

Abstract
Mammalian DNA replication is a highly organized and regulated process. Large, Mb-sized regions are replicated at defined times along S-phase. Replication Timing (RT) is thought to play a role in shaping the mammalian genome by affecting mutation rates. Previous analyses relied on somatic RT profiles. However, only germline mutations are passed on to offspring and affect genomic composition. Therefore, germ cell RT information is necessary to evaluate the influences of RT on the mammalian genome. We adapted the RT mapping technique for limited amounts of cells, and measured RT from two stages in the mouse germline - primordial germ cells (PGCs) and spermatogonial stem cells (SSCs). RT in germline cells exhibited stronger correlations to both mutation rate and recombination hotspots density than those of RT in somatic tissues, emphasizing the importance of using correct tissues-of-origin for RT profiling. Germline RT maps exhibited stronger correlations to additional genetic features including GC-content, transposable elements (SINEs and LINEs), and gene density. GC content stratification and multiple regression analysis revealed independent contributions of RT to SINE, gene, mutation, and recombination hotspot densities. Together, our results establish a central role for RT in shaping multiple levels of mammalian genome composition.
INTRODUCTION
DNA replication follows a highly regulated temporal program consisting of reproducible RT of different genomic regions (1–9). RT is conserved across species (2,10–12), and within a species ∼50% of genomic regions have stable RT across cell types, while the other 50% have variable RT between cell types (13,14). The importance and role of this temporal organization are still unclear.
RT correlates with many genomic and epigenomic features including transcription (2,15–17), gene density (18), chromatin state (19,20), retrotransposon density (17,21), lamina proximity (19), topological state (22–24), and GC content (2,24–26). RT is also associated with mutation rates both in cancer (27,28) and in the germline (29,30). Late replicating regions are enriched with point mutations (30,31), whereas the association between copy number variations (CNVs) and RT is more subtle and depends on the mechanism of CNV generation (32) and on the organism (reviewed in (33)). We recently investigated the correlation between RT and GC content and found that different substitution types have different associations with RT: late-replicating regions tend to gain both As and Ts along evolution. whereas early replicating regions tend to lose them (24). Measuring the levels of free dNTPs at different time points along S phase revealed an increase in the dATP + dTTP to dCTP + dGTP ratio along S, suggesting that a replication timing-dependent deoxynucleotide imbalance may underlie this mutation bias.
The association between RT and germline mutation frequency points to the importance of RT in shaping the genome sequence. To fully understand this association would require profiles of replication timing in germ cells. However, all previous studies used somatic tissue RT profiles as proxies for the investigation of the evolutionary impacts of RT. Thus, it is crucial to measure the RT in germ cells.
Germ cells refer to all the cells in an organism that pass on their genetic material to progeny. Mouse oogenesis and spermatogenesis involve 25 and 37–62 cell divisions, respectively (34). Mutations occurring at each step of this process are inherited by the next generation and thus all steps in this process are important from an evolutionary standpoint. RT has been measured in an in vitro model of the early stages of this process (embryonic stem cells (ESCs) to epiblast stem cells (EpiSCs) (13)), but there is no data regarding replication timing at later stages during which the majority of cell divisions occur (34) and during which a high percentage of germline mutations likely accumulate. In order to start filling this gap, we have measured RT at two different stages along the germline: primordial germ cells (PGCs, isolated directly from gonads of E13.5 mouse embryos) and spermatogonial stem cells (SSCs, isolated directly from testes of p5 pups). While SSCs can be grown in culture, the most relevant germline cells are those directly derived from animals, such as PGCs. However, only small amounts of such cells can readily be obtained. The current methods for measuring genome wide RT (reviewed in (35) and (20)), are usually applied to millions of growing cells (2,36), which is not feasible for many cell types including in vivo germ cells.
By improving the RT mapping method, we were able to generate reliable RT maps from as few as 1000 S-phase cells. We first demonstrated the reliability of this method on small populations of mouse embryonic fibroblasts (MEFs). We then measured the RT of in vivo PGCs and of in vitro grown SSCs. RT patterns of germ cells were highly correlated to each other, and were more similar to early embryonic tissues than to somatic cells. Both germline mutation and recombination hotspot densities correlated more strongly with the RT of the germ cell compared to that of somatic tissues, as expected. Mapping RT in the germline enabled us to similarly explore other genomic features such as GC content, LINE, SINE and gene density, all of which correlated better with germ cell RT. GC content stratification, as well as multiple regression analyses revealed that germ cell RT contributes to SINE, gene and recombination hot spot densities as well as to mutation rates, independently from the contributions of GC content. Taken together, our results suggest a role for germ cell RT in shaping multiple features of the genome sequence.
MATERIALS AND METHODS
Tissue culture
Mouse embryonic fibroblasts (MEFs) were cultured in DMEM medium (BI) supplemented with penicillin, streptomycin, l-glutamine and 20% (v/v) heat-inactivated (56°C, 30 min) FBS (BI). L1210 were cultured in L-15 medium (BI) supplemented with penicillin, streptomycin, l-glutamine and 10% v/v heat-inactivated FBS (BI). Cells isolated from the bone marrow of female C57BL6 mouse (10 weeks old) were grown in RPMI 1640 media (Gibco) supplemented with 10% fetal bovine serum (Hyclone), penicillin–streptomycin (Gibco), l-glutamine (Gibco) and 50 μM of β-mercaptoethanol (Gibco) on irradiated ST2 feeder cells. IL-7 conditioned medium (collected from J558L-IL7 secreting cells provided by A. Rolink) was added to the cells to select for pre-B cell populations for 14 days.
Isolation and activation of CD8+ cells
C57Bl/6J mouse spleen was processed and erythrocytes were lysed (155 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA). For each experiment, 3 × 106 splenocytes were moved to a 24-well plate and activated for 24–48 h in RPMI medium (BI) containing 1μg/ml anti CD3 (2C11, BioLegend) and supplemented with penicillin, streptomycin, and 10% (v/v) heat-inactivated (56°C, 30 min) FBS (BI). Activated splenocytes were stained with anti CD8-Pacific blue (J3.6.7, BioLegend) 1:500 and anti CD90.2-APC (H12, BioLegend) 1:500. Cells were then fixated as described below.
