

Abstract
The regulatory mechanism of Helicobacter pylori-induced gastric carcinogenesis remains unclear. Autophagy has previously been identified as an effective method of regulating carcinogenesis. In the present study, microRNA (miR)-99b levels increased in H. pylori-infected gastric cancer tissues and the BGC-823 gastric cancer cell line. Overexpression of miR-99b significantly enhanced autophagy, decreased intracellular bacterial loads and blocked cell proliferation. The effect on autophagy was demonstrated to be triggered by mammalian target of rapamycin inhibition. These results indicate that miR-99b expression serves a key role in preventing H. pylori-associated gastric cancer formation and this may provide potential targets for the future treatment of H. pylori-associated diseases.
Keywords: Helicobacter pylori, autophagy, microRNA, mTOR
Introduction
Helicobacter pylori infection is associated with an increased risk of gastric cancer (1) and results inchanges in the expression of inflammatory molecules, including cytokines and other immune mediators (2). Immune mediators activated by H. pylori have been demonstrated to promote neoplastic transformation (3).
MicroRNAs (miRNAs/miRs) are small noncoding RNAs that have been implicated in a number of physiological and pathological responses as post-transcriptional repressors of gene expression (4). miRNAs can specifically bind to the 3′ untranslated region (UTR) of target cellular mRNAs, in turn triggering mRNA degradation or inhibition of translation (4). Previous studies have suggested an association between miRNAs and H. pylori-induced gastric carcinogenesis; Zhang et al (5) demonstrated that miR-21 was significantly upregulated in human gastric cancer tissues and H. pylori-infected gastric mucosa, and that this upregulation enhanced cell proliferation and invasion in a gastric cancer cell line. Miao et al (6) demonstrated that miR-375 inhibits H. pylori-induced gastric carcinogenesis by blocking the Janus kinase 2-signal transducer and activator of transcription 3 signaling pathway.
Autophagy is an important cellular function that enables the recycling of proteins or damaged organelles (7). Previous studies have revealed that H. pylori can induce autophagy in gastric epithelial cells (8), and autophagy serves an important functionin the development and progression of cancer (9). Therefore H. pylori-dependent autophagy maybe carcinogenic; however, the molecular mechanisms underlying the association between autophagy and H. pylori-induced gastric carcinogenesis remain unclear.
Mammalian target of rapamycin (mTOR) is an inhibitor of autophagy (8–10). Inhibitors of mTOR and miRNAs that target mTOR have been demonstrated to promote autophagy and therefore suppress the proliferation of a number of types of cancer cell (10). In the present study, miR-99b enhanced autophagy by targeting mTOR in gastric epithelial cells and promoted H. pylori-induced gastric carcinogenesis.
Materials and methods
Antibodies and reagents
The GFP-microtubule-associated protein 1A/1B light chain 3 (LC3) plasmid was kindly provided by Dr Di Wang (Department of Clinical Laboratory, The People's Liberation Army No. 309 Hospital, Beijing, China). Rapamycin (Rapa; cat. no. R8781), bafilomycin A1 (cat. no. b1793) and 3-methyladenine (3-MA; cat. no. 5142-23-4) were purchased from Sigma-Aldrich; Merck KGaA, Darmstadt, Germany. Antibodies against mTOR (cat. no. 2972), LC3 (cat. no. 4108) and GAPDH (cat. no. 5174) were obtained from Cell Signaling Technology, Inc., Danvers, MA, USA. Anti-rabbit (cat. no. sc-2054) and anti-mouse (cat. no. sc-358914) secondary antibodies were purchased from Santa Cruz Biotechnology, Inc., Dallas, TX, USA. ELISA kits for tumor necrosis factor-α (TNF-α; cat. no. MTA00B) and interleukin-8 (IL-8; cat. no. D8000C) were purchased from R&D Systems, Inc., Minneapolis, MN, USA.
Cell and bacterial culture
The human embryonic kidney cell line HEK-293T and human gastric cancer BGC-823 cell line were purchased from the American Type Culture Collection (Manassas, VA, USA). HEK-293T was cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) in a humidified incubator at 37°C with 5% CO2. The human gastric cancer cell line BGC-823 was cultured in RPMI-1640 medium (Invitrogen; Thermo Fisher Scientific, Inc.) supplemented with 10% FBS and 100 U/ml penicillin in a humidified incubator at 37°C with 5% CO2. The wild-type H. pylori strain 26695 was obtained from American Type Culture Collection (Manassas, VA, USA) and grown as previously described (11). Subsequently, cells were seeded inthe wells of a 12-well plate and grown to 80% confluency. The medium was then replaced with antibiotic-free medium. H. pylori was added to cells at a multiplicity of infection (MOI) of 100:1. The infection model was monitored by the release of IL-8 and TNF-α, as measured by the DuoSet® ELISA Development System (R&D Systems, Inc., Minneapolis, MN, USA).
