

Abstract
Autosomal-recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) is caused by mutations of the SACS gene, characterized by late-infantile-onset spastic ataxia and other neurological features. ARSACS has a high prevalence in northeastern Quebec, Canada. Recently, several ARSACS cases have been reported from outside Canada. We report typical clinical and neuroimaging features in a Turkish child, which confirmed genetic diagnosis of ARSACS.
KEYWORDS: Autosomal recessive spastic ataxia of Charlevoix-Saguenay, ataxia, genetic study
INTRODUCTION
Autosomal-recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) is a hereditary neurological disorder mostly manifested with a classical triad: progressive early-onset cerebellar ataxia, lower limb pyramidal signs, and peripheral neuropathy.[1] It is related to progressive degeneration of the cerebellum and spinal cord. The ARSACS syndrome has shown a consistent pattern of clinical symptoms and electrophysiologic and morphologic findings that have led to the identification of the responsible gene localized on chromosome 13q.[2]
It was originally described in the late 1970s, has a high prevalence in northeastern Quebec, Canada.[1] In recent years, several cases have been described in countries other than Canada.[3,4,5,6]
We present a Turkish child from consanguineous parents whose genetic analysis revealed ARSACS.
CASE REPORT
A 10-year-old girl first presented with early onset of low progressive difficulty in walking and slurred speech. She walked at the age of 18 months but was clumsy, fell frequently, had problems with her balance, and had poor fine motor skills. Neurologic examination at 4 years showed an action tremor of her hands, spasticity in the legs, with hyperactive knee reflexes, and ankle clonus. A diagnosis of spastic paraparesis was suggested in another hospital. First magnetic resonance imaging (MRI) scan of the brain and spinal cord was normal at 4 years. Her history revealed a normal pregnancy and delivery in the mother. Her parents were consanguineous. A second MRI scan of the brain at the age of 6 years showed atrophy of the cerebellar vermis.
On neurological examination in our hospital, she had ataxia, dysarthria, nystagmus, spasticity in lower limbs, global hyperreflexia, and pes cavus. Plantar responses were extensor. Her cognitive function and ophthalmologic evaluation were normal. The MRI showed atrophy of the superior cerebellar vermis and linear hypodensity in the pons [Figure 1]. Electroneuromyography showed sensorial and motor axonal peripheral neuropathy. Previous investigations in other clinics and our hospital showed negative genetic test for Friedreich’s ataxia, normal vitamin E status, alpha-fetoprotein, immunoglobulins, urine amino acids, and organic acids and negative molecular tests for spinocerebellar ataxias type 1, 2, 3, 6, 7, and 10. The clinical and radiological findings suggested ARSACS. We detected homozygous mutation (c.2182C>T) (p.Arg728) (p.R728) in the SACS gene. Written informed consent was obtained from the parents of the patient who participated in this case.
Figure 1.
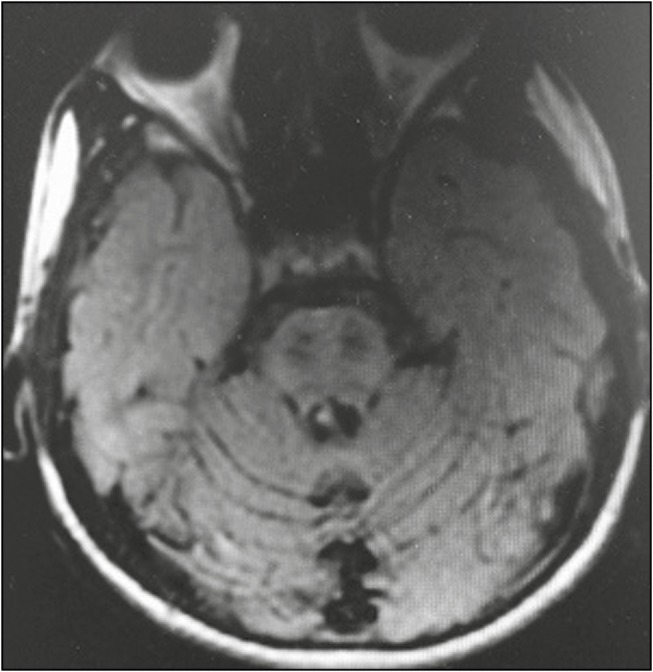
Cerebral magnetic resonance imaging showing atrophy of the superior cerebellar vermis and linear hypodensity in the pons
DISCUSSION
ARSACS is an inherited neurodegenerative disorder characterized by early-onset spastic ataxia, dysarthria, nystagmus, distal muscle wasting, finger and foot deformities, mixed sensorimotor neuropathy, and retinal hypermyelination. The ARSACS gene is located on chromosome 13q12.12 and encodes the large protein sacsin.[2] Sacsin is most highly expressed in large neurons, including cerebellar Purkinje cells. It is an otoxic protein and a regulator of the Hsp70 chaperone machinery that is implicated in the processing of other ataxia-linked proteins was changed as it is an ataxia protein and a regulator of the Hsp70 chaperone machinery that is implicated in the processing of other ataxia-linked proteins.