Isolation of PGCs
Either Oct4-GFP+/+ (129/b6 strain; Jackson Labs) or Sox2-GFP+/– (129 strain; Jackson Labs) mice were used for the isolation of PGC cells. These mice were bred to WT mouse strains (B6 M2rtTA+/+ mice; Jackson Labs) and females were sacrificed on day 13.5 of pregnancy. GFP positive cells were isolated from E13.5 gonads of either Oct4-GFP+/+ or Sox2-GFP+/– mouse embryos, resulting in a pure population of PGCs (37), according to the following procedure. Embryos were dissected in PBS under the microscope, GFP-positive gonads were chosen by fluorescent microscopy and 4–8 embryos were processed per experiment. Gonads and mesonephros were first dissected and then separated into single cells using trypsin (BI) and 700 μg/ml DNaseI (sigma), followed by neutralization using FBS (BI). Cells were washed with PBS (BI), and filtered through 35μm mesh into 5ml polystyrene tubes (BD). GFP+ cells were isolated using FACSARIA III (BD) using cold conditions, into new 5 ml polystyrene tubes, and fixated as described below. The identity of the PGCs was assessed by RT-PCR (Supplementary Figure S1). Most PGC samples were isolated from Oct4-GFP mice. The only sample derived from Sox2-GFP mice (male 2) was very similar to all other samples (Supplementary Figure S2).
Preparation and growing of SSCs
SSC culture was prepared from the testis of 4–7 days old F1 C57/Bl6 crossed with DBA male mice, according to Kubota et al. (38) with minor modifications. Testis cells suspensions were obtained using trypsin (BI) and DNaseI (Sigma). Thy1+ cells were isolated using magnetic microbeads conjugated with anti-Thy-1 antibody (Miltenyi Biotec). Cells were examined for their replenishment potential in busulfan treated NODSCID mice.
SSCs were seeded on irradiated MEFs and grown in StemPro-34 medium (Invitrogen) as described by Kanatsu-Shinohara et al. (39). Cells were supplemented with 1% FBS (BI), human GDNF 20 ng/ml (R&D systems), human LIF 50ng/ml (PeproTech), human FGF basic 1 ng/ml (PeproTech) and mouse EGF 20 ng/ml (PeproTech). Cells were cultured for 4–6 weeks and split every 5 days. The identity of the SSCs was assessed by RT-PCR (Supplementary Figure S1).
Fixation
MEFs and SSCs were washed with ice-cold PBS (BI), detached using trypsin (BI), and neutralized using the growth medium. Pre-B cells in suspension were carefully collected with their growth medium. All cells were moved to a 5ml Polystyrene tube (BD). All following reactions until filtration were done in the same tube and samples were kept at 4°C along the entire process. Cells were gently washed twice with ice-cold PBS, and resuspended in 250 μl cold PBS. PGCs were diluted with up to 250 μl cold PBS. CD8+ cells were diluted to 1.0 × 106 cells per 250μl cold PBS. For all cells, 100% high purity EtOH (Gadot) was added dropwise while slowly vortexing to a final volume of 75% EtOH. Cells were then incubated for 1–24 h at 4 °C.
Propidium iodide staining
Fixed cells were washed twice with 1ml cold PBS and spun down at 500 g for 10 min at 4 °C after each wash. Cells were resuspended in 0.2 ml PI-mix (PBS with 50 μg/ml propidium iodide (PI) (sigma) and 50 μg/ml RNAse-A (sigma)) and filtered through a 35 μm mesh into a new 5ml polystyrene tube (BD). In order to enhance cell recovery another 0.2 ml PI-mix was added and filtered to the new tube. For higher amounts of cells, we kept a concentration of 2.0 × 106 cells per ml of PI-mix. PI-stained cells were incubated for 15–30 min in the dark before sorting.
Flow cytometry
Cells were sorted using FACSARIA III (BD) based on their PI– intensity to G1 and S phases (40), using a flow rate of 1. Sorted cells were collected into 1.5 ml Protein-LoBind tubes (Eppendorf) and moved to ice.
DNA elution
DNA was extracted using DNeasy-kit (QIAGEN) and eluted twice with 2 × 200 μl of the kit elution buffer (AE). DNA was moved to a 1.7 ml MaxyClear tube (Axygen) which is compatible with the PureProteom Magnetic Stand (Milipore). 1.8x Agencourt AMPure XP beads (Beckman Coulter) were used to lower the elution buffer volume and gDNA was eluted from beads in 50 μl EB (Qiagen). DNA amounts were measured using Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific).
Sonication
Samples of 50 μl gDNA were transferred to a microTUBE Screw-Cap (520096, Covaris). Sonication was performed in the M220 Focused-ultrasonicator (Covaris) using 50 W, 20% Duty Factor at 20°C for 120 s, in order to reach an average target peak size of 250 bp. Sonication was verified using the D1000 or D1000 High Sensitivity ScreenTape using the Electrophoresis 2200 TapeStation system (Agilent).
Library preparation for whole genome sequencing
Library preparation was done similar to Blecher-Gonen et al. (41) with some changes. Briefly, Sonicated DNA was subjected to a 50 μl end repair reaction using 1 μl End repair mix (E6050L, NEB), cleaned by 1.8× Ampure XP beads, followed by a 50μl A-tail reaction using 2 μl Klenow fragment exo- (M0212L, NEB). The products were cleaned by 1.8× beads and were ligated by 2 μl quick ligase (M2200, NEB) to 0.75 μM illumina compatible forked indexed adapters. Ligation products were size selected by 0.7× PEG (considering the PEG in the ligation buffer) in order to remove free adaptors. 12–19 cycles of amplification were performed by PFU Ultra II Fusion DNA polymerase (600670, Agilent) with the following Primers:
P7 5′AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGAC 3′,
P5 5′ CAAGCAGAAGACGGCATACGAGAT 3′.
Amplified DNA was size selected for 300–700 bp fragments by taking the supernatant after using 0.5× beads (which removed fragments greater than 700 bp) followed by a 1.0× beads cleaning (which removed remaining primers and adapter dimers). The final quality of the library was assessed by Qubit and TapeStation. Libraries were pooled and sequenced on NextSeq (illumina) for 75 bp paired-end sequencing, generating 10M reads per each library.
Generation of RT maps
RT measurements were performed as described (40). Briefly, sequencing reads were mapped to the mm9 genome using Bowtie 2. Discordant reads and PCR duplicates were removed. Every 200 G1 phase reads were binned in order to establish genomic windows. Corresponding S phase reads were counted in order to determine an S/G1 ratio for each window. Ratio data was normalized by subtracting the mean and dividing by the standard deviation. Continuous data was smoothed and interpolated using the Matlab csaps function (with a smoothing parameter of 10−16) at a resolution of 100 kb (approximate average size of the windows). Continuous segments containing less than 15 informative windows were removed from the analysis.