Cell transfection
All oligonucleotides were synthesized byShanghai GenePharma Co., Ltd. (Shanghai, China). Sequences of all the oligonucleotides were as follows: miR-99b mimic, 5′-CACCCGUAGAACCGACCUUGCG-3′; miR-99b inhibitor, 5′-CACCCGUAGAACCGACCUUGCG-3′; and scrambled miR-control, 5′-CAGUACUUUUGUGUAGUACAA-3′. Transfections were performed using Lipofectamine® 2000 (Invitrogen; Thermo Fisher Scientific, Inc.), according to the manufacturer's protocol. Cells were transfected with 50 nM miRNA mimics, inhibitors, or scrambled miR-control (negative control) for 24 h prior to subsequent experiments.
Clinical samples
Patients undergoing gastrointestinal endoscopy at The People's Liberation Army No. 309 Hospital (Beijing, China) between January 1, 2014 and December 30, 2015, were enrolled. In total, 38 patients with H. pylori+ gastric cancer (28 male and 10 female; age range, 33–64 years), 10 patients with H. pylori− gastric cancer (6 male and 4 female; age range, 27–63 years), 22 patients with H. pylori+ non-gastric cancer (17 male and 5 female; age range, 36–66 years) and 9 healthy controls (6 male and 3 female; age range, 26–60 years) were included in the present study. Gastric cancer tissues from the patients and gastric mucosa tissuefrom non-cancer controls were retrieved from all the subjects who received endoscopic sub-mucosal dissection and stored at −80°C prior to subsequent experiments. The diagnoses of all the specimens were histopathologically confirmed. The present study was approved by the Ethics Committee of Dalian Medical University. Written informed consent was obtained from all patients.
Bacterial colony-forming unit (CFU) determination
Following infection, the cell bacterium co-culture was washed three times with 1 ml of warm PBS per well to remove non-adherent bacteria. To determine the CFU count corresponding to intracellular bacteria, the cell monolayers were treated with gentamicin (100 µg/ml; Sigma-Aldrich; Merck KGaA; G1272) at 37°C in 5% CO2 for 1 h, washed three times with warm PBS, and then incubated with 1 ml of 0.5% saponin (Sigma-Aldrich; Merck KGaA; 47036) in PBS at 37°C for 15 min. The treated monolayers were homogenized and viable bacteria were grown on Mueller-Hinton agar (BD Biosciences, Franklin Lakes, NJ, USA) containing sterile defibrinated sheep blood (Shanghai YIJI industries Co., Ltd., Shanghai, China; cat. no. P0041) (5% v/v) and enumerated by the pour plate method following serial dilution (11).
Bioinformatics approach
The Target Scan database (http://www.targetscan.org) was used to identify putative human target genes for miR-99b by their 3′UTRs.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) in accordance with the manufacturer's protocol. RT-qPCR analysis for miRNAs was performed using TaqMan miRNA assays (Ambion; Thermo Fisher Scientific, Inc.) and has-miR-99b RT-PCR primer set (cat. no., abx096835; Abbexa Ltd., Cambridge, UK) in an iQ5 Real-Time PCR Detection System (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Reverse transcription reactions were performed using TAQMAN® microRNA RT kit (Ambion; Thermo Fisher Scientific, Inc.) under the following conditions: 16°C for 30 min, 42°C for 30 min and 84°C for 5 min. PCR reactions were performed using the following conditions: 95°C for 2 min followed by 40 cycles of 95°C for 15 sec and 60°C for 30 sec. U6 small nuclear RNA was used as an endogenous control for data normalization. Relative expression was calculated using the 2−ΔΔCq method (12).
