


Abstract
Advances in stem cell engineering, gene therapy and molecular medicine often involve genome engineering at a cellular level. However, functionally large or multi transgene cassette insertion into the human genome still remains a challenge. Current practices such as random transgene integration or targeted endonuclease-based genome editing are suboptimal and might pose safety concerns. Taking this into consideration, we previously developed a transgenesis tool derived from phage λ integrase (Int) that precisely recombines large plasmid DNA into an endogenous sequence found in human Long INterspersed Elements-1 (LINE-1). Despite this advancement, biosafety concerns associated with bacterial components of plasmids, enhanced uptake and efficient transgene expression remained problematic. We therefore further improved and herein report a more superior Int-based transgenesis tool. This novel Int platform allows efficient and easy derivation of sufficient amounts of seamless supercoiled transgene vectors from conventional plasmids via intramolecular recombination as well as subsequent intermolecular site-specific genome integration into LINE-1. Furthermore, we identified certain LINE-1 as preferred insertion sites for Int-mediated seamless vector transgenesis, and showed that targeted anti-CD19 chimeric antigen receptor gene integration achieves high-level sustained transgene expression in human embryonic stem cell clones for potential downstream therapeutic applications.
INTRODUCTION
Precise and safe genome engineering is critical for the development of innovative cell-based products and has an impact on biotechnology, gene/cell therapy, regenerative medicine and synthetic biology (1,2). However, with currently available transgenesis platforms employing random genome integration, e.g. viral-/transposon-based, or sequence-specific endonuclease-based tools such as CRISPR, TALENs or ZFNs (3–7), it remains a difficult task to routinely achieve functional insertion of larger DNA payloads. Furthermore, the efficiency of genome targeting by these methods is inversely proportional to insert size. For insertion strategies that involve linear donor DNA, and in particular those comprising > 5kb multi-gene constructs, illegitimate genome integration and/or incomplete integrants are frequent byproducts (8–10). This is further compounded by safety concerns, such as insertional mutagenesis, transgene silencing, off-targets and uncontrolled genomic double-strand breaks (DSBs) (11–15). Hence, there is a continuous strong demand for improved methods for non-viral, sequence-specific genome insertion in particular for large transgene cassettes.
With plasmid-based transgenesis, co-inserted prokaryotic sequences can lead to interference with proper transgene expression, elicit immune responses, and raise biosafety concerns (16–21). Seamless vector transgenesis therefore would serve as a progressive and tractable approach for various downstream applications. Ideally, coupling of large seamless vector production with specific genome targeting could lead to more cost effective, safer, long-lasting expression of multi-transgenes or large therapeutic gene cassettes, preferably in combination with targeted recombination at ‘safe harbour sites’ for clinical applications (9,22).
To this end, designer endonucleases, such as ZFNs, TALENs and CRISPR/Cas9 enable targeted transgene insertions (2,15,23–26), but the endonuclease-centric mechanistic principle of transgene insertion relies on the generation of genomic DSBs at selected endogenous target sequences, followed by insertion of donor DNA templates through either host cell-mediated homology-directed DNA repair synthesis or non-homologous DNA end-joining (3,8,23,27). Hence, this approach lacks control over the insertion reaction and can lead to the introduction of indels at non-targeted alleles, DSBs at off-target locations, DNA damage signalling, chromosomal aberrations and genotoxic effects (28–31).
Members of the integrase and resolvase/invertase site-specific recombinase families, which do not rely on host cell machineries to achieve transgenesis, are attractive alternatives to designer endonucleases for use in higher eukaryotes (32–36). However, only a few recombinase systems demonstrated potential for transgene insertion into pre-existing endogenous human genome sequences. In this context, the bacteriophage phiC31 integrase is perhaps the most prominent example, which can recombine circular target vectors into so-called pseudo-attachment (pseudo-att) sites already present in the human genome (37,38). There exist, however, several problems associated with phiC31 integrase-catalyzed recombination at human genomic pseudo-att sites, which include generation of indels flanking the inserted transgenic DNA, chromosomal aberrations triggered by recombinase-induced DSBs at multiple pseudo-att sites, low targeting efficiency and debatable true site-specific recombination events (39–41).
We previously reported a novel transgenesis tool for human genome engineering derived from the integration system of phage λ (42). A highly active, co-factor independent λ integrase (Int) variant (43,44) now presents itself as a simple transgenesis tool for sequence-specific functional transgene cassette insertion into the human genome (42). The minimal 18bp-encompassing endogenous human target sequence resides within Long INterspersed Elements-1 (LINE-1) and is present in 933 copies scattered throughout the human genome. This transgenesis system, which so far employed bacterial plasmids as target vectors, has been validated in various cell lines including human embryonic stem cells (hESCs). At least some of the identified LINE-1 integration sites were considered ’safe harbour sites’ that enabled sustained and regulated transgene expression in hESCs (42).
In this study, we present an advanced and comprehensive λ-Int platform that utilizes Int variants for both in vitro and successive intracellular ex vivo recombination reactions. Specifically, seamless target vectors devoid of bacterial sequences are first generated in vitro by intramolecular recombination and are subsequently integrated into LINE-1 sequences inside various human cell types through Int-mediated intermolecular recombination. Using this platform, we identified several ‘hotspot’ LINE-1 targets. Furthermore, we achieved high level expression of therapeutic anti-CD19 chimeric antigen receptor (CAR) in hESCs. Hence, our novel platform technology serves as a versatile basis for future biotechnological and therapeutic applications.
MATERIALS AND METHODS
Cell lines
This study used human embryonic stem cell (hESC) line ‘Genea 047’ (Genea Biocells, Sydney, Australia) and cancer cell line of HT1080 (fibrosarcoma).
Plasmids
Standard molecular cloning techniques were employed to generate plasmids used in this work. High fidelity Pfu polymerase (Thermo Fisher Scientific) was used for PCR amplifications and E. coli DH5α was used for plasmid DNA amplifications.
pattP4X-PGKssEGFP-attH4X was generated by PCR amplification of the EGFP gene from pCMVssEGFP (in-house), using the primers PstI_EGFP and NotI_attH4X_NheI_bpa (includes attH4X sequence) (all the primer sequences are listed in Supplementary Table S1), followed by cloning into pattP4X-PGKssPuro between PstI and NotI sites, thus replacing the Puro cassette with the EGFP and attH4X sequence.
pattP4X-PGKssPuro-attH4X was generated by PCR amplification of the Puro and polyA cassette from pattP4X-PGKssPuro, using the primers Puro_fwd_PstI and Puro_rev_XbaI, followed by cloning into pattP4X-PGKssEGFP-attH4X between PstI and XbaI sites, thus replacing the EGFP with the Puro cassette.
pattP4X-PGKssCAR-attH4X was generated by PCR amplification of the CAR and poly A cassette from pVAX1-αCD19BBZ (provided by Prof. Dario Campana), using the primers CAR_fwd_NcoI and CAR_rev_XbaI, followed by cloning into pattP4X-PGKss EGFP-attH4X between NcoI and XbaI sites, thus replacing the EGFP with the CAR cassette.
pattP4X-PGKssCAR-EF-GFP-attH4X was generated by PCR amplification of the EF-EFGP cassette from pEF1-EGFP (in-house), using the primers HR_NheI_EF fwd and HR_NheI_Bgh rev that add homologous arms to PCR product. PCR products were cloned into pattP4X-PGKssCAR-attH4X between NheI sites by using In-Fusion HD cloning kit (Takara Bio USA), thus adding EF-EGFP cassette downstream of PGKssCAR cassette.
pattP4X-PGKssCAR-EF-Puro-attH4X was generated by cleaving the GFP cassette from pattP4X-PGKssCAR-EF-GFP-attH4X with the enzymes BamHI and EcoRI and replacing it with PCR amplified puromycin cassette using In-Fusion HD cloning kit (Takara Bio USA). Puromycin cassette was amplified from pattP4X-PGKssPuro-attH4X using primers Puro fwd_BamHI_HR and Puro rev_EcoRI_HR.