Published Replication Timing profiles were obtained from replicationdomain.com accessions: Int26004257, Int62905691, Int3190888, Int20705995, Int93235019, Int83562596, Int52548116, Int87752970, Int17857752, Int62150809, Int88652090. Data was smoothed and interpolated similar to smoothing of RT profiles generated in our lab.
Determination of differential regions
Differential regions were determined using the likelihood ratio test at each genomic window. The null model assumes that at a given point, all six RT measures come from the same distribution with a given mean. The alternative model assumes that the replicates of each sample belong to two separate distributions each with its own mean. Probabilities were calculated using the normal distribution probability density function. The variances used were estimated as the average of the normally distributed genome-wide variance of each sample.
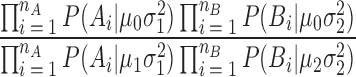 |
A chi squared P value was calculated for −2ln value of the ratio with 1 df. FDR correction (Benjamini-Hochberg) was used to control for multiple testing. All regions with a q value below 0.01 were selected as differential and extended until the q value exceeded 0.05. Regions were further filtered to include only those that contain at least one window with a mean RT difference >0.5 between the two samples.
Statistical analyses
For analyses including multiple datasets, RT data was filtered to only include windows containing informative data in all datasets. In addition, sex chromosomes were excluded from the analyses, resulting in ∼20000 windows or 2Gb. Where applicable, RT data was filtered to include only the RT switching regions as determined by Hiratani et al. (13). Clustering was performed using the python seaborn clustermap using the correlation metric and the average method. Correlations were calculated according to Spearman and confidence intervals were calculated by bootstrapping the data (n = 1000). LINE, SINE and GENEID data were obtained from the UCSC genome browser. Gene content was calculated as the percentage of bases covered by genes (from start to end of transcription) for each window. For inter-mammalian divergence, we used published mouse-rat divergence data in which exons, splicing junctions and CpGs were excluded (30). Recombination hot spots were taken from (42). All genomic features were analyzed in 100 kb windows besides for recombination hot spots which are sparser and thus were analyzed in 1 Mb windows. Chromatin accessibility data was downloaded from the GEO database (GSM2442671, GSM2098124, GSM1014153, GSM1014149) (43–45) and calculated in 100 kb windows by counting the number of peaks overlapping each window. Chromatin marks were downloaded from the GEO database (GSM936100, GSM1586501, GSM2067718) (46–48) and calculated in 100 kb windows by counting the number of peaks overlapping each window. SSC and PGC expression was downloaded from the GEO database (GSM1911697, GSE79552 – using the 11.5d PGC) (44,49) and calculated as the FPKM sum over 100 kb windows.
Principal component analysis (PCA) was performed using the python sklearn PCA function. For stratification analyses utilizing fixed GC content, genomic windows were sorted according to GC content and split into four equally sized bins. Genomic windows were similarly split into RT bins by binning the genome into five equally sized bins. Any intersection of RT and GC bins containing fewer than 50 or 15 windows for the 100 kb or megabase windows data respectively, were removed from analysis. Multiple regression analysis was performed using the python statsmodels OLS function. Autocorrelations were performed using the plot_acf function from the python statsmodels package. Partial correlations were calculated using a custom script based on the Matlab partialcorr function.
DATA ACCESS
The data have been deposited in NCBI’s Gene Expression Omnibus (50) and are accessible through GEO Series accession number GSE109804.
RESULTS
RT maps from small amounts of cells
One of the major limitations in RT mapping is the requirement of many cells. Current protocols usually require at least 105 S-phase cells (40). In order to overcome this limitation and to extend the technology for mapping RT from in vivo samples we optimized the RT profiling technique to minimize cell loss by optimizing fixation conditions, avoiding material transfer between tubes, using a slow flow rate during cell sorting and optimizing DNA extraction and library preparation protocols (see supplementary methods). Using this improved technique, we measured RT of MEFs using 103, 104 or 105 S-phase cells.
Triplicates were highly similar for 104 and 105 samples (R > 0.91 and R > 0.95, respectively) and quite similar even in the 103 triplicates (R > 0.76). Moreover, the RT maps from all cell numbers were quite similar to each other and to published RT map (13) and distinct from RT profiles generated from other tissues (Figure 1). Moreover, autocorrelation analyses performed on the different samples were almost identical (Supplementary Figure S3). Taken together, our results demonstrate the ability to obtain reliable RT maps from as little as 1000 S phase cells.
Figure 1.

RT mapping of small populations of cells. ( A) RT maps of 103,104 and 105 MEF cells, in triplicates, along a ∼80 Mb region on chromosome 1. (B) PCA analysis of RT profiles. Plot of PC1 vs PC2 for RT profiles of multiple MEF samples (described in A or published) and other somatic cells either sequenced by us (L1210, Pre-B and CD8) or published (CH12 and CD4). The MEF samples mapped in this paper are color coded as in A. CD8 samples, also achieved from small populations of cells are colored in gray. (C) A heatmap of spearman correlation coefficients between different RT profiles.
In order to further demonstrate the usefulness of our methodology for measuring RT of cell types for which the biological material is limited, we applied our technique to in vivo cell population of CD8+ cells. To this end, we isolated CD8+ cells from mouse peripheral blood, activated them in vitro, isolated 2000–5000 G1 and S phase cells and measured their RT. As expected, the RT of these cells was distinct from the RT of MEF cells and resembled the published RT profile of CD4+ cells (Figure 1), further supporting the accuracy of our methodology.
Replication timing profiles of the mammalian germline
To evaluate germ cell RT we concentrated on the two stages in mouse germline development in which most germ-cell divisions occur: PGC and SSC (Figure 2A). Following the double FACS procedure and our cell recovery improvements (see supplementary methods) we were able to isolate ∼1000–2000 G1 and S phase PGCs for each experiment (from 4–8 embryos). We used G1 and S phase PGCs in triplicates from both male and female embryos, and 104 G1 and S phase SSCs in triplicates, in order to generate RT maps (see methods). Despite the small amounts of PGC cells used, PGC and SSC RT maps showed high reproducibility (R > 0.8 and R > 0.85, respectively) and correlated with many genomic features as well as gene expression (Supplementary Figure S4), as had been shown for other RT maps (20). Interestingly, we found high similarity between the different PGC samples regardless of their sex (Supplementary Figure S2). Moreover, as expected, PGC and SSC RT showed specific association with chromatin accessibility in germ cells (Supplementary Figure S5), further supporting their accuracy.
Figure 2.