Western blot analysis
Cells were washed with PBS and then lysed with protein lysis buffer (Pierce; Thermo Fisher Scientific, Inc.). Following centrifugation at 5,000 × g for 15 min at 4°C, the protein concentration was measured with a bicinchoninic acid protein assay kit (Pierce; Thermo Fisher Scientific, Inc.). A total of 50 µg protein was loaded per lane and run on a 10% SDS-PAGE gel. The proteins were transferred to polyvinylidene difluoride membranes. The membranes were blocked with 5% non-fat dry milk in Tris-buffered saline (pH 7.4) containing 0.05% Tween-20 at room temperature for 1 h, and were incubated with primary antibodies (1:200; Cell Signaling Technology, Inc.) and horseradish peroxidase-conjugated secondary antibodies (1:5,000; Santa Cruz Biotechnology, Inc.) at 4°C overnight in accordance with the manufacturer's protocol. The protein of interest was visualized using an enhanced chemiluminescence western blotting substrate (Pierce; Thermo Fisher Scientific, Inc.) and the Chemidoc XRS Gel Documentation System (Bio-Rad Laboratories, Inc.). Image J (version 2.1.4.7; National Institutes of Health, Bethesda, MD, USA) was used for quantification of the results.
Cell proliferation assay
An MTT assay, which tests for cell proliferation and viability, was used in this study as previously described (13). Cells were seeded at a density of 5×103/well and incubated in 96-well culture plates for 72 h at 37°C. A total of 50 µl MTT solution was added to each well prior to incubation at 37°C for 4 h. Following incubation, the MTT reagent was aspirated and 150 µl/well of DMSO was added to each well to dissolve the formazan precipitate. Subsequently, the DuoSet® ELISA Development System (R&D Systems, Inc., Minneapolis, MN, USA) was used to measure the optical densities of the plates at 570 nm. Cell number was calculated as follows: Cell numbertarget=(ODtarget-ODblank)/(ODcontrol-ODblank) × cell numbercontrol.
Confocal microscopy
Cells were grown on collagen-precoated glass cover slips in 24-well plates at a density of 105 cells/well. Cells were transfected with control, miR-99b mimics or inhibitor, together with GFP-LC3 using Lipofectamine® 2000. Cells were washed three times for 3 min with PBS, fixed with 4% paraformaldehyde at 37°C for 15 min and further washed with PBS. The cells were then mounted with vecta-shield reagent (Vector Laboratories, Inc., Burlingame, CA, USA). A Radiance 2000 laser scanning confocal microscope was used for confocal microscopy, followed by analysis with Laser Sharp 2000 software (Bio-Rad Laboratories, Inc.). Images were captured in the sequential scanning mode.
Plasmid constructs and luciferase reporter assay
To construct the luciferase report vector, mTOR 3′UTR and its flanking sequence were PCR-amplified using primers: 5′-CGGGGTACCAGATGTGCCCATCACGTTTT-3′ (forward) and 5′-CCGGAATTCTGGTGTCTAGACATGGCTACACTT-3′ (reverse). The amplified fragment was cloned into the PGL3 vector (Promega Corporation, Madison, WI, USA). Similarly, a mutated mTOR 3′UTR fragment, in which the miR-99b (5′-UACGGGU-3′ to 5′-AUGCCCA-3′) binding site was mutated, was PCR-amplified using primers: 5′-CCATAACTTTAGAAAGCTACACTTTGACTTAACTCAC-3′ (forward) and 5′-GTGAGTTAAGTCAAAGTGTAGCTTTCTAAAGTTATGG-3′ (reverse). The PCR product was cloned into the vector PGL3. Luciferase activity assay was performed according to the instruction manual (Promega Corporation).
Statistical analysis
All data are expressed as the mean ± standard deviation from ≥3 independent experiments performed in triplicate. Statistically significant differences between groups were determined using a two-tailed Student's t-test using SPSS software (version 17.0; SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Expression of miR-99b is increased in H. pylori+cancer samples compared with H. pylori− cancer samples
The expression of miR-99b was evaluated in 38 H. pylori+ and 10 H. pylori−gastric cancer samples. The expression of miR-99b was significantly increased in H. pylori+ gastric cancer samples compared with that in H. pylori− gastric cancer samples (P<0.05; Fig. 1A). To confirm these clinical findings, miR-99b expression was evaluated in H. pylori-infected BGC-823 human gastric carcinoma cells. Expression of miR-99b was significantly increased following H. pylori infection compared with control cells (P<0.01; Fig. 1B). Simultaneously, the expression of the pro-inflammatory cytokines IL-8 and TNF-α was significantly increased following H. pylori infection compared with the control (both P<0.01; Fig. 1C and D). These data suggested that H. pylori infection promoted the upregulation of miR-99b, which may be implicated in neoplastic transformation.
