, Hohmann AG. Small molecule inhibitors of PSD95-nNOS protein-protein interactions suppress formalin-evoked Fos protein expression and nociceptive behavior in rats. Neuroscience 2017; 349: 303–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D'Mello R, Marchand F, Pezet S, McMahon SB, Dickenson AH. Perturbing PSD-95 interactions with NR2B-subtype receptors attenuates spinal nociceptive plasticity and neuropathic pain. Mol Ther 2011; 19: 1780–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Florio SK, Loh C, Huang SM, Iwamaye AE, Kitto KF, Fowler KW, Treiberg JA, Hayflick JS, Walker JM, Fairbanks CA, Lai Y. Disruption of nNOS-PSD95 protein-protein interaction inhibits acute thermal hyperalgesia and chronic mechanical allodynia in rodents. Br J Pharmacol 2009; 158: 494–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee WH, Xu Z, Ashpole NM, Hudmon A, Kulkarni PM, Thakur GA, Lai YY, Hohmann AG. Small molecule inhibitors of PSD95-nNOS protein-protein interactions as novel analgesics. Neuropharmacology 2015; 97: 464–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith AE, Xu Z, Lai YY, Kulkarni PM, Thakur GA, Hohmann AG, Crystal JD. Source memory in rats is impaired by an NMDA receptor antagonist but not by PSD95-nNOS protein-protein interaction inhibitors. Behav Brain Res 2016; 305: 23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee WH, Li LL, Chawla A, Hudmon A, Lai YY, Courtney MJ, Hohmann AG. Disruption of nNOS-NOS1AP protein-protein interactions suppresses neuropathic pain in mice. Pain 2018; 159: 849–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Otvos L, Jr, Wade JD. Current challenges in peptide-based drug discovery. Front Chem 2014; 2: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov Today 2015; 20: 122–128. [DOI] [PubMed] [Google Scholar]
- 14.Zhu LJ, Li TY, Luo CX, Jiang N, Chang L, Lin YH, Zhou HH, Chen C, Zhang Y, Lu W, Gao LY, Ma Y, Zhou QG, Hu Q, Hu XL, Zhang J, Wu HY, Zhu DY. CAPON-nNOS coupling can serve as a target for developing new anxiolytics. Nat Med 2014; 20: 1050–1054. [DOI] [PubMed] [Google Scholar]
- 15.Li LL, Melero-Fernandez de Mera RM, Chen J, Ba W, Kasri NN, Zhang M, Courtney MJ. Unexpected heterodivalent recruitment of NOS1AP to nNOS reveals multiple sites for pharmacological intervention in neuronal disease models. J Neurosci 2015; 35: 7349–7364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li LL, Cisek K, Courtney MJ. Efficient binding of the NOS1AP C-terminus to the nNOS PDZ pocket requires the concerted action of the PDZ ligand motif, the internal ExF site and structural integrity of an independent element. Front Mol Neurosci 2017; 10: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cata JP, Weng HR, Lee BN, Reuben JM, Dougherty PM. Clinical and experimental findings in humans and animals with chemotherapy-induced peripheral neuropathy. Minerva Anestesiol 2006; 72: 151–169. [PubMed] [Google Scholar]
- 18.Li LL, Ginet V, Liu X, Vergun O, Tuittila M, Mathieu M, Bonny C, Puyal J, Truttmann AC, Courtney MJ. The nNOS-p38MAPK pathway is mediated by NOS1AP during neuronal death. J Neurosci 2013; 33: 8185–8201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melero-Fernandez de Mera RM, Li LL, Popinigis A, Cisek K, Tuittila M, Yadav L, Serva A, Courtney MJ. A simple optogenetic MAPK inhibitor design reveals resonance between transcription-regulating circuitry and temporally-encoded inputs. Nat Comms 2017; 8: 15017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindqvist C, Holmberg C, Oetken C, Courtney M, Stahls A, Akerman KE. Rapid Ca2+ mobilization in single LGL cells upon interaction with K562 target cells–role of the CD18 and CD16 molecules. Cell Immunol 1995; 165: 71–76. [DOI] [PubMed] [Google Scholar]
- 21.Cao J, Viholainen JI, Dart C, Warwick HK, Leyland ML, Courtney MJ. The PSD95-nNOS interface: a target for inhibition of excitotoxic p38 stress-activated protein kinase activation and cell death. J Cell Biol 2005; 168: 117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Semenova MM, Maki-Hokkonen AM, Cao J, Komarovski V, Forsberg KM, Koistinaho M, Coffey ET, Courtney MJ. Rho mediates calcium-dependent activation of p38alpha and subsequent excitotoxic cell death. Nat Neurosci 2007; 10: 436–443. [DOI] [PubMed] [Google Scholar]
- 23.von Ossowski L, Li L-L, Möykkynen T, Coleman S K, Courtney M J, Keinänen K. Cysteine 893 is a target of regulatory thiol modifications of GluA1 AMPA receptors. PLoS One 2017; 12: e0171489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Slivicki RA, Xu Z, Kulkarni PM, Pertwee RG, Mackie K, Thakur GA, Hohmann AG. Positive allosteric modulation of cannabinoid receptor type 1 suppresses pathological pain without producing tolerance or dependence. Biol Psychiatry Epub ahead of print 8 July 2017. DOI: 10.1016/j.biopsych.2017.06.032. [DOI] [PMC free article] [PubMed]
- 25.Di Veroli GY, Fornari C, Wang D, Mollard S, Bramhall JL, Richards FM, Jodrell DI. Combenefit: an interactive platform for the analysis and visualization of drug combinations. Bioinformatics. 2016; 32: 2866–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guindon J, Guijarro A, Piomelli D, Hohmann AG. Peripheral antinociceptive effects of inhibitors of monoacylglycerol lipase in a rat model of inflammatory pain. Br J Pharmacol 2011; 163: 1464–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsou K, Lowitz KA, Hohmann AG, Martin WJ, Hathaway CB, Bereiter DA, Walker JM. Suppression of noxious stimulus-evoked expression of Fos protein-like immunoreactivity in rat spinal cord by a selective cannabinoid agonist. Neuroscience 1996; 70: 791–798. [DOI] [PubMed] [Google Scholar]
- 28.Nackley AG, Makriyannis A, Hohmann AG. Selective activation of cannabinoid CB(2) receptors suppresses spinal fos protein expression and pain behavior in a rat model of inflammation. Neuroscience 2003; 119: 747–757. [DOI] [PubMed] [Google Scholar]
- 29.Nackley AG, Suplita RL, Hohmann AG. A peripheral cannabinoid mechanism suppresses spinal fos protein expression and pain behavior in a rat model of inflammation. Neuroscience 2003; 117: 659–670. [DOI] [PubMed] [Google Scholar]
- 30.Carey LM, Slivicki RA, Leishman E, Cornett B, Mackie K, Bradshaw H, Hohmann AG. A pro-nociceptive phenotype unmasked in mice lacking fatty-acid amide hydrolase. Mol Pain 2016; 12: 1–23. DOI: 10.1177/1744806916649192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Presley RW, Menetrey D, Levine JD, Basbaum AI. Systemic morphine suppresses noxious stimulus-evoked Fos protein-like immunoreactivity in the rat spinal cord. J Neurosci 1990; 10: 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guindon J, Lai Y, Takacs SM, Bradshaw HB, Hohmann AG. Alterations in endocannabinoid tone following chemotherapy-induced peripheral neuropathy: effects of endocannabinoid deactivation inhibitors targeting fatty-acid amide hydrolase and monoacylglycerol lipase in comparison to reference analgesics following cisplatin treatment. Pharmacol Res 2013; 67: 94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deng L, Guindon J, Cornett BL, Makriyannis A, Mackie K, Hohmann AG. Chronic cannabinoid receptor 2 activation reverses paclitaxel neuropathy without tolerance or cannabinoid receptor 1-dependent withdrawal. Biol Psychiatry 2015; 77: 475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ward SJ, Ramirez MD, Neelakantan H, Walker EA. Cannabidiol prevents the development of cold and mechanical allodynia in paclitaxel-treated female C57Bl6 mice. Anesth Analg 2011; 113: 947–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 4th ed San Diego, CA: Academic Press, 1998. [Google Scholar]
- 36.Hillier BJ, Christopherson KS, Prehoda KE, Bredt DS, Lim WA. Unexpected modes of PDZ domain scaffolding revealed by structure of nNOS-syntrophin complex. Science 1999; 284: 812–815. [PubMed] [Google Scholar]
- 37.Jaffrey SR, Snowman AM, Eliasson MJ, Cohen NA, Snyder SH. CAPON: a protein associated with neuronal nitric oxide synthase that regulates its interactions with PSD95. Neuron 1998; 20: 115–124. [DOI] [PubMed] [Google Scholar]
- 38.Ma QP, Woolf CJ. Noxious stimuli induce an N-methyl-D-aspartate receptor-dependent hypersensitivity of the flexion withdrawal reflex to touch: implications for the treatment of mechanical allodynia. Pain 1995; 61: 383–390. [DOI] [PubMed] [Google Scholar]
- 39.South SM, Kohno T, Kaspar BK, Hegarty D, Vissel B, Drake CT, Ohata M, Jenab S, Sailer AW, Malkmus S, Masuyama T, Horner P, Bogulavsky J, Gage FH, Yaksh TL, Woolf CJ, Heinemann SF, Inturrisi CE. A conditional deletion of the NR1 subunit of the NMDA receptor in adult spinal cord dorsal horn reduces NMDA currents and injury-induced pain. J Neurosci 2003; 23: 5031–5040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci 2003; 26: 696–705. [DOI] [PubMed] [Google Scholar]