Germ cell RT. (A) Schema of the germline including the main stages from the zygote to the gonads, both for male and female mice. The number of cell divisions in each stage is shown (data taken from (34)). (B) RT map of triplicates of PGCs, SSCs and MEFs (105) along ∼14 Mb region of mouse Chr2. Below, three graphs are shown depicting the FDR-corrected q values of the likelihood ratio test for a pairwise comparison between two cell types. Differential regions are highlighted in green. (C) Bar graphs showing the total size (in megabases) of the differential regions, each bar graph is divided into two portions depicting the size of the regions that are earlier in the SSC (Blue), PGC (light blue) and MEF (red). (D) Boxplots showing the distribution of PGC chromatin accessibility data (number of NOME-seq peaks per window; (44)) in regions showing differential RT between SSC or PGC and MEF. Chromatin accessibility distribution was separated into MEF early versus germ cells early. P values (two sided t test) are shown above the box plots. (E) RT profiles from the current work along with published embryonic tissues (13) were hierarchically clustered. Only switching RT regions (see methods) were included. (F) PCA of RT profiles, plot of PC1 versus PC2 for RT profiles of different types, using the same color code and regions as in (E). Similar results were obtained by comparing germ cells to Pre-B cells (Supplementary Figure S6).
The PGC and SSC profiles were very similar to each other (R = 0.86), as expected due to their being from the same lineage, but showed significantly less similarity to the MEF RT profile (R = 0.74 for both PGC and SSC) (Figure 2B, C and Supplementary Figure S4B). We identified statistically significant differential RT regions (see Materials and Methods) between SSCs, PGCs and MEFs (Figure 2B, C), referring to the subset of genomic regions in which the RT of two tissues differ. Overall, we found ∼400 Mb and 370 Mb of differential RT between MEFs and SSC or PGC, respectively (Figure 2C). On the other hand, the SSC and the PGC profiles were very similar (Figure 2B, C), with only 14 Mb of differential RT. We confirmed the accuracy of these differential regions by analyzing their chromatin accessibility using published PGC data (44). Indeed, earlier-replicating regions in PGC or SSC were significantly more accessible than regions replicating later in the germ cells (Figure 2D). Differential regions between germline tissues and another somatic tissue (pre-b cells) showed similar results (Supplementary Figure S6).
In order to put the germ cell RT maps in a broader context, PGCs and SSCs were compared to many published RT maps, expanding the work of Hiratani et al. (13). As was previously reported, embryonic tissues RT clustered into early and late embryonic stages (13). The germ cells clustered as a third embryonic cells cluster, distinct from terminally differentiated cells (Figure 2E and F).
Mutation rate and recombination hotspot density correlate most strongly with the RT of germ cells
Although the mechanism(s) responsible for the association between mutation rates and replication timing is still under investigation, it is clear that it stems from differences between early and late replicating regions, either in mutation rates directly or in DNA repair rates (33). As inter-mammalian divergence (defined as the amount of sequence differences between mouse and rat per genomic window) (30) reflects germline mutation rates, we expected it to more strongly correlate with germ cell RT than with somatic cell RT. Indeed, we found stronger correlations between inter-mammalian divergence and PGC or SSC RT than MEF RT (R = −0.63 and −0.65 versus −0.52; Supplementary Figure S7A). To further emphasize this trend, we used published data that divided the mouse genome into two types of regions—those that show similar RT across 28 mouse RT datasets (constitutive RT) and those that show variability between tissues (developmental or switching RT) (13). As expected, in switching RT regions the correlation was much stronger in germ cells than in somatic cells (Figure 3A–C). Further analysis of the differential regions between MEFs and germ cells revealed that Germ-Earlier MEF-Later regions have significantly lower mutation rates than Germ-Later MEF-Earlier regions (Figure 3D), further demonstrating that the mutation rates follow germ cell RT more strongly than somatic cell RT. Analysis using differential regions from pre-b cells versus germ cells showed similar results (Supplementary Figure S8).
Figure 3.

Mutation rate and recombination hotspots correlate better with germ cell RT. The stronger association between inter-mammalian divergence (A–D) and recombination hot spots (E–H) with germ cell RT is shown as RT maps (A, E), box plots in 5 RT bins (B, F), and bar graphs capturing the Spearman correlation coefficients along with confidence intervals (bars) for multiple cell types (C,G) and boxplots (as in Figure 2D) showing the distribution in differential regions (D,H). B, C, E and F were calculated using only the switching RT regions of the genome (13). Similar results were obtained by comparing germ cells to Pre-B cells (Supplementary Figure S8).
Another germ cell related feature is meiotic recombination hotspots (42). In order to analyze its association with germ cell RT, we took advantage of the recently published dataset depicting the genome-wide recombination hotspots using Spo11 pull-down in mouse sperm cells (42). Analyzing the recombination hot spots data (in 1 Mb windows) revealed a stronger correlation with germ cell RT for both PGC and SSC (Figure 3E–H and Supplementary Figure S7B) compared to somatic cell RT. Taken together, these results emphasize the centrality of replication timing in determining germline mutation and recombination rates, and establish a resource for further studies of the influence of replication timing on germline genetic and epigenetic events.
Germline RT is associated with GC content and gene and transposon densities
Having demonstrated that germline RT provides the best proxy, so far, for germline mutation and recombination rates, we turned to search for additional genetic properties that specifically relate to germline RT. Finding such features, would suggest that they originated in the germ cells possibly as a consequence of RT. On the other hand, finding a feature that is associated with the RT in all tissues to the same extent, would suggest that this feature is most probably affecting the RT and thus it has the same effect in all tissues.
We explored four additional genomic features that are known to be associated with RT but for which the causative relationship with RT has been unclear – GC content, and SINE, LINE and gene density. Early replicating regions tend to have higher GC content (2,21). LINEs are known to populate mainly late replicating regions (17), whereas SINEs and genes are known to populate mainly early replicating regions (21,26). We have previously shown that the genomic GC distribution (GC content) depends on RT, in a mechanism by which RT affects the type of mutations that occur at early and late S (24). According to this explanation, we expect to obtain higher correlations to GC content when using germ cell RT data. Indeed, using the same strategy as with mutation rates, we found stronger correlations of GC content with germ cell RT than with somatic cells RT (Figure 4A and Supplementary Figure S9). Interestingly, using the same approach, we found that LINE, SINE and gene density also correlate better with RT in germ cells (Figure 4B–D and Supplementary Figure S9). Similar results were found using pre-B cells instead of MEFs (Supplementary Figure S10).Thus, our findings suggest that it is less likely that either gene or retrotransposon densities affect RT, but rather point to an influence of RT on those features through a germline-related mechanism (see Discussion).
Figure 4.