Cell culture
HT1080 were cultured in Dulbecco's Modified Eagle Medium (DMEM) growth medium supplemented with 10% FBS, 1% l-glutamine and 100 U/ml of Penicillin and Streptomycin each (Gibco, Life technologies) at 37°C under 5% CO2 in humidified condition. For selection of puromycin-resistant recombinants, puromycin (Gibco, Life technologies) was added in the growth medium (1μg/ml). Trypsin-EDTA (Gibco, Life technologies) was used for detaching the adherent cells for passaging.
Human Embryonic Stem Cells (GENEA 047) were cultured at 37°C under 5% CO2 and 5% O2 on Collagen I coated cell culture dishes (Biocoat, Corning) in Genea M2 Medium, (Genea Biocells, Sydney, Australia), supplemented with Penicillin and Streptomycin at 25 U/ml each (Gibco, Life technologies). For selection of recombinants and maintenance of targeted clones, Puromycin (300ng/ml) (Gibco, Life technologies) was included in the growth medium. For passaging or preparing cell suspension for reverse transfections, adherent hESCs were rinsed with 1× PBS, detached by incubating at 37°C for 3 min with passaging solution (Genea Biocells) (with a volume of 100μl per well of a six-well plate or 1 ml per 10 cm dish), dislodging cells by tapping and re-suspending the cells with at least 3× volume of Neutralization solution (Genea Biocells). After counting the cells in a haemocytometer (Neubauer), they were pelleted by centrifuging at 300 × g for 4 min and resuspended in Genea M2 Medium to the required cell density and added drop-wise to Collagen I-coated dishes.
Transfections
For transfections in HT1080, 3 × 105 or 3 × 106 cells were seeded per well of six-well plate (IWAKI, Japan) or per 10 cm tissue culture dishes (TPP, Switzerland), respectively, in DMEM growth medium a day before transfection to obtain 70–90% confluence at the time of transfection. Transfections were carried out by employing Lipofectamine 2000 (Invitrogen, Life technologies) with DNA to Lipofectamine2000 ratio of 1 μg:3 μl. For every transfection per well, DNA and Lipofectamine 2000 were incubated separately in 100 ul of Opti-MEM medium (Life Technologies). The complexes were then prepared by mixing DNA and Lipofectamine 2000 reagent and incubating for 20 min at room temperature. The transfection mix was added drop wise onto the cells (under DMEM growth medium without antibiotics) and transfection was allowed to proceed for 4–6 h before complexes were removed by fresh DMEM medium.
For transfections in hESCs, FuGENE HD Transfection reagent (Promega) was used in a reverse transfection protocol. DNA to FuGENE ratio of 1μg:3μl was used. Transfection mixes were prepared by first diluting DNA in 100 μl of Opti-MEM and 5 min later the FuGENE reagent was added to the DNA dilution, mixed and incubated for 15 min at room temperature for the complexes to form. During the incubation period, hESCs were harvested (as described above) and resuspended in Genea M2 Medium (without antibiotics). The transfection complexes were added drop-wise to Collagen 1-coated plates and incubated at culturing conditions for 5 min after which the harvested cells were gently pipetted to the dishes at 5 × 105 cells per well of six-well plate and 5 × 106 cells per 10cm dish. Transfections were kept overnight under standard culture conditions for hESCs, and media containing transfection complex was replaced with fresh M2 media.
Antibiotic selection and screening for targeted cell clones
Forty eight hours post-transfection, selection with the respective antibiotic in growth medium at the concentrations indicated above was initiated. Selection medium was replaced once in 2 days until colonies expanded to ∼0.3–0.4 cm in diameter. At this stage, the colonies were picked by carefully scraping patches of cells with a pipette tip and transferred to 96-well plates for clonal expansion. The clones were sequentially expanded from 96 wells to 24 wells and subsequently in six-well plates. Genomic DNA was extracted using DNeasy Blood & Tissue Kit (Qiagen, GmbH) as per manufacturer's protocol.
Identification of recombination events by PCR screening
PCR was performed using GoTaq Flexi DNA polymerase (Promega) to amplify both the junctions using primers listed in the figure descriptions and 500 ng of genomic DNA from each recombinant clone or parental cells as template in 50 μl reactions. The thermal cycling parameters used for PCRs were as follows: initial denaturation at 95°C for 5 min, 35 cycles of denaturation at 95°C for 1 min, annealing at 56°C for 30 s and extension at 72°C for 1 min, and a final step of 72°C for 5 min. The PCR samples were analyzed by electrophoresis in 1% agarose (Seakem Agarose, Lonza, USA) gels in 1× TBE (Tris-boric acid-EDTA buffer) containing 0.5 μg/ml ethidium bromide and PCR-amplified products were compared with DNA standard markers and digitally documented under UV illumination (Quantum Vilber Lourmat, Germany). PCR-amplified products were analyzed by sequencing.
Purification of Int-h/218
Plasmid pINT (provided by Farid John Ghadessy) coding for C-terminally histidine-taged Int-h/218 (44) was transformed in Escherichia coli Rosetta (DE3) pLysS cells and positive clones were screened using LB plus antibiotics (250 μg/ml ampicillin and 34 μg/ml chloramphenicol) plates. Overnight culture of Rosetta (DE3) pLysS cells overexpressing Int-h/218 was sub-cultured in 200 ml of LB containing ampicillin (250 μg/ml) and chloramphenicol (34 μg/ml) and incubated at 37°C until optical density reached 0.6 at 650 nm. At this point, the culture was induced with 0.5 mM IPTG for Int-h/218 overexpression and the induced culture was grown for 6 h at 30°C (44). The cell pellet was collected and sonicated in Lysis Buffer (50 mM Tris pH 8.0, 2 mM EDTA pH 8.0, 1 M KCl, 3 mM PMSF, 5 mM DTT, 10 mM β-mercarptoethanol, 1× protease inhibitor) (45). After removing cell debris, the cell extract was mixed with Ni-NTA slurry at 4°C overnight. The Ni-NTA slurry was loaded on the column, and the cell extract was collected as a flow through. The slurry was then washed twice with Wash Buffer (50 mM Tris pH 8.0, 2 mM EDTA pH 8.0, 1 M KCl, 3 mM PMSF, 5 mM DTT, 10 mM β-mercarptoethanol, 20 mM Imidazole, 1× protease inhibitor). The protein was first eluted in Elution Buffer 1 (50 mM Tris pH 8.0, 2 mM EDTA pH 8.0, 1 M KCl, 3 mM PMSF, 5 mM DTT, 10 mM β-mercarptoethanol, 50 mM Imidazole, 1× protease inhibitor) followed by Elution Buffer 2 (50 mM Tris pH 8.0, 2 mM EDTA pH 8.0, 1 M KCl, 3 mM PMSF, 5 mM DTT, 10 mM β-mercarptoethanol, 100 mM Imidazole, 1× protease inhibitor). Eluted fractions were screened for protein, and the appropriate fractions were desalted using desalting prepacked gravity flow columns (BioRad) in Desalting Buffer (50 mM Tris pH 8.0, 2 mM EDTA pH 8.0, 1 M KCl, 3 mM PMSF, 5 mM DTT, 10 mM β-mercarptoethanol, 10% glycerol, 1× protease inhibitor). The desalted fractions were screened for protein and appropriate fractions were tested for in vitro recombination activity.