Figure 1.

miR-99b expression is increased in H. pylori+ compared with H. pylori− gastric cancer tissue samples. Reverse transcription-quantitative polymerase chain reaction results of (A) miR-99b expression in H. pylori+ and H. pylori− gastric cancer tissue samples, (B) miR-99b expression in BGC-823 cells infected with H. pylori, (C) IL-8 expression in BGC-823 cells infected with H. pylori and (D) TNF-α expression in BGC-823 cells infected with H. pylori. **P<0.01 vs. control. miR, microRNA; IL-8, interleukin-8; TNF-α, tumor necrosis factor-α.
miR-99b enhances intracellular H. pylori elimination and decreases BGC-823 cell proliferation
BGC-823 gastric cancer cells were transiently transfected with miR-99b mimics, inhibitors, or negative control and then challenged with H. pylori at an MOI of 100. The intracellular survival rate of H. pylori was examined at different time points. The results demonstrated that transfection with miR-99b mimics significantly reduced the survival rate of intracellular H. pylori compared with the negative control (P<0.05; Fig. 2A). Transfection with a miR-99b inhibitor had no significant effect on H. pylori survival, with the exception of at 48 h following infection, when the survival rate of H. pylori was significantly increased compared with the negative control (P<0.05; Fig. 2A). To verify whether miR-99b serves a rolein tumor initiation, the effects miR-99b on cell proliferation were assessed. An MTT analysis demonstrated that the BGC-823 cell number was markedly decreased following transfection with miR-99b mimics compared with the negative control group, whereas transfection with an miR-inhibitor markedly increased the cell number compared with the control group at 12, 24 and 48 h post-infection (Fig. 2B).
Figure 2.
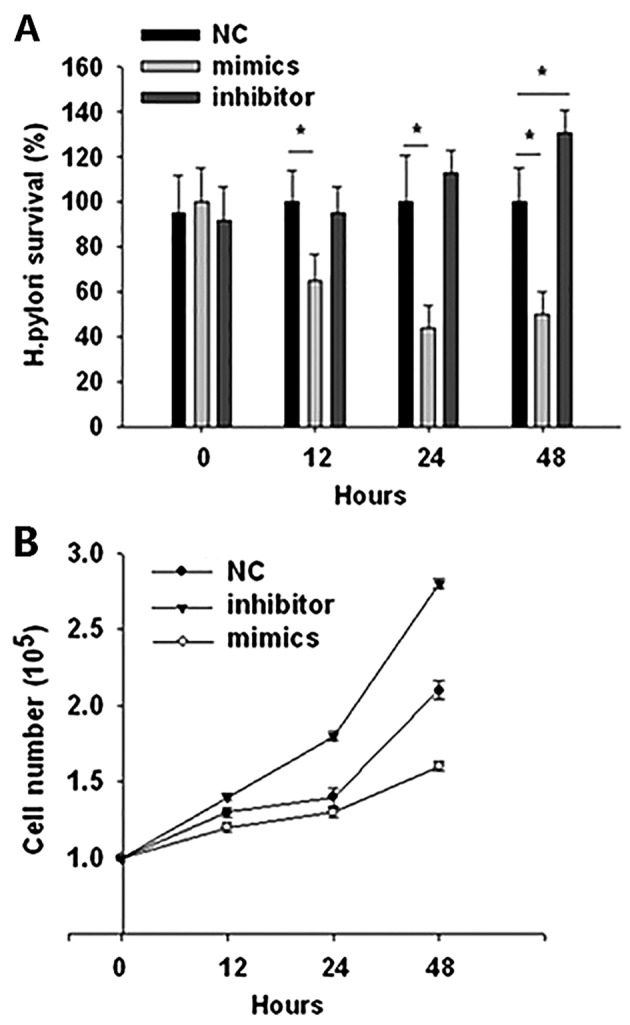
miR-99b overexpression decreases intracellular bacteria load and negatively regulates gastric cancer cell proliferation. BGC-823 cells were transfected with miR-99b mimics, inhibitor or negative control and then challenged with H. pylori at a multiplicity of infection of 100. (A) Intracellular bacteria loads at different time points post-infection with H. pylori. (B) Cell numbers at different time points as measured using an MTT assay. *P<0.05 vs. NC. miR, microRNA; NC, negative control.