Cell-type specific association of RT with additional genomic features. RT association with GC content (A), LINE counts (B), SINE counts (C) and gene coverage (D) in 100 kb windows. Left: Bar plots showing the spearman correlation coefficients (along with confidence intervals) with the RT of different cell types, in the switching RT regions of the genome; Right: Box plots (as in Figure 2D) showing the distribution of these features in differential RT regions. Similar results were obtained by comparing germ cells to Pre-B cells (Supplementary Figure S10).
RT directly associates with SINE, gene, mutation and recombination hotspot densities
The aforementioned correlations between germ cell RT and the density of various genetic features do not necessarily imply independent associations between them. We have previously shown that RT has a causative role in determining the genomic GC content (24). Therefore, we wanted to assess the unique contribution of RT to five genomic features independently from the contribution of GC content. To this end, we stratified the RT data according to GC content and analyzed the association between RT and each genomic feature in each bin (Figure 5A and Supplementary Figure S11A). We found that for LINE density, the contribution of RT was small relative to the contribution of GC content. On the other hand, for SINE density, mutation rate and recombination hotspot density, germ cell RT was a major contributor even after accounting for GC content. Gene density showed an intermediate pattern in which RT contributed only in genomic regions of low GC content, and was not important in other parts of the genome.
Figure 5.

Independent association of RT with genomic features. (A) Scatter plot showing the association between RT and GC content and its stratification into four GC content groups; associations between multiple genomic features and RT stratified by GC content, are shown using the same colors as in the scatter plot. (B) Barplots depicting relative contribution of RT and GC content to percent variance explained for LINE, SINE, gene coverage, mutation rate and recombination hot spots. For each predicted feature, the model was created twice beginning with either RT or GC content. The order of the addition of the predictors to the model is from bottom to top. Similar results were obtained with SSC RT data (Supplementary Figure S11).
To corroborate this point further, we built a multiple regression model, which allowed us to see the additional contribution of RT over the contribution of GC content. We simultaneously built two models either starting with GC content or with germ cell RT. These models revealed that for LINE the additional contribution to the percent variance explained (PVE) of RT beyond GC content was very small. On the contrary, when predicting SINE density, gene density, mutation rate and recombination hotspots density, adding RT as a predictor increased the PVE by a factor of 20%, 25%, 34% and 35%, respectively, relative to the PVE from using only GC content (Figure 5B and Supplementary Figure S11B). Further confirmation of this conclusion was obtained by partial correlation analysis (Supplementary Figure S12). Taken together, these results demonstrated the independent association between RT and multiple genomic features, suggesting it may has a causative role in their formation.
DISCUSSION
By improving the RT profiling technique, we were able for the first time, to map the RT of in vivo mouse germline cells. We have profiled both E13.5 PGCs and in vitro-grown SSCs, and found that their RT profiles are similar. Our results add a new dimension to previous efforts to map the RT of multiple mouse developmental stages (13). We found that the two stages of germ cell development clustered together, and to a lesser extent, clustered with other embryonic tissues while remaining distinct from terminally differentiated cells. The similarity between the RT maps of PGCs and embryonic tissues is not surprising since PGCs are taken from an early embryonic stage prior to terminal differentiation. Though SSCs are isolated from young mice and therefore may reflect later developmental stages, our finding of their similarity to embryonic stages most probably stems from their germline origin. Further research is needed to expand our study to additional germ cells models (51–54), which reflect other stages in germ cells development.
It is well established now that RT is associated with both germline and somatic mutations (reviewed in (33)), however, due to a lack of information regarding germ cell RT, previous studies of germline mutations used somatic cell replication profiles as a proxy. By profiling RT in germ cells, we showed that the correlation between mutation rate and RT is stronger than when using somatic cell RT profiles. More generally, obtaining germline-specific RT data is very important for understanding the regional variation in mutation rate (RViMR) along the genome (55–57). It has been shown that RViMR is dependent mainly on RT and transcription activity (27), which both differ between tissues. Using the correct tissue data improves RViMR estimation (58,59) and accordingly, germ cell RT data is especially important for estimation of germline RViMR. Obtaining a correct cancer related RViMR turned out to be crucial for the identification of new cancer-associated genes (27). Similarly, obtaining a reliable germline RViMR is important for understanding the selection forces acting on various genes (60,61), for interpreting the importance of genetic variation and de novo mutations for diseases (62), and for reliably performing inter species alignments (63). Performing similar experiments in human germ cells will be even more informative, since i) there is more data regarding mutations and CNVs in humans than in mice, and ii) getting a better estimate for the local mutation frequency in humans may allow better understanding of disease-related mutations.
We found that the strongest correlations for germline mutation rate and for recombination hotspots density are found with germ cell RT profiles. This suggests that the strength of the correlation is indicative of the tissue of origin of the studied association. Indeed, correlation between RT and an epigenetic feature (like chromatin accessibility) is found to be stronger when both the RT and the chromatin accessibility data are from the same tissue (Supplementary Figure S5). Interestingly, the improvement of the correlation between RT and recombination hot spots in the germ cells was not as strong as the improvement seen with mutation rates. This can be explained by the fact that the actual tissue of origin for recombination hot spots is not the SSC or the PGCs but rather later stages in the germline – when cells enter meiosis. Expanding our study to the meiosis-associated replication may resolve this point.
We found the strongest correlation between RT and GC content in germ cells, supporting our previous finding that the mutation spectrum in genomic late replication domains shapes mammalian GC content (24). Expanding this idea to other genomic features such as SINE, LINE and gene density, revealed that all these features also correlate more strongly with germ cell RT profiles, suggesting that germline tissues are indeed the relevant tissues of origin for these correlations. This finding implies that it is less likely that these features are involved in affecting RT, either directly or indirectly, since in that case we would expect them to influence RT in all tissues similarly. Rather, our findings suggest that the association of these features with RT occurs in the germline, probably through RT’s effect on genome stability (33). Nevertheless, it does not necessarily imply that RT is directly affecting these features, since it can be that other processes, associated with RT, like certain chromatin modifications, chromatin accessibility, or the association of certain proteins with chromatin in germ cells, are the direct effectors. Recently, ChIP-seq experiments were performed on PGCs and SSCs using antibodies against a number of histone modifications, including the H3K27me3 and H3K9me2, that mark closed chromatin (46–48). Multiple regression analysis of these modifications (summed in 100Kb windows) revealed that these marks have small or no contribution to the prediction of all other genomic features over RT (Supplementary Figure S13). Additional germ cell chromatin data, such as ATAC-seq, would be required for further evaluating this point.