In vitro recombination using Int-h/218
The in vitro recombination was carried out in a reaction mixture comprised of TE buffer, 600 ng of supercoiled substrate plasmid, 17 ng/μl of purified single chain IHF (46), 200 mM KCl and 15 ng/μl of partially purified Int-h/218. The reaction mixture was incubated at 37°C for 2 h, and DNA was purified using PCR Purification Kit (Qiagen, GmbH). The in vitro recombination leads to formation of two catenane rings. One ring is digested with an appropriate restriction enzyme(s) that will cleave in the bacterial backbone and leaves the supercoiled seamless vector intact. The entire reaction mixture was separated on 1% agarose gel containing 1μg/ml ethidium bromide at 40 V overnight. The seamless vector being covalently closed migrates ahead of the linearized bacterial part of plasmid. The seamless vectors were purified from agarose gels using the QIAquick Gel Extraction Kit (Qiagen, GmbH).
Insertion mapping
The first step of insertion mapping involves using a single biotinylated primer that binds in the known sequence of the inserted vector. The primer will extend, amplify and generate many molecules of the biotinylated ssDNA including the vector sequence and its point of integration into the genome. This ssDNA PCR was performed on genomic DNA from parental HT1080 cells, bulk cultures obtained from co-transfections of seamless vector attL4X-PGKssPuro with pCMVssIna and bulk cultures obtained from co-transfections of seamless vector attL4X-PGKssPuro with pCMVssInt-C3CNLS. Biotinylated primer Puro Fwd 127 was used to generate the ssDNA extending from puro cassette to the junction of insertion of vector into the human genome (Figure 3A). PCR was performed using Bioline Taq polymerase with Tris-based buffer (Vivantis) and the reaction conditions include 95°C, 120 s; 95°C, 5 s; 50°C, 30 s, 72°C, 120 s; 50 cycles.
Figure 3.

Seamless vector genomic insertion site mapping via deep sequencing. (A) An illustration depicting the PCR strategy for mapping insertion sites of seamless vector attL4X-PGKssPuro in human HT1080 cells. This includes generation of targeted ssDNA using a biotinylated primer complementary to the inserted vector sequence (step 1), capture of the ssDNA with streptavidin beads, ligation of adenylated oligonucleotide (adaptor) and PCR amplification and sequencing analysis (steps 2–6). Positions of the relevant primers used to map attH4X (right junction) are indicated in step 5. (B) PCR analysis for mapping insertion sites from HT1080 bulk cell culture. PCR with the primers bpa1 and cs_attH4X_R1 (see step 5 in Figure 3A), using bulk genomic DNA from co-transfections of 100ng of attL4X-PGKssPuro and pCMVssInt-C3CNLS resulted in a specific band (highlighted), that was not observed with bulk genomic DNA from control co-transfections of 100 ng of attL4X-PGKssPuro and pCMVssIna. M1, 1 kb ladder; HT, genomic DNA from parental HT1080 cells; INA_100: bulk genomic DNA from co-transfection of 100ng of attL4X-PGKsspuro and 1 μg Inactive Int; C3_100: bulk genomic DNA from co-transfection of 100 ng of attL4X-PGKsspuro and 1 μg Int-C3. W: no DNA template control.
The second step includes capture of ssDNA onto streptavidin dynabeads (M-280 Streptavidin, Thermo Fisher Scientific), which includes washing of dynabeads with 1× PCR buffer (Tris-based) followed by incubation of PCR product generated in the first step with dynabeads for 3 h on a roller at room temperature. A phosphorylated primer (pETF2-PH) was used as an adaptor and for 5′-adenylation (NEB E2610S). The ligation of the 5′ adenylated oligo with captured ssDNA was catalyzed by 5′ AppDNA ligase (NEB) at 65°C for an hour. Semi-nested PCR was performed on beads with forward primer Puro Fwd 128 and reverse primer petF2RC (Figure 3A) using Taq Polymerase (Bioline) and non-Tris based buffer (Bioline). PCR reaction conditions were 95°C, 120 s; 95°C, 5 s; 55°C, 30 s, 72°C, 120 s; 34 cycles. Further, nested PCR was carried with forward primer bpa1 and cs_attH4X_R1 (Figure 3A), and PCR products were analyzed on 1% agarose gels in 0.5× TBE buffer.
Detection of anti-CD19 CAR expression
hESCs were stained with biotin-conjugated AffiniPure F(ab')2 Fragment Goat-anti-Mouse IgG (Jackson Immunoresearch, West Grove, PA, USA) that reacted with the scFv portion of anti-CD19, followed by streptavidin conjugated to Allophycocyanin (APC; Jackson Immunoresearch, West Grove, PA, USA). For detection of anti-CD19 CAR expression on hESCs, 0.25 × 106 cells were harvested and washed twice with PBSA (0.1% BSA; 0.1% sodium azide in 1× PBS). This was followed by blocking with 2 μl of Normal Rabbit Serum (Thermo Fischer Scientific) and staining with biotin-conjugated AffiniPure F(ab')2 Fragment Goat-anti-Mouse IgG at room temperature for 10 min in the dark. Cells were washed with PBSA twice and incubated with streptavidin conjugated to APC at room temperature for 10 min in the dark. After washing cells twice with PBSA, the cells were fixed with 300 μl of 0.5% formaldehyde. APC-stained hESCs were detected with BD LSRFortessa™ X-20 (Becton Dickson, USA).
Flow cytometry
BD LSRFortessa™ X-20 (Becton Dickson, USA) was used to analyze and quantify anti-CD19 CAR+ cells. hESCs were harvested, centrifuged and suspended in M2 media. Dot plot of side scatter (SSC) versus forward scatter (FSC) was used to gate live cells in order to separate them from aggregated and dead cells. For gated cells, a histogram plot of APC (Allophycocyanin) was constructed to identify anti-CD19 CAR+ cells. The data was analyzed using FlowJo software and represented as an overlay histogram of stained non-transfected control population onto the stained transfected population that clearly showed the percentage of anti-CD19 CAR expressing population.