miR-99b induces intracellular H. pylori elimination and BGC-823 cell death through autophagy upregulation
It has previously been demonstrated that H. pylori-induced cell proliferation was acarcinogenic factor (6), and autophagy serves a critical role in intracellular H. pylori elimination and regulation of cell proliferation and carcinogenesis (8). Therefore miR-99b may mediate H. pylori-induced cell proliferation by regulating autophagy. LC3 conversion from LC3-I to LC3-II indicates activation of autophagy (8,9). To test our hypothesis, LC-3 expression was measured in BGC-823 cells following transfection with miR-99b mimics, inhibitors or a negative controlfor 24 h. Transfection with miR-99b mimics significantly increased the expression of LC3-II compared with the negative control (P<0.05), whereas transfection with miR-99b inhibitor had no clear effect on the expression of LC-II (Fig. 3A). As a positive control, treatment with rapamycin (200 nM; autophagy inducer) significantly increased the expression of LC3-II compared with the control (P<0.05). In addition, treatment with bafilomycin A1 (200 nM; LC3-II degradation inhibitor) resulted in a significant increase in LC3-II expression compared with the control, indicating that the autophagic flux was unobstructed (P<0.05; Fig. 3A). To confirm that miR-99b triggers autophagy, a GFP-LC3-II puncta formation assay was used to monitor autophagy. Transfection with miR-99b significantly increased the percentage of cells with GFP-LC3-II-positive puncta compared with the control cells (P<0.05), whereas transfection with miR-99b inhibitor had no effect (Fig. 3B). Taken together, these results demonstrated that miR-99b promotes autophagy in gastric cancer cells.
Figure 3.
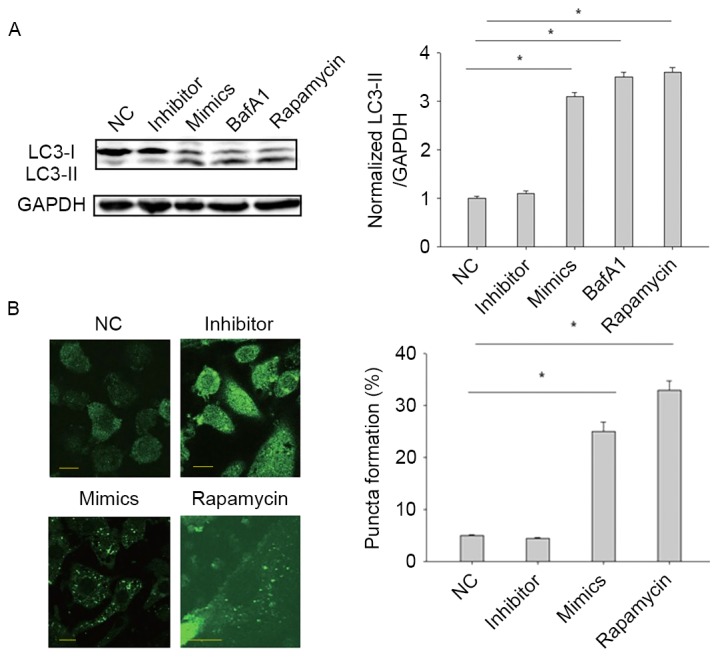
miR-99b promotes autophagy. (A) BGC-823 cells were treated with rapamycin, BafA1, miR-99b mimics or miR-99b inhibitor. The LC3-II/GAPDH level was determined by western blot analysis. (B) BGC-823 cells were transfected with plasmid expressing GFP-LC3B, together with either negative control, miR-99b mimics or miR-99b inhibitor. After 24 h, following fixation, cells were immediately visualized using confocal microscopy (scale bar=10 µm). The number of GFP-LC3B puncta in each cell was counted. *P<0.05 vs. NC. miR, microRNA; LC3, microtubule-associated protein 1A/1B light chain 3; NC, negative control; BafA1, bafilomycin A1.
It was further investigated whether miR-99b induces intracellular H. pylori elimination and gastric carcinoma cell death through autophagy upregulation. BGC-823 cells were pre-treated with miR-99b mimics or not, and either left untreated or pre-treated with the autophagy inhibitor 3-MA (5 mM), followed by exposure to H. pylori for 12, 24 and 48 h. Treatment with 3-MA reversed the effect of miR-99b mimics on the H. pylori survival rate (Fig. 4A) and BGC-823 cell number (Fig. 4B). These data demonstrate that miR-99b decreases the survival of intracellular H. pylori and BGC-823 cell numberby inducing autophagy.
Figure 4.