By using the germ cell RT data we were able, for the first time, to address the relative contribution of RT and GC content to multiple genomic features. Interestingly, we found an independent contribution of RT to all examined genomic features besides LINE density. It was shown that L1 elements (LINE) are associated with AT-rich, late-replicating regions, whereas Alu elements (SINE) are associated with GC rich, early-replicating regions. Detailed analysis of old and new SINE and LINE elements revealed that both integrate preferably into AT rich regions, but SINEs are preferentially deleted from those regions and thus old SINEs are enriched in high GC, early replicating regions whereas new SINEs are enriched in low GC, late replicating regions (64,65). Our results, showing GC-content independent RT association only with SINEs but not LINEs densities, suggest that RT plays a role in the deletion process rather than in the integration process. This conclusion is consistent with the finding that both point mutations and deletions are more prevalent in late replicating regions (33). We assume that a similar mechanism involving differences in genome stability in early and late regions is involved in determining gene distribution.
Previous work has shown that the strongest correlation between RT and both GC content and retroelement density is obtained when using ectoderm tissue RT profiles (13), but the reason for this phenomenon remained obscure. Although we cannot explain these results, they are consistent with the similarity we found between germ cell RT profiles and ectodermal tissue RT profiles (Supplementary Figure S14). The independent association between mutation rate and RT has been reported before using somatic cells RT data (30). Our new germ cell RT data confirms previous results and further demonstrates the importance of RT in determining germline mutation rates.
The association between recombination hot spots and somatic cells RT was studied in humans and revealed a stronger association in females than in males (32). This study estimated recombination events by analyzing 15000 Icelandic parent-offspring pairs. Using a direct measurement of the locations of the DNA recombination associated double strand breaks (Spo11 oligo mapping) in mouse sperm cells, we were able to show a moderate, yet stronger, correlation between germ cell RT and recombination hotspots density (in 1Mb windows). Interestingly, our results differ from the previous report in two aspects: (i) We found a much stronger correlation with male recombination hotspots than previously reported. (ii) In the current study, we found that RT contributes to recombination hotspots even after controlling for GC content whereas the previous report suggested that the association between RT and recombination strongly depends on GC content. Differences between the studies in (a) the organism studied (mouse versus human); (b) the source of the RT data (germ cell versus somatic cells) and (c) the definition of a recombination hotspot (DSB versus recombination events) may explain this discrepancy.
By improving the RT profiling protocol (see supplementary methods) we were able to produce reliable RT maps from as little as 1000 G1 and S phase cells. This development paves the way for similar experiments in which RT can be determined for other samples with limited numbers of cells, in particular in vivo cell populations. As far as we know, RT profiling of in vivo vertebrate cells was done only in zebrafish (66); this is the first time it has been performed in mammalian cells. This technique is especially relevant in the field of cancer, in which it was shown that using the correct tissue of origin RT can best explain mutation rate (58,59). Currently, there are no RT profiles of primary tumors and the association between RT and mutation rates has been based so far on tissue culture cells. Measuring RT from in vivo tumors may help elucidate the correct mutation rate and aid in understanding the mutational spectrum in a given cancer.
In summary, by optimizing the RT profiling methodology we were able to determine the RT of two types of mouse germ cells. These novel RT profiles allow the identification of the tissue of origin of many genomic features. Moreover, they suggest a fundamental role for RT in determining multiple facets of genomic composition. Further research is needed for understanding the precise mechanisms by which this is achieved.
DATA AVAILABILITY
The data was deposited to GEO - accession number GSE109804. The major scripts were uploaded in GitHub - https://github.com/britnyblu/germline-RT.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Dan Lehmann and Dr Eleonora Medvedev for assistance with the FACS; Dr Idit Shiff, Dr Abed Nasereddin and Alexia Azoulay for help in sequencing; Dr. Sharon Schlesinger and Dr. Sheera Adar for critical reading of the manuscript; and Prof. George Iliakis for the MEF cells.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Israel Science Foundation [184/16]; ISF-NSFC joint research program [2555/16]; ERC Starting Grants [281306]; Israel Cancer Research Fund (ICRF). Funding for open access charge: The Israel Science Foundation grants.
Conflict of interest statement. None declared.
REFERENCES
- 1. Hiratani I., Takebayashi S., Lu J., Gilbert D.M.. Replication timing and transcriptional control: beyond cause and effect–part II. Curr. Opin. Genet. Dev. 2009; 19:142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Farkash-Amar S., Lipson D., Polten A., Goren A., Helmstetter C., Yakhini Z., Simon I.. Global organization of replication time zones of the mouse genome. Genome Res. 2008; 18:1562–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Goren A., Cedar H.. Replicating by the clock. Nat. Rev. Mol. Cell Biol. 2003; 4:25–32. [DOI] [PubMed] [Google Scholar]
- 4. MacAlpine D.M., Rodriguez H.K., Bell S.P.. Coordination of replication and transcription along a Drosophila chromosome. Genes Dev. 2004; 18:3094–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Karnani N., Taylor C., Malhotra A., Dutta A.. Pan-S replication patterns and chromosomal domains defined by genome-tiling arrays of ENCODE genomic areas. Genome Res. 2007; 17:865–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Desprat R., Thierry-Mieg D., Lailler N., Lajugie J., Schildkraut C., Thierry-Mieg J., Bouhassira E.E.. Predictable dynamic program of timing of DNA replication in human cells. Genome Res. 2009; 19:2288–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Norio P., Kosiyatrakul S., Yang Q., Guan Z., Brown N.M., Thomas S., Riblet R., Schildkraut C.L.. Progressive activation of DNA replication initiation in large domains of the immunoglobulin heavy chain locus during B cell development. Mol. Cell. 2005; 20:575–587. [DOI] [PubMed] [Google Scholar]
- 8. Schwaiger M., Schubeler D.. A question of timing: emerging links between transcription and replication. Curr. Opin. Genet. Dev. 2006; 16:177–183. [DOI] [PubMed] [Google Scholar]