Western blotting
Cell lysates were prepared by re-suspending the cells in RIPA buffer (150 mM sodium chloride, 1.0% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris, pH 8.0) containing Protease Inhibitor Cocktail (Roche), followed by incubation on ice for 20 mins. The lysates were sonicated on ice (5 W, 10 × 3 s), and insoluble components were removed by centrifugation at 12 000 × g for 15 min. The supernatant was collected and protein concentrations were determined with Bio-Rad Protein Assay Dye Concentrate. Proteins were separated by SDS PAGE gels (10–15% acrylamide) and then transferred onto PVDF membranes with 0.2 μm pore size (Bio-Rad). Nonspecific binding was blocked by 4% skim milk in 1× PBST (1× PBS, 0.05% Tween20) for 1 h at room temperature and then incubated overnight at 4°C with mouse anti-human CD3ζ primary antibody (1:1000 dilution; clone 8D3; Pharmingen) in 1× PBST containing 5% BSA. Amounts of ß-actin protein were determined by monoclonal antibodies raised against human ß-actin (1:10 000 dilution; A1978, Sigma). Blots were washed with 1× PBST and incubated for 2 h at room temperature with goat anti-mouse IgG HRP conjugate secondary antibody (1:3000, R&D systems) in 4% skim milk. After washing with 1× PBST, immunoreactive bands were detected using the Western HRP substrate (Luminata Forte, Millipore) in an infrared Imager (LAS-4000, Fuji).
Southern analysis
Genomic DNA was purified using DNeasy Blood & Tissue Kit (Qiagen, GmbH). 20 μg of genomic DNA was subjected to restriction digestion using 50 U of the respective enzyme in 200 μl overnight at 37°C. DNA was ethanol precipitated and dissolved in 20 μl TE buffer (pH 8.0). Targeting vectors were linearized with single cutter restriction enzyme and diluted to 107, 108 copies per μl. Digested genomic DNA samples were resolved overnight on a 0.8% agarose gel in 1× TAE (Tris-acetate–boric acid) buffer, with 1 kb DNA ladder (Thermo Scientific) and 1 μl of positive control samples. Southern blotting employing the respective probes, as indicated, was performed using the DIG-High Prime DNA Labeling and Detection Starter Kit II (Roche) as per the manufacturers’ protocol. The probe-target hybrids on the blots were detected by chemiluminescent assay followed by exposure to an X-ray film (Kodak MXG film, Kodak) and developed on a Kodak X-OMAT 2000 Processor.
RESULTS
Generation of seamless transgene target vectors through in vitro recombination
Our basic human genome transgenesis technology embodies highly active λ-Int variant Int-C3 and two distinct nucleotide sequences as partners for intermolecular recombination (42–44). The two sequences are termed attachment (att) sites: attH4X, which comprised of 18 bp present in a small sub-set of primate-specific LINE-1, and the more complex 241 bp attP4X, which we placed on circular target vectors. We recently demonstrated that intracellular integrative recombination between genomic attH4X and plasmid-borne attP4X catalyzed by Int-C3 leads to hybrid attL4X and attR4X sites, which flank an entire bacterial plasmid after integration into LINE-1(42) (Figure 1A).
Figure 1.

Generation of seamless vectors for λ-Int-mediated genomic recombination. (A) A schematic diagram showing λ-Int-mediated recombination between genomic attH4X (located in LINE-1 elements) and attP4X (present on a plasmid target vector), leading to site-specific integration of the entire plasmid into the human LINE-1 elements, then flanked by attL4X (left junction) and attR4X (right junction). (B) An illustration depicting in vitro intra-molecular attH4X x attP4X recombination of a substrate plasmid, resulting in the formation of a seamless vector carrying the attL4X sequence after releasing the undesired bacterial DNA catenane ring by endonucleolytic digest. The seamless vector is subsequently targeted at genomic attH4X, leading to the insertion of payload through a second λ-Int-mediated recombination event. (C) SDS PAGE gel image depicting different desalted fractions of partially purified C-terminally histidine-tagged Int-h/218 (41 kDa). L, Prestained protein ladder (10–250 kDa); E, first eluted fraction from desalting column; 1, 2, 3, 4, different desalted fractions of partially purified C-terminal histidine-tagged Int-h/218. (D) Representative analysis of an in vitro intra-molecular attH4X x attP4X recombination with reaction buffers containing increasing KCl concentrations as indicated (left). Supercoiled seamless vector (SC-SV) and open circular seamless vector (OC-SV) obtained after λ-Int-mediated recombination were resolved via agarose gel electrophoresis in the presence of ethidium bromide. This resulted in DNA bands for SC-SV and OC-SV, which migrated at ∼1.2 and ∼2.2 kb, respectively, whereas the linearized second catenane DNA (linear bacterial DNA) and the unrecombined substrate plasmid migrated at ∼3 and ∼5.0 kb, respectively, as indicated. Purified seamless vector is shown at the right.
A problematic feature of this strategy resulted from the requirement that target vectors for Int-mediated recombination into attH4X need to be circular. Hence, the attP4X-carrying vectors produced in bacterial cells contained prokaryotic origins of replication, selection markers and other functionally irrelevant sequences, in addition to the payload to be delivered to the human genome. These prokaryotic elements unnecessarily increased the size of target vectors and are known to interfere with transgene expression and elicit innate immune responses (16,47,48).
In our previous study, we provided preliminary evidence that a 120 bp attL4X hybrid recombination site can functionally replace the plasmid-borne attP4X for Int-mediated recombination with attH4X in LINE-1 (42). We therefore set out to improve our technology by eliminating bacterial sequences from target vectors through in vitro attH4X x attP4X intramolecular recombination that results in seamless vectors carrying attL4X, which can be isolated and subsequently recombined with genomic attH4X inside human cells (Figure 1B).
We established a simple and efficient protocol for partial purification of C-terminally histidine-tagged variant Int-h/218 (43) from E. coli (Materials and Methods), which readily generates milligrams of highly enriched active Int-h/218 (Figure 1C). The partially purified recombinase efficiently catalyzed in vitro intramolecular recombination between directly repeated attP4X and attH4X on a supercoiled plasmid. This reaction was more efficient at higher salt concentrations. Recombination resulted in a 3.2 kb catenane ring containing the unwanted bacterial genetic elements that was subsequently linearized (Figure 1B, D). The released supercoiled circular seamless target vector (1.8 kb) only carried the payload as well as the hybrid attL4X site as recombination partner for genomic attH4X. Starting, for example, with 10 μg of plasmid DNA, we routinely yielded several μg of purified, mostly supercoiled seamless target vector within 24 h (Figure 1D, right panel).
Site-specific seamless vector insertion into human LINE-1
We employed the isolated 1.8 kb seamless vector attL4X-PGKssPuro to demonstrate intracellular site-specific recombination with genomic attH4X. Different amounts of seamless DNA were co-transfected into human HT1080 cells with an expression plasmid for either Int-C3 or a recombination-inactive Int variant (Int-INA). The latter carries an alanine substitution for the scissile bond-forming tyrosine 342 (42). Puromycin selection was applied 48 h post transfection, and genomic DNA was extracted 2 weeks later and subjected to PCR/sequence analysis (Figure 2A).
Figure 2.