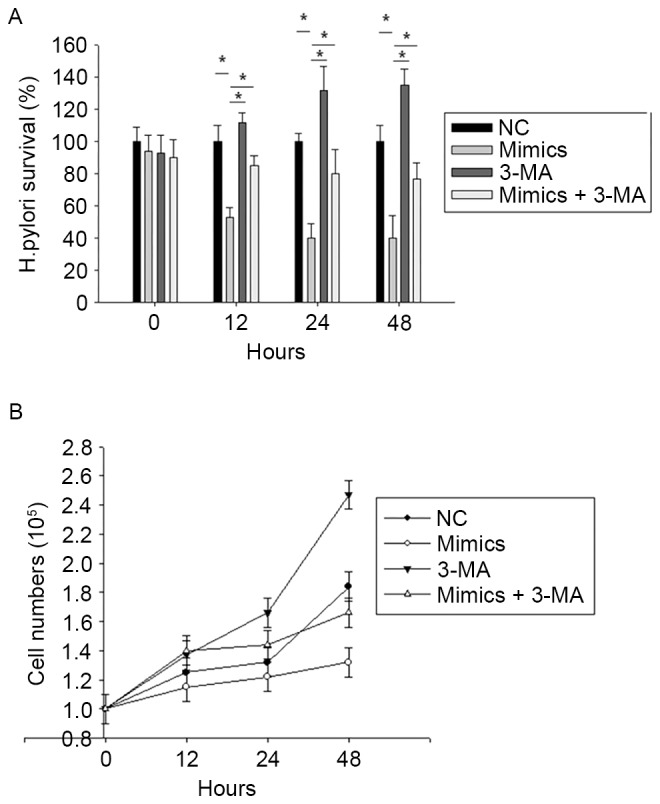
miR-99b decreases gastric cancer cell proliferation by suppressing autophagy. BGC-823 cells were pretreated with miR-99b mimics or negative control, and either left untreated or pre-treated with the autophagy inhibitor 3-MA, followed by exposing to H. pylori for 12, 24 and 48 h. (A) Intracellular bacteria loads at different time points. (B) Cell numbers at different time points as determined using an MTT assay. *P<0.05 vs. NC. miR, microRNA; 3-MA, 3-methyladenine; NC, negative control.
miR-99b promotes autophagy by targeting mTOR
To determine the mechanism underlying the effect of miR-99b on autophagy, a bioinformatics approach was used to identify potential miR-99b targets, and miRNA target identification quality was improved through theuse of prediction programs. mTOR was identified as a putative miR-99b target gene (Fig. 5A). To directly address whether miR-99b binds to the 3′UTR of target mRNAs, luciferase report vectors were constructed that contained the putative miR-99b binding sites within the mTOR 3′UTR. In addition, a vector containing the mTOR 3′UTR mutated within the putative miR-99b binding site (3′MUT) was generated. A significant reduction in luciferase activity was observed in HEK-293T cells following transfection with the mTOR 3′UTR luciferase vector and miR-99b mimics compared with cells transfected with the luciferase vector alone (P<0.05; Fig. 5B). However, no significant difference in luciferase activity was observed in cells transfected with 3′MUT and miR-99b mimics compared with cells transfected with 3′MUT alone.
Figure 5.

miR-99b inhibits autophagy by targeting mTOR. (A) The region of the human mTOR mRNA 3′UTR predicted to be targeted by miR-99b. (B) HEK-293T cells were transiently co-transfected with luciferase reporter vectors, and either miR-99b mimics, inhibitor or negative control. Luciferase activities were normalized to the activity of Renilla luciferase. (C) BGC-823 cells were transfected with miR-99b mimics, inhibitor or negative control, then the protein level of mTOR was determined by western blot analysis. (D) Quantification of (C). *P<0.05 vs. NC. miR, microRNA; mTOR, mammalian target of rapamycin; UTR, untranslated region; NC, negative control.
Next, the protein expression of mTOR inBGC-823 cells transfected with miR-99b mimics or inhibitors was measured. Treatment with miR-99b mimics significantly reduced the protein expression of mTOR (P<0.05), whereas treatment with miR-99b inhibitor significantly increased the protein expression of mTOR (P<0.05; Fig. 5C and D). As the mTOR inhibitor rapamycin significantly enhanced autophagy in gastric cancer cells (Fig. 3), taken together these results indicate that miR-99b decreases intracellular bacterial load and gastric cancer cell proliferation by promoting autophagy through the inhibition of mTOR expression.