- 9. Schwaiger M., Stadler M.B., Bell O., Kohler H., Oakeley E.J., Schubeler D.. Chromatin state marks cell-type- and gender-specific replication of the Drosophila genome. Genes Dev. 2009; 23:589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pope B.D., Chandra T., Buckley Q., Hoare M., Ryba T., Wiseman F.K., Kuta A., Wilson M.D., Odom D.T., Gilbert D.M.. Replication-timing boundaries facilitate cell-type and species-specific regulation of a rearranged human chromosome in mouse. Hum. Mol. Genet. 2012; 21:4162–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yaffe E., Farkash-Amar S., Polten A., Yakhini Z., Tanay A., Simon I.. Comparative analysis of DNA replication timing reveals conserved large-scale chromosomal architecture. PLos Genet. 2010; 6:e1001011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ryba T., Hiratani I., Lu J., Itoh M., Kulik M., Zhang J., Schulz T.C., Robins A.J., Dalton S., Gilbert D.M.. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010; 20:761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hiratani I., Ryba T., Itoh M., Rathjen J., Kulik M., Papp B., Fussner E., Bazett-Jones D.P., Plath K., Dalton S. et al. Genome-wide dynamics of replication timing revealed by in vitro models of mouse embryogenesis. Genome Res. 2010; 20:155–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rivera-Mulia J.C., Gilbert D.M.. Replication timing and transcriptional control: beyond cause and effect-part III. Curr. Opin. Cell Biol. 2016; 40:168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Braunstein J.D., Schulze D., DelGiudice T., Furst A., Schildkraut C.L.. The temporal order of replication of murine immunoglobulin heavy chain constant region sequences corresponds to their linear order in the genome. Nucleic Acids Res. 1982; 10:6887–6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gilbert D.M. Temporal order of replication of Xenopus laevis 5S ribosomal RNA genes in somatic cells. Proc. Natl. Acad. Sci. U.S.A. 1986; 83:2924–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hiratani I., Ryba T., Itoh M., Yokochi T., Schwaiger M., Chang C.W., Lyou Y., Townes T.M., Schubeler D., Gilbert D.M.. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008; 6:e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cohen S.M., Cobb E.R., Cordeiro-Stone M., Kaufman D.G.. Identification of chromosomal bands replicating early in the S phase of normal human fibroblasts. Exp. Cell Res. 1998; 245:321–329. [DOI] [PubMed] [Google Scholar]
- 19. Farkash-Amar S., David Y., Polten A., Hezroni H., Eldar Y.C., Meshorer E., Yakhini Z., Simon I.. Systematic determination of replication activity type highlights interconnections between replication, chromatin structure and nuclear localization. PLoS One. 2012; 7:e48986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Farkash-Amar S., Simon I.. Genome-wide analysis of the replication program in mammals. Chromosome Res. 2010; 18:115–125. [DOI] [PubMed] [Google Scholar]
- 21. Woodfine K., Fiegler H., Beare D.M., Collins J.E., McCann O.T., Young B.D., Debernardi S., Mott R., Dunham I., Carter N.P.. Replication timing of the human genome. Hum. Mol. Genet. 2004; 13:191–202. [DOI] [PubMed] [Google Scholar]
- 22. Dileep V., Ay F., Sima J., Vera D.L., Noble W.S., Gilbert D.M.. Topologically associating domains and their long-range contacts are established during early G1 coincident with the establishment of the replication-timing program. Genome Res. 2015; 25:1104–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pope B.D., Ryba T., Dileep V., Yue F., Wu W., Denas O., Vera D.L., Wang Y., Hansen R.S., Canfield T.K. et al. Topologically associating domains are stable units of replication-timing regulation. Nature. 2014; 515:402–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kenigsberg E., Yehuda Y., Marjavaara L., Keszthelyi A., Chabes A., Tanay A., Simon I.. The mutation spectrum in genomic late replication domains shapes mammalian GC content. Nucleic Acids Res. 2016; 44:4222–4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. White E.J., Emanuelsson O., Scalzo D., Royce T., Kosak S., Oakeley E.J., Weissman S., Gerstein M., Groudine M., Snyder M. et al. DNA replication-timing analysis of human chromosome 22 at high resolution and different developmental states. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:17771–17776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Woodfine K., Beare D.M., Ichimura K., Debernardi S., Mungall A.J., Fiegler H., Collins V.P., Carter N.P., Dunham I.. Replication timing of human chromosome 6. Cell Cycle. 2005; 4:172–176. [DOI] [PubMed] [Google Scholar]
- 27. Lawrence M.S., Stojanov P., Polak P., Kryukov G.V., Cibulskis K., Sivachenko A., Carter S.L., Stewart C., Mermel C.H., Roberts S.A. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013; 499:214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Donley N., Thayer M.J.. DNA replication timing, genome stability and cancer: late and/or delayed DNA replication timing is associated with increased genomic instability. Semin. Cancer Biol. 2013; 23:80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stamatoyannopoulos J.A., Adzhubei I., Thurman R.E., Kryukov G.V., Mirkin S.M., Sunyaev S.R.. Human mutation rate associated with DNA replication timing. Nat. Genet. 2009; 41:393–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen C.L., Rappailles A., Duquenne L., Huvet M., Guilbaud G., Farinelli L., Audit B., d’Aubenton-Carafa Y., Arneodo A., Hyrien O. et al. Impact of replication timing on non-CpG and CpG substitution rates in mammalian genomes. Genome Res. 2010; 20:447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cui P., Ding F., Lin Q., Zhang L., Li A., Zhang Z., Hu S., Yu J.. Distinct contributions of replication and transcription to mutation rate variation of human genomes. Genomics Proteomics Bioinformatics. 2012; 10:4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Koren A., Polak P., Nemesh J., Michaelson J.J., Sebat J., Sunyaev S.R., McCarroll S.A.. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am. J. Hum. Genet. 2012; 91:1033–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Blumenfeld B., Ben-Zimra M., Simon I.. Perturbations in the replication program contribute to genomic instability in cancer. Int. J. Mol. Sci. 2017; 18:E1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Drost J.B., Lee W.R.. Biological basis of germline mutation: comparisons of spontaneous germline mutation rates among drosophila, mouse, and human. Environ. Mol. Mutagen. 1995; 25:48–64. [DOI] [PubMed] [Google Scholar]
- 35. Gilbert D.M. Evaluating genome-scale approaches to eukaryotic DNA replication. Nat. Rev. Genet. 2010; 11:673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dileep V., Didier R., Gilbert D.M.. Genome-wide analysis of replication timing in mammalian cells: troubleshooting problems encountered when comparing different cell types. Methods. 2012; 57:165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yabuta Y., Kurimoto K., Ohinata Y., Seki Y., Saitou M.. Gene expression dynamics during germline specification in mice identified by quantitative single-cell gene expression profiling. Biol. Reprod. 2006; 75:705–716. [DOI] [PubMed] [Google Scholar]