Targeting of seamless vector to LINE-1 elements in HT1080 cells. (A) Experimental strategy demonstrating the λ-Int-mediated targeting of seamless vectors for various cell lines. This includes co-transfection of seamless vectors and Int-expressing plasmids, followed by selection, expansion and characterization of the targeted clones using PCR, sequencing and functional studies. (B) A schematic illustration showing the targeting of genomic attH4X site with seamless vector attL4X-PGKssPuro by Int-C3, leading to integration of the PGK-Puro cassette. Positions of primers used to identify successful integration at attH4X sites in LINE-1 elements are indicated. (C) PCR analysis revealing integration of seamless vector attL4X-PGKssPuro at attH4X site in LINE-1 elements. PCR was performed with primers cs_attH4X_F1/F2 and Puro Rev 24 (left junction) and cs_attH4X_R1/R2 and Puro Fwd 127 (right junction), using genomic DNA from three independent transfections with 50, 100 ng and 200 ng of attL4X-PGKssPuro seamless vector. PCR amplified products of the expected size (∼1000 bp; for the left junction) were detected in bulk C3_50, C3_100 and C3_200 (left panels) and (∼950 bp; for the right junction) in bulk C3_50, C3_100 and C3_200 (right panels). W, no DNA template control; HT, control with genomic DNA from HT1080 parental cells. Ina_100/Ina_200: bulk genomic DNA from puromycin resistant clones obtained through co-transfection of 100 and 200 ng attL4X-PGKssPuro seamless vector and 1 and 2 μg pCMVssIna, respectively. C3_50, C3_100, C3_200: bulk genomic DNA from puromycin resistant HT1080 clones obtained through co-transfection of 50, 100 and 200 ng of attL4X-PGKssPuro seamless vector with 500 ng, 1 and 2 μg pCMVssInt-C3CNLS, respectively; M1, 1kb DNA ladder.
For each amount of transfected seamless vector, bulk cultures were expanded and subsequently screened by genomic PCR using four diagnostic primer pairs as described previously (42) (Figure 2B, C). Expected products were detectable even with the smallest amount (50 ng) of transfected seamless vector and were absent from all controls (Figure 2C). Sequence analysis of these PCR products confirmed targeted seamless vector insertion into various LINE-1 (Supplementary Table S2). Because DNA templates used for this PCR came from bulk cultures, we expected that the identified left and right junction sequences can be assigned to different genomic LINE-1 loci, possibly due to PCR amplification biases.
In parallel to the bulk culture analysis, we performed PCR/sequencing on 92 isolated HT1080 colonies that resulted from transfections with 50 ng of seamless vector. By using three established primer pairs specific for amplification of recombination products (42), we identified 45 cell clones (∼50%) that resulted from targeted recombination events, as evidenced by at least one positive junction-PCR (Supplementary Figure S1). Only two colonies could be expanded from control transfection with catalytically inactive Int, but no specific PCR products were detected in these cases. Based on the PCR/sequencing data, we were able to identify the respective loci of targeted LINE-1 in the majority of the 45 clones (Supplementary Table S3). Some clones, e.g. # 27, 41, 45, most likely carried single copy integrants, since all junction PCR sequences could be mapped to the same genomic LINE-1 locus. However, a substantial fraction of the remaining clones most likely harboured more than one inserted seamless vector.
Next, we were interested in determining seamless vector transgenesis pattern across different targeting attempts. This was important because we employed here attL4X instead of the attP4X as recombination partner sequence described earlier (42). For this, we first designed a novel PCR-based strategy to identify individual targeted LINE-1 loci from bulk HT1080 cell cultures (Figure 3A). Using bulk genomic DNA after transfection of 100 ng of the 1.8 kb seamless vector and selection, we obtained expected PCR products only with templates from co-transfections with active Int-C3 (Figure 3B). Deep paired-end sequencing resulted in 208,936 reads, which contained the expected junction sequence between vector and attH4X. This corresponded to 26,380 unique sequences, which were mapped to the human genome (GEO accession: GSE99883). The top hit for each read was tabulated and merged where they overlapped. This yielded 434 different LINE-1 loci where genomic integration had occurred in bulk cells (Supplementary Table S4). A large fraction of these loci, however, were counted only thrice or less, which could indicate that these loci were less frequently targeted.
We next expanded our approach of targeting LINE-1 with seamless vectors and employed hESCs. Using the same 1.8 kb seamless vector, we identified 25 out of 32 hESC clones, which carried targeted LINE-1, as determined by PCR products obtained for at least one junction (Supplementary Figure S2). Based on subsequent sequence analysis, we identified the loci of targeted LINE-1 in all clones (Supplementary Table S5). As mentioned above in the context of HT1080 transgenesis, targeted hESCs contained multiple copy integrants.
Targeted seamless CAR vector insertion and sustained expression in hESCs
With an ultimate aim of delivering a functional anti-CD19 CAR gene cassette in hESCs and other cell types using our novel λ-int platform for immunotherapy of B-cell leukemia and lymphoma, we intended to insert the multi-reporter pPGKssCAR-EF-Puro cassette that expressed an anti-CD19-41BB-CD3zeta CAR (49) into LINE-1. First, the substrate plasmid pattP4X-PGKssCAR-EF-Puro-attH4X was recombined in vitro to produce supercoiled seamless vector attL4X-PGKssCAR-EF-Puro (Figure 4A). Co-transfection of the 5.5 kb purified seamless CAR vector (Figure 4A, left) with expression plasmid for Int-C3 into hESCs resulted in 48 puromycin resistant colonies, while co-transfections with expression vector for Int-INA yielded only 10 viable colonies. In order to verify LINE-1 targeting, semi-nested PCR was carried out on all 58 clones (Figure 4A and Supplementary Figure S3). The PCR analysis resulted in a total of 12 positive clones, detectable only with genomic DNA isolated from Int-C3 co-transfected cells (Supplementary Figure S3). Subsequent sequence analysis of the clones confirmed the presence of the recombinant left junction (Supplementary Table S6) and an example of such analysis has been shown in Figure 4B. These 12 clones were further expanded to test anti-CD19-CAR expression through flow cytometry. Out of 12 clones, clone#19 and clone#34 had high level of anti-CD19-CAR expression. However, clone #34 did not survive long-term cultures for unknown reasons. We found that 87.1% of cells expressed anti-CD19-CAR in clone#19 (after 8 weeks of culturing) in comparison to only 0.8% anti-CD19-CAR positive cells in a clone obtained from random vector integration (Figure 4C, left and middle panel). The expression of anti-CD19-CAR in clone#19 remained remarkably stable over a period of 6 months (Figure 4C, right panel) and, interestingly, the fraction of low anti-CD19-CAR expressing cells substantially decreased over 6 months, perhaps indicating that some sort of suppression of negative epigenetic modulation of transgene expression at this locus over time. Western Blotting further confirmed the anti-CD19-CAR expression in clone#19 (Figure 4D). Southern blot analysis of targeted clone#19 using the probes complementary to CAR (left panel) or Puro (right Panel) revealed that it carried single-copy transgene of indicated sizes (Figure 4E). However, the sequence information obtained from the UCSC browser shows discrepancy with the corresponding genomic fragment size obtained by Southern analysis. We have encountered such discrepancies in our previous study (42) and had proposed two possible explanations. First, the genome annotation of the corresponding LINE-1 elements may not be entirely correct perhaps due to the repetitive nature of these sequences. Second, sequence polymorphism surrounding these sequences could generate different restriction patterns in different genetic backgrounds (50–54).