Discussion
H. pylori promotes inflammation of the gastric mucosa, which can then lead to gastric atrophy and intestinal metaplasia, resulting in the development of gastric cancer. H. pylori may therefore represent acritical risk factor for gastritis and gastric cancer transformation. Previous studies have indicated that cell proliferationis an important indicator of gastric carcinogenesis; therefore controlling cell proliferation may be an effective method to prevent and treat gastric cancer (6).
Among the mediators induced in response to H. pylori infection, miRNAs may serve a major rolein the outcome of the bacteria-host interaction by acting as immune mediators that link inflammatory responses with gastric carcinogenesis; Li et al (14) demonstrated that H. pylori may function as an initiator in the process of carcinogenesis by up-regulating miR-222, which promotes cell proliferation and inhibits the expression of reversion-inducing-cysteine-rick protein with kazal motifs. Ma et al (15) demonstrated that miR-223 contributes to cancer cell proliferation and migration during H. pylori infection. In addition, Chang et al (16) identified a series of miRNAs (miR-99b-3p, −564 and −638) that are aberrantly expressed in gastric cancer in a H. pylori-dependent manner, further demonstrating that certain miRNAs are differentially expressed between H. pylori+ and H. pylori− gastric cancer tissues.
miR-99b has been demonstrated to serve as a tumor suppressor by inhibiting cancer cell proliferation by targeting a number of proteins, including homeobox-A1, SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 5 (SMARC) subfamily A member 5, SMARC subfamily D member 1 and mTOR (17). mTOR serves a crucial role in cancer biology and has emerged as a potential target for drug development (18).
The results from the present study demonstrated that H. pylori infection induces cell proliferation, which is regulated by miR-99b. Challenging with miR-99b inhibitor increased mTOR expression and promoted BGC-823 cell proliferation, the latter of which was reversed by treatment with the mTOR inhibitor rapamycin. H. pylori elimination may block cancer cell proliferation (6), indicating that miR-99b may suppress carcinogenesis partly through bacterial elimination. As treatment with miR-99b mimics in the absence of H. pylori infection also decreased cell proliferation in the current study, there may be an additional regulatory pathway of miR-99b that affects carcinogenesis.
mTOR has previously been identified as a key regulatory factor in the autophagic pathway due to is role as a sensor for amino acids and ATP depletion, and its role in integrating hormonal stimuli via the class I phosphoinositide 3-kinase/protein kinase B signaling pathway (8). In the immune system, mTOR is induced by inflammatory cytokines and growth factors, and inhibits inflammation-associated gastrointestinal tumorigenesis (19). A previous study suggested that H. pylori suppresses autophagy, mediates inflammation and facilitates intracellular survival of the pathogen itself, while also generating an environment that favors carcinogenesis (20). The results from the present study suggest that the induction of autophagy by miR-99b negatively regulates cancer cell proliferation and therefore prevents H. pylori-induced carcinogenesis. These findings provide a basis for novel treatments for cancer based on miRNA-dependent induction of autophagy.
Acknowledgements
The authors of the present study would like to thank Dr Qi Yu and Dr Chaohui Zhu from The People's Liberation Army No. 309 Hospital (Beijing, China) for providing the clinical tissues samples of the patients.