- 38. Kubota H., Brinster R.L.. Culture of rodent spermatogonial stem cells, male germline stem cells of the postnatal animal. Methods Cell Biol. 2008; 86:59–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kanatsu-Shinohara M., Shinohara T.. Germline modification using mouse spermatogonial stem cells. Methods Enzymol. 2010; 477:17–36. [DOI] [PubMed] [Google Scholar]
- 40. Yehuda Y., Blumenfeld B., Lehmann D., Simon I.. Genome-wide determination of mammalian replication timing by DNA content measurement. J. Vis. Exp. 2017; 119:e55157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Blecher-Gonen R., Barnett-Itzhaki Z., Jaitin D., Amann-Zalcenstein D., Lara-Astiaso D., Amit I.. High-throughput chromatin immunoprecipitation for genome-wide mapping of in vivo protein-DNA interactions and epigenomic states. Nat. Protoc. 2013; 8:539–554. [DOI] [PubMed] [Google Scholar]
- 42. Lange J., Yamada S., Tischfield S.E., Pan J., Kim S., Zhu X., Socci N.D., Jasin M., Keeney S.. The landscape of mouse meiotic Double-Strand break formation, processing, and repair. Cell. 2016; 167:695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yue F., Cheng Y., Breschi A., Vierstra J., Wu W., Ryba T., Sandstrom R., Ma Z., Davis C., Pope B.D. et al. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014; 515:355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guo H., Hu B., Yan L., Yong J., Wu Y., Gao Y., Guo F., Hou Y., Fan X., Dong J. et al. DNA methylation and chromatin accessibility profiling of mouse and human fetal germ cells. Cell Res. 2017; 27:165–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li D., Liu J., Yang X., Zhou C., Guo J., Wu C., Qin Y., Guo L., He J., Yu S. et al. Chromatin accessibility dynamics during iPSC reprogramming. Cell Stem Cell. 2017; 21:819–833. [DOI] [PubMed] [Google Scholar]
- 46. Kurimoto K., Yabuta Y., Hayashi K., Ohta H., Kiyonari H., Mitani T., Moritoki Y., Kohri K., Kimura H., Yamamoto T. et al. Quantitative dynamics of chromatin remodeling during germ cell specification from mouse embryonic stem cells. Cell Stem Cell. 2015; 16:517–532. [DOI] [PubMed] [Google Scholar]
- 47. Ng J.H., Kumar V., Muratani M., Kraus P., Yeo J.C., Yaw L.P., Xue K., Lufkin T., Prabhakar S., Ng H.H.. In vivo epigenomic profiling of germ cells reveals germ cell molecular signatures. Dev. Cell. 2013; 24:324–333. [DOI] [PubMed] [Google Scholar]
- 48. Liu Y., Giannopoulou E.G., Wen D., Falciatori I., Elemento O., Allis C.D., Rafii S., Seandel M.. Epigenetic profiles signify cell fate plasticity in unipotent spermatogonial stem and progenitor cells. Nat. Commun. 2016; 7:11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jo J., Hwang S., Kim H.J., Hong S., Lee J.E., Lee S.G., Baek A., Han H., Lee J.I., Lee I. et al. An integrated systems biology approach identifies positive cofactor 4 as a factor that increases reprogramming efficiency. Nucleic Acids Res. 2016; 44:1203–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Edgar R., Domrachev M., Lash A.E.. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002; 30:207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hikabe O., Hamazaki N., Nagamatsu G., Obata Y., Hirao Y., Hamada N., Shimamoto S., Imamura T., Nakashima K., Saitou M. et al. Reconstitution in vitro of the entire cycle of the mouse female germ line. Nature. 2016; 539:299–303. [DOI] [PubMed] [Google Scholar]
- 52. Seandel M., James D., Shmelkov S.V., Falciatori I., Kim J., Chavala S., Scherr D.S., Zhang F., Torres R., Gale N.W. et al. Generation of functional multipotent adult stem cells from GPR125+ germline progenitors. Nature. 2007; 449:346–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Geijsen N., Horoschak M., Kim K., Gribnau J., Eggan K., Daley G.Q.. Derivation of embryonic germ cells and male gametes from embryonic stem cells. Nature. 2004; 427:148–154. [DOI] [PubMed] [Google Scholar]
- 54. Mitsunaga S., Odajima J., Yawata S., Shioda K., Owa C., Isselbacher K.J., Hanna J.H., Shioda T.. Relevance of iPSC-derived human PGC-like cells at the surface of embryoid bodies to prechemotaxis migrating PGCs. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E9913–E9922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hardison R.C., Roskin K.M., Yang S., Diekhans M., Kent W.J., Weber R., Elnitski L., Li J., O’Connor M., Kolbe D. et al. Covariation in frequencies of substitution, deletion, transposition, and recombination during eutherian evolution. Genome Res. 2003; 13:13–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hodgkinson A., Eyre-Walker A.. Variation in the mutation rate across mammalian genomes. Nat. Rev. Genet. 2011; 12:756–766. [DOI] [PubMed] [Google Scholar]
- 57. Makova K.D., Hardison R.C.. The effects of chromatin organization on variation in mutation rates in the genome. Nat. Rev. Genet. 2015; 16:213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Polak P., Karlic R., Koren A., Thurman R., Sandstrom R., Lawrence M., Reynolds A., Rynes E., Vlahovicek K., Stamatoyannopoulos J.A. et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature. 2015; 518:360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Supek F., Lehner B.. Differential DNA mismatch repair underlies mutation rate variation across the human genome. Nature. 2015; 521:81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Martincorena I., Luscombe N.M.. Non-random mutation: the evolution of targeted hypermutation and hypomutation. Bioessays. 2013; 35:123–130. [DOI] [PubMed] [Google Scholar]
- 61. Caporale L.H. Mutation is modulated: implications for evolution. Bioessays. 2000; 22:388–395. [DOI] [PubMed] [Google Scholar]
- 62. Samocha K.E., Robinson E.B., Sanders S.J., Stevens C., Sabo A., McGrath L.M., Kosmicki J.A., Rehnstrom K., Mallick S., Kirby A. et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 2014; 46:944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li J., Miller W.. Significance of interspecies matches when evolutionary rate varies. J. Comput. Biol. 2003; 10:537–554. [DOI] [PubMed] [Google Scholar]
- 64. Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W. et al. Initial sequencing and analysis of the human genome. Nature. 2001; 409:860–921. [DOI] [PubMed] [Google Scholar]
- 65. Deininger P.L., Batzer M.A.. Mammalian retroelements. Genome Res. 2002; 12:1455–1465. [DOI] [PubMed] [Google Scholar]
- 66. Siefert J.C., Georgescu C., Wren J.D., Koren A., Sansam C.L.. DNA replication timing during development anticipates transcriptional programs and parallels enhancer activation. Genome Res. 2017; 27:1406–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data was deposited to GEO - accession number GSE109804. The major scripts were uploaded in GitHub - https://github.com/britnyblu/germline-RT.