Figure 4.

Anti-CD19 CAR expression from seamless vectors in hESCs. (A) A schematic drawing showing in vitro generation and ex vivo targeting of a two-reporter seamless vector (attL4X-PGKssCAR-EF-Puro) at genomic attH4X. Positions of relevant screening primers (PGK Rev 187, attP Rev and cs_attH4X_F1/F2), restriction sites and the probes used for Southern blotting are indicated. The purified 5.5 kb seamless vector resulting from in vitro recombination was analyzed by agarose gel electrophoresis shown at the left; supercoiled seamless vector: SC-SV; open circular seamless vector: OC-SV. (B) Sequence analysis of the targeted hESC clone #19. A chromatogram representation of the DNA sequence of the PCR product obtained with the cs_attH4X_F2 and attP Rev primer pair using genomic DNA from targeted hESC clone#19. The recombinant attL4X site (HOP’), resulting from λ-Int-mediated recombination between genomic attH4X (chr2:159357384-159357546) and attL4X (present on an attL4X-PGKssCAR-EF-Puro seamless vector), is boxed. (C) Flow cytometric analysis of anti-CD19 CAR expression in targeted hESC clone#19. A single parameter histogram overlay plots representation of stained non-transfected control hESCs onto the anti-CD19 CAR-stained population. An overlay obtained with cells from co-transfection of anti-CD19 CAR seamless expression vector with inactive Int expression plasmid and non-transfected control hESCs is shown in the left panel, indicating negligible expression. An overlay obtained with cells from co-transfection of anti-CD19 CAR seamless expression vector with Int-C3 expression plasmid and control hESCs clearly differentiate the anti-CD19 CAR-expressing population; clone#19 (after 8 weeks of culturing) is shown in middle panel. Long term and stable anti-CD19 CAR expression of clone#19 over a period of 6 months is shown in the right panel. (D) Western blot showing anti-CD19 CAR expression. Western analysis using anti-human CD3ζ primary antibody and a goat anti-mouse IgG horseradish peroxidase-conjugated secondary antibody shows anti-CD19 CAR expression in clone #19. (Top Panel). ß-actin was used as loading control (bottom panel). Lanes: ES, lysate from non-transfected hESCs; 19, lysate from clone #19 (a puromycin resistant clone obtained from targeting of seamless vector attL4X-PGKssCAR-EF-Puro and plasmid expressing Int C3); IN, lysate from an inactive clone (a puromycin resistant clone obtained from targeting of seamless vector attL4X-PGKssCAR-EF-Puro and plasmid expressing inactive integrase); JUR, a bulk culture of Jurkat T cells obtained from targeting attL4X-PGKss-CAR-EF-GFP seamless vector (expressing anti-CD19 CAR) along with plasmid expressing Int C3 was used as a positive control. Note: hESC clone #19 and Jurkat T cells show two bands at ∼55 kDa suggesting post translational modifications of anti-CD19 CAR whereas a ∼15kDa band in Jurkat T cells indicates the expression of native monomer of endogenous CD3ζ. (E) Southern blot analysis of the targeted hESC clone#19. A southern blot analysis showing the bands indicated by arrows obtained using PCR-derived digoxigenin-labeled probe complementary to CAR (left panel) or Puro (right Panel) with genomic DNA purified from targeted hESC clone#19 digested with BsrGI and/or NcoI. Lanes: pPCEP (108, 107), copies of linearized vector pattP4X-PGKssCAR-EF-Puro-attH4X loaded as positive control; ES, genomic DNA from parental DNA; IN, genomic DNA obtained from inactive clone (a puromycin resistant clone obtained from co-transfection of attL4X-PGKssCAR-EF-Puro and plasmid expressing inactive integrase); 19, genomic DNA from targeted hESC clone. hESC clone#19 (carrying a single-copy transgene) is a puromycin resistant clone obtained through co-transfection of attL4X-PGKssCAR-EF-Puro and plasmid expressing active Int C3.
Identification of LINE-1 as preferred target loci
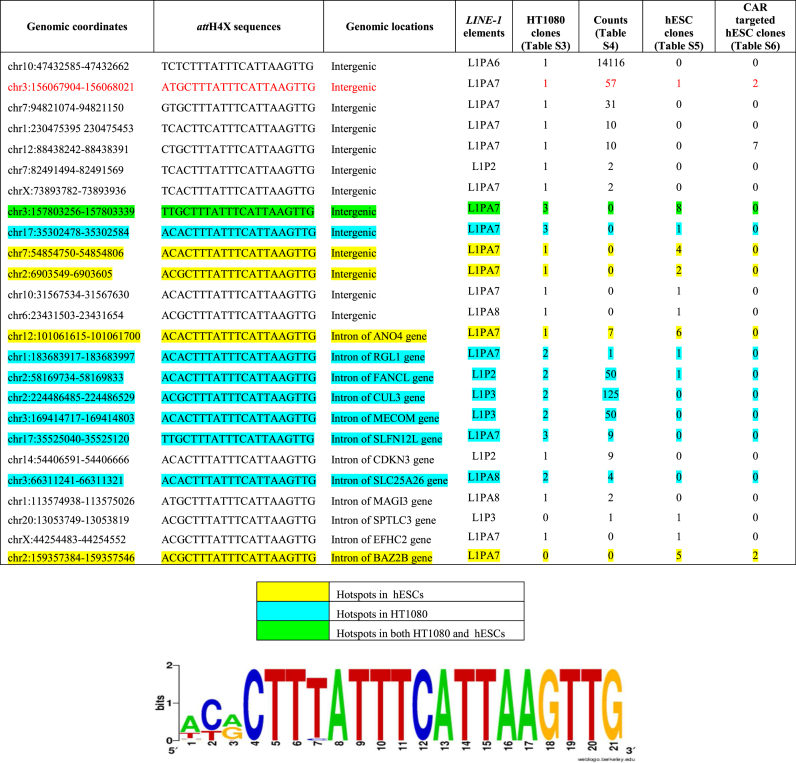
By analyzing data from four lists of targeted attH4X sequences obtained with Int-C3 in HT1080 cells and hESCs (Supplementary Tables S3–S6), we identified 25 loci which were targeted at least twice in four independent cell transfections (Table 1). Furthermore, taking into account the possible variability in preferential integration sites in different cell types, we analysed the data for each cell line separately and obtained different hotspots for HT1080 and hESCs as highlighted by different colours in Table 1. About half of them are in intergenic regions of the human genome, while the rest appear in LINE-1 which contribute to introns. Because only one-third of the 933 attH4X-containing LINE-1 are actually located in introns (Supplementary Table S7), this could indicate a slight preference for recombination reactions at attH4x sequences of LINE-1 that are located in introns. However, we identified one intergenic attH4X locus, which was targeted in all four independent co-transfections in different genetic backgrounds (highlighted in red, Table 1).
Table 1.