References
- 1.Lee YC, Chiang TH, Chou CK, Tu YK, Liao WC, Wu MS, Graham DY. Association between Helicobacter pylori eradication and gastric cancer incidence: A systematic review and meta-analysis. Gastroenterology. 2016;150:1113–1124. doi: 10.1016/S0016-5085(16)33759-3. [DOI] [PubMed] [Google Scholar]
- 2.Wessler S, Krisch LM, Elmer DP, Aberger F. From inflammation to gastric cancer-the importance of Hedgehog/GLI signaling in Helicobacter pylori-induced chronic inflammatory and neoplastic diseases. Cell Commun Signal. 2017;15:15. doi: 10.1186/s12964-017-0171-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adamsson J, Lundin SB, Hansson LE, Sjovall H, Svennerholm AM. Immune responses against Helicobacter pylori in gastric cancer patients and in risk groups for gastric cancer. Helicobacter. 2013;18:73–82. doi: 10.1111/j.1523-5378.2012.00991.x. [DOI] [PubMed] [Google Scholar]
- 4.Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Z, Li Z, Gao C, Chen P, Chen J, Liu W, Xiao S, Lu H. MiR-21 plays a pivotal role in gastric cancer pathogenesis and progression. Lab Invest. 2008;88:1358–1366. doi: 10.1038/labinvest.2008.94. [DOI] [PubMed] [Google Scholar]
- 6.Miao L, Liu K, Xie M, Xing Y, Xi T. miR-375 inhibits Helicobacter pylori-induced gastric carcinogenesis by blocking JAK2-STAT3 signaling. Cancer Immunol Immunother. 2014;63:699–711. doi: 10.1007/s00262-014-1550-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fulda S. Autophagy in cancer therapy. Front Oncol. 2017;7:128. doi: 10.3389/fonc.2017.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang JC, Chien CT. A new approach for the prevention and treatment of Helicobacter pylori infection via upregulation of autophagy and downregulation of apoptosis. Autophagy. 2009;5:413–414. doi: 10.4161/auto.5.3.7826. [DOI] [PubMed] [Google Scholar]
- 9.Zhan Z, Li Q, Wu P, Ye Y, Tseng HY, Zhang L, Zhang XD. Autophagy-mediated hmgb1 release antagonizes apoptosis of gastric cancer cells induced by vincristine via transcriptional regulation of mcl-1. Autophagy. 2012;8:109–121. doi: 10.4161/auto.8.1.18319. [DOI] [PubMed] [Google Scholar]
- 10.Kim YC, Guan KL. mTOR: A pharmacologic target for autophagy regulation. J Clin Invest. 2015;125:25–32. doi: 10.1172/JCI73939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu K, Yang L, Li C, Zhu CH, Wang X, Yao Y, Jia YJ. MicroRNA-146a enhance Helicobacter pylori induced cell apoptosis in human gastric cancer epithelial cell. Asian Pac J Cancer Prev. 2014;15:5583–5586. doi: 10.7314/APJCP.2014.15.14.5583. [DOI] [PubMed] [Google Scholar]
- 12.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCt method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 13.Wang D, Zhu X, Cui C, Dong M, Jiang H, Li Z, Liu Z, Zhu W, Wang JG. Discovery of novel acetohydroxyacid synthase inhibitors as active agents against Mycobacterium tuberculosis by virtual screening and bioassay. J Chem Inf Model. 2013;53:343–353. doi: 10.1021/ci3004545. [DOI] [PubMed] [Google Scholar]
- 14.Li N, Tang B, Zhu ED, Li BS, Zhuang Y, Yu S, Lu DS, Zou QM, Xiao B, Mao XH. Increased mir-222 in H. pylori-associated gastric cancer correlated with tumor progression by promoting cancer cell proliferation and targeting reck. FEBS Lett. 2012;586:722–728. doi: 10.1016/j.febslet.2012.01.025. [DOI] [PubMed] [Google Scholar]
- 15.Ma L, Chen Y, Zhang B, Liu G. Increased microRNA-223 in Helicobacter pylori-associated gastric cancer contributed to cancer cell proliferation and migration. Biosci Biotechnol Biochem. 2014;78:602–608. doi: 10.1080/09168451.2014.895661. [DOI] [PubMed] [Google Scholar]
- 16.Chang H, Kim N, Park JH, Nam RH, Choi YJ, Lee HS, Yoon H, Shin CM, Park YS, Kim JM, et al. Different MicroRNA expression levels in gastric cancer depending on Helicobacter pylori infection. Gut Liver. 2014;9:188–196. doi: 10.5009/gnl13371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen D, Chen Z, Jin Y, Dragas D, Zhang L, Adjei BS, Wang A, Dai Y, Zhou X. MicroRNA-99 family members suppress homeobox a1 expression in epithelial cells. PLoS One. 2013;8:e80625. doi: 10.1371/journal.pone.0080625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Francipane MG, Lagasse E. mTOR pathway in colorectal cancer: An update. Oncotarget. 2014;5:49–66. doi: 10.18632/oncotarget.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thiem S, Pierce TP, Palmieri M, Putoczki TL, Buchert M, Preaudet A, Farid RO, Love C, Catimel B, Lei Z, et al. mTORC1 inhibition restricts inflammation-associated gastrointestinal tumorigenesis in mice. J Clin Invest. 2013;123:767–781. doi: 10.1172/JCI65086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenfield LK, Jones NL. Modulation of autophagy by Helicobacter pylori and its role in gastric carcinogenesis. Trends Microbiol. 2013;21:602–612. doi: 10.1016/j.tim.2013.09.004. [DOI] [PubMed] [Google Scholar]