ARSACS was originally described in the late 1970s with a high prevalence among inhabitants of northeastern Quebec, Canada.[1] In recent years, ARSACS has since been recognized in patients around the world.[3,4,5,6] Ouyang et al.[4] reported four patients with ARSACS in the first known Belgian family. In Tunisia, four unrelated families carrying four distinct mutations have been already described in a study by El Euch-Fayache et al.[3] In another study, Pedroso et al.[5] described a Brazilian family with ARSACS. To the best of our knowledge, Turkish patients with ARSACS have been previously reported.[6,7,8] Detected mutations in previously reported Turkish patients were different from that of our patient. We identified the same mutation that was reported in a study by Synofzik et al.[9]
In patients with ARSACS, electrophysiological studies show axonal and demyelinating neuropathy. Sometimes, even severe peripheral nerve degeneration may have masked any spasticity. Atrophy of the superior cerebellar vermis is always present. Retinal hypermyelinated fibers have been observed in patients from Quebec, but absent or mild in patients from France, Tunisia, and Turkey.[10] Similar to previous reports, our patient has polyneuropathy and cerebellar vermis atrophy, but not retinal hypermyelinated fibers.
In conclusion, ARSACS has been described in patients around the world in the recent years. We suggest that in patients suspected of having a recessive ataxia with hyperreflexia, spasticity, and cerebellar vermis atrophy, studies of nerve conduction and ophthalmological examination may be informative. The prevalence of ARSACS in the world may be greater than previously suspected, and screening for mutations in the SACS gene may be useful.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Bouchard JP, Barbeau A, Bouchard R, Bouchard RW. Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Can J Neurol Sci. 1978;5:61–9. [PubMed] [Google Scholar]
- 2.Richter A, Rioux JD, Bouchard JP, Mercier J, Mathieu J, Ge B, et al. Location score and haplotype analyses of the locus for autosomal recessive spastic ataxia of Charlevoix-Saguenay, in chromosome region 13q11. Am J Hum Genet. 1999;64:768–75. doi: 10.1086/302274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.El Euch-Fayache G, Lalani I, Amouri R, Turki I, Ouahchi K, Hung WY, et al. Phenotypic features and genetic findings in sacsin-related autosomal recessive ataxia in Tunisia. Arch Neurol. 2003;60:982–8. doi: 10.1001/archneur.60.7.982. [DOI] [PubMed] [Google Scholar]
- 4.Ouyang Y, Segers K, Bouquiaux O, Wang FC, Janin N, Andris C, et al. Novel SACS mutation in a Belgian family with sacsin-related ataxia. J Neurol Sci. 2008;264:73–6. doi: 10.1016/j.jns.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 5.Pedroso JL, Braga-Neto P, Abrahão A, Rivero RL, Abdalla C, Abdala N, et al. Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS): typical clinical and neuroimaging features in a Brazilian family. Arq Neuropsiquiatr. 2011;69:288–91. doi: 10.1590/s0004-282x2011000300004. [DOI] [PubMed] [Google Scholar]
- 6.Gücüyener K, Ozgül K, Paternotte C, Erdem H, Prud’homme JF, Ozgüç M, et al. Autosomal recessive spastic ataxia of Charlevoix-Saguenay in two unrelated Turkish families. Neuropediatrics. 2001;32:142–6. doi: 10.1055/s-2001-16616. [DOI] [PubMed] [Google Scholar]
- 7.Oguz KK, Haliloglu G, Temucin C, Gocmen R, Has AC, Doerschner K, et al. Assessment of whole-brain white matter by DTI in autosomal recessive spastic ataxia of Charlevoix-Saguenay. AJNR Am J Neuroradiol. 2013;34:1952–7. doi: 10.3174/ajnr.A3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kurt S, Kartal E, Aksoy D, Cevik B, Eken AG, Sahbaz I, et al. Coexistence of autosomal recessive spastic ataxia of Charlevoix-Saguenay and spondyloepiphyseal dysplasia in a Turkish patient. J Neurol Sci. 2015;357:290–1. doi: 10.1016/j.jns.2015.06.050. [DOI] [PubMed] [Google Scholar]
- 9.Synofzik M, Soehn AS, Gburek-Augustat J, Schicks J, Karle KN, Schüle R, et al. Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS): expanding the genetic, clinical and imaging spectrum. Orphanet J Rare Dis. 2013;8:41. doi: 10.1186/1750-1172-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palau F, Espinós C. Autosomal recessive cerebellar ataxias. Orphanet J Rare Dis. 2006;1:47. doi: 10.1186/1750-1172-1-47. [DOI] [PMC free article] [PubMed] [Google Scholar]