List of 25 ‘hotspot’ LINE-1. Genomic coordinates, corresponding attH4X sequences, locations and LINE-1 elements of the ‘hotspots’ compiled from Supplementary Tables S3–S6. A sequence logo obtained from the 25 ‘hotspot’ sequences is shown at the bottom. See text for further details
|
A sequence logo analysis on these 25 ‘hot-spot’ attH4X sequences revealed that within the variable three nucleotides at the 5′-end of the 21 bp attH4X sequence (42), adenine and cytosine at the first and second position, respectively, seemed to be overrepresented (Table 1, bottom). Interestingly, the cytosine at the second position within attH4X appears to play a more important role in genomic recombination reactions with attL4X, when compared with the previously described attP4X as recombination partner (42).
DISCUSSION
Emphasis in the field of genome transgenesis has shifted towards developing better solutions which achieve more precise and safer delivery of multi-transgene cassettes into the human genome. In this context, non-viral vectors have substantial advantages over viral vectors in terms of production costs, safety and capacity to accommodate larger transgene cassettes. Hence, efforts have increased to improve and optimize plasmid-based gene delivery for expansion into the clinics (55).
Multiple recombinase-based systems, such as phiC31, Cre, and Flp recombinase, have been utilized to remove unwanted prokaryotic sequences and minimize target vector length. This resulted in seamless vectors, which could make gene delivery clinically safer and more practical (56–62). In fact, several studies have shown enhanced transgene expression and significantly reduced immune responses of seamless vectors for a variety of applications, such as DNA vaccines, cancer immunotherapy, stem cell reprogramming and gene therapy (19,47,63,64). However, none of these platforms exhibit a dual capability of producing as well as targeting seamless multi-cassette vectors specifically to the human genome using the same recombination system.
Our new λ-Int platform presented in this study provides, to our knowledge, the first such example related to human genome-editing. Its components are very easy and cost-effective to produce, and can yield stable transgenic clonal cell lines with sustained transgene expression patterns from single or multiple integrant(s) within less than three weeks, depending on the growth properties of the targeted cell type. Our study demonstrates a simple method for in vitro production and purification of seamless vectors followed by site-specific genomic integration. Further optimization may lead to the production of seamless target vectors using engineered E. coli strains, potentially at industrial scales, for in vivo applications (65,66). However, for most routine ex vivo applications, our simple in vitro seamless vector production pipeline produces sufficient amounts to achieve cell line engineering.
Our new platform substantially simplifies and improves sustained transgene expression, because the delivered DNA lacks unwanted nucleotide sequences such as bacterial or viral regulatory elements, which are known to promote transgene silencing (16,18). It has also led to increased specificity and reduced random transgene insertion events, as shown here with HT1080 and hESCs. This can be exemplified using the targeting data obtained with HT1080, where 45/92 clones were positive for attH4x targeting with attL4x-PGK-Puro seamless vector compared to 4/33 clones with our previous pattP4x-PGK-Puro plasmid based targeting (42). In addition, with seamless vectors, we obtained only 2 clones positive with the expression of inactive integrase, also indicating lower rates of off-targets and increased specificity. The elimination of unwanted prokaryotic sequences has given us the leeway to further increase the payload size ultimately to be delivered to the human genome. It, therefore, will enable integration of large payloads including more complex genetic regulatory control regions, such as silencers and enhancers, specifically into the genome.
We demonstrated the use of our novel platform to express anti-CD19 CAR protein in hESCs. By targeting the corresponding two-reporter cassette (anti-CD19 CAR and puromycin resistance) to LINE-1 in hESCs and achieving sustained expression of the therapeutic CAR protein in the vast majority of cells from two clones, we provided a compelling example of the usefulness of the λ-Int technology to engineer multi-transgene cassettes that could be used to overcome various clinical problems in the therapeutics space (67). Hence, our study serves as an alternative basis for future CAR-T-based immunotherapy, where Int-mediated recombination could be used to first engineer autologous, self-renewable T-cell-derived iPSCs with multiple CARs, that can be differentiated ex vivo to CAR-T-cell lineage (68). Therefore, high targeting specificity combined with highly sustained transgene expression patterns without integration of undesired extra nucleotide sequences increases the value of our novel platform in the context of future therapeutic applications and should, in principle, be expandable to in vivo gene therapy using nano-carriers as transgene vehicle (69).
We have identified in this study several LINE-1 that can be targeted in different cell lines and independent transfections via attL4X recombination with attH4X. It is possible that target site selectivity is due to variations in local chromatin structure and thus accessibility for Int-C3. In fact, about half of these ‘hotspots’ have been identified in introns that are composed of LINE-1, while only one-third of the 933 attH4X-containing LINE-1 are actually located in introns. Hence, it is likely that the transcriptional and/or epigenetic status of the respective LINE-1 plays an important role for the recombinogenic potential of Int-C3 with the human chromatin. (42). Interestingly, however, the LINE-1 ‘hotspot’ that was targeted in all independent cell transfections is in an intergenic region on chromosome 3 and could potentially be further developed into a preferred safe harbour site by employing Int variants which exhibit increased site specificity. This also raises the interesting possibility of designing seamless target vectors containing splice acceptors, which will result in transgene expression driven by an endogenous gene promoter after genome insertion. In addition, the sequence logo analysis of ‘hot spot’ attH4X sequences pointed out that an adenine and cytosine at the first and second position, respectively, could play an important role in genomic target site recognition and/or catalysis by Int-C3 with attL4X as recombination partner on the seamless vector. Therefore, future in vitro evolution strategies could yield Int variants with enhanced specificity for this subset of attH4X sequences (44,70).
We envision that our technology has great potential for both therapeutic and biotechnological applications. However, realizing the fragile nature of human stem cells’ genome and the devastating effect of any random insertion or insertion in a ‘not so good’ LINE-1 in a clinical set-up, future studies and experiments will be aimed at further optimizing and improving the current λ-Int technology. This would primarily include minimizing off-target effects, refinement of mutant integrases and choosing the more ‘neutral’ LINE-1 elements that would allow the expression of transgenes both at undifferentiated and differentiated cellular states.
In summary, the novel λ-Int mediated seamless vector transgenesis platform presented in this study is capable of producing as well as inserting multi-transgene seamless vectors into a specific endogenous sequence present in a subset of primate LINE-1 elements. This technological advancement, along with the inherent advantages of λ-Int mediated targeting in terms of cytotoxicity, genome integrity and capability of choosing the best performing clones from various targeting attempts (42), makes our platform attractive for utility at an intersection of molecular medicine, gene therapy and systems/synthetic biology for various biomedical applications.
DATA AVAILABILITY
The sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE99883.
Supplementary Material
ACKNOWLEDGEMENTS
Human ESCs were provided by GENEA, Sydney, Australia. H.M. and P.D. designed the study. H.M. and S.R. performed cell-based targeting assays and analyzed data. S.H. performed sequence and bioinformatic analyses. F.J.G. developed PCR strategies and provided plasmids. D.W. and D.C. provided the anti-CD19 CAR construct and technical advice. R.J. provided advice on Int purification and bioinformatics. H.M., S.R. and P.D. wrote the paper.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NTUitive and Singapore/MIT Alliance for Research and Technology [SMART to P.D.]; National Medical Research Council Fund [NMRC/StaR/0025/2015 to D.C.]; Agency for Science, Technology and Research, Singapore [to F.J.G. and S.H.]. Funding for open access charge: Singapore-MIT Alliance for Research and Technology (SMART) [M4062198.080].
Conflict of interest statement. H.M. and P.D. declare competing financial interests.
REFERENCES
- 1. Cheng A.A., Lu T.K.. Synthetic biology: an emerging engineering discipline. Annu. Rev. Biomed. Eng. 2012; 14:155–178. [DOI] [PubMed] [Google Scholar]
- 2. Cheng J.K., Alper H.S.. The genome editing toolbox: a spectrum of approaches for targeted modification. Curr. Opin. Biotechnol. 2014; 30:87–94. [DOI] [PubMed] [Google Scholar]
- 3. Doudna J.A., Charpentier E.. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014; 346:1258096. [DOI] [PubMed] [Google Scholar]
- 4. Gabriel R., von Kalle C., Schmidt M.. Mapping the precision of genome editing. Nat. Biotechnol. 2015; 33:150–152. [DOI] [PubMed] [Google Scholar]
- 5. Chamberlain K., Riyad J.M., Weber T.. Expressing transgenes that exceed the packaging capacity of Adeno-Associated virus capsids. Hum. Gene Ther. Methods. 2016; 27:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kumar M., Keller B., Makalou N., Sutton R.E.. Systematic determination of the packaging limit of lentiviral vectors. Hum. Gene Ther. 2001; 12:1893–1905. [DOI] [PubMed] [Google Scholar]
- 7. Wu Z., Yang H., Colosi P.. Effect of genome size on AAV vector packaging. Mol. Ther. 2010; 18:80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Byrne S.M., Ortiz L., Mali P., Aach J., Church G.M.. Multi-kilobase homozygous targeted gene replacement in human induced pluripotent stem cells. Nucleic Acids Res. 2015; 43:e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Holkers M., Maggio I., Henriques S.F., Janssen J.M., Cathomen T., Goncalves M.A.. Adenoviral vector DNA for accurate genome editing with engineered nucleases. Nat. Methods. 2014; 11:1051–1057. [DOI] [PubMed] [Google Scholar]
- 10. Li K., Wang G., Andersen T., Zhou P., Pu W.T.. Optimization of genome engineering approaches with the CRISPR/Cas9 system. PLoS One. 2014; 9:e105779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Modlich U., Bohne J., Schmidt M., von Kalle C., Knoss S., Schambach A., Baum C.. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood. 2006; 108:2545–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Montini E., Cesana D., Schmidt M., Sanvito F., Ponzoni M., Bartholomae C., Sergi Sergi L., Benedicenti F., Ambrosi A., Di Serio C. et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat. Biotechnol. 2006; 24:687–696. [DOI] [PubMed] [Google Scholar]
- 13. Nayak S., Herzog R.W.. Progress and prospects: immune responses to viral vectors. Gene Ther. 2010; 17:295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fischer A., Hacein-Bey-Abina S., Cavazzana-Calvo M.. 20 years of gene therapy for SCID. Nat. Immunol. 2010; 11:457–460. [DOI] [PubMed] [Google Scholar]
- 15. Ma Y., Zhang L., Huang X.. Genome modification by CRISPR/Cas9. FEBS J. 2014; 281:5186–5193. [DOI] [PubMed] [Google Scholar]
- 16. Chen Z.Y., He C.Y., Ehrhardt A., Kay M.A.. Minicircle DNA vectors devoid of bacterial DNA result in persistent and high-level transgene expression in vivo. Mol. Ther. 2003; 8:495–500. [DOI] [PubMed] [Google Scholar]
- 17. Darquet A.M., Cameron B., Wils P., Scherman D., Crouzet J.. A new DNA vehicle for nonviral gene delivery: supercoiled minicircle. Gene Ther. 1997; 4:1341–1349. [DOI] [PubMed] [Google Scholar]
- 18. Chen Z.Y., He C.Y., Meuse L., Kay M.A.. Silencing of episomal transgene expression by plasmid bacterial DNA elements in vivo. Gene Ther. 2004; 11:856–864. [DOI] [PubMed] [Google Scholar]
- 19. Jechlinger W. Optimization and delivery of plasmid DNA for vaccination. Expert Rev. Vaccines. 2006; 5:803–825. [DOI] [PubMed] [Google Scholar]
- 20. Klinman D.M. Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat. Rev. Immunol. 2004; 4:249–258. [DOI] [PubMed] [Google Scholar]
- 21. Krieg A.M., Yi A.K., Matson S., Waldschmidt T.J., Bishop G.A., Teasdale R., Koretzky G.A., Klinman D.M.. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995; 374:546–549. [DOI] [PubMed] [Google Scholar]
- 22. Sadelain M., Papapetrou E.P., Bushman F.D.. Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer. 2011; 12:51–58. [DOI] [PubMed] [Google Scholar]
- 23. Carroll D. Genome engineering with targetable nucleases. Annu. Rev. Biochem. 2014; 83:409–439. [DOI] [PubMed] [Google Scholar]
- 24. Sander J.D., Joung J.K.. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014; 32:347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miller J.C., Tan S., Qiao G., Barlow K.A., Wang J., Xia D.F., Meng X., Paschon D.E., Leung E., Hinkley S.J. et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011; 29:143–148. [DOI] [PubMed] [Google Scholar]
- 26. Bibikova M., Beumer K., Trautman J.K., Carroll D.. Enhancing gene targeting with designed zinc finger nucleases. Science. 2003; 300:764. [DOI] [PubMed] [Google Scholar]
- 27. Christian M., Cermak T., Doyle E.L., Schmidt C., Zhang F., Hummel A., Bogdanove A.J., Voytas D.F.. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010; 186:757–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guilinger J.P., Pattanayak V., Reyon D., Tsai S.Q., Sander J.D., Joung J.K., Liu D.R.. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat. Methods. 2014; 11:429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pattanayak V., Guilinger J.P., Liu D.R.. Determining the specificities of TALENs, Cas9, and other genome-editing enzymes. Methods Enzymol. 2014; 546:47–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D.. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013; 31:822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lin Y., Cradick T.J., Brown M.T., Deshmukh H., Ranjan P., Sarode N., Wile B.M., Vertino P.M., Stewart F.J., Bao G.. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014; 42:7473–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Karow M., Calos M.P.. The therapeutic potential of PhiC31 integrase as a gene therapy system. Expert Opin. Biol. Ther. 2011; 11:1287–1296. [DOI] [PubMed] [Google Scholar]
- 33. Calos M.P. The phiC31 integrase system for gene therapy. Curr. Gene Ther. 2006; 6:633–645. [DOI] [PubMed] [Google Scholar]
- 34. Shah R., Li F., Voziyanova E., Voziyanov Y.. Target-specific variants of Flp recombinase mediate genome engineering reactions in mammalian cells. FEBS J. 2015; 282:3323–3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Farruggio A.P., Bhakta M.S., du Bois H., Ma J., Calos M.P.. Genomic integration of the full-length dystrophin coding sequence in Duchenne muscular dystrophy induced pluripotent stem cells. Biotechnol. J. 2017; 12:1600477. [DOI] [PubMed] [Google Scholar]
- 36. Stark W.M. Making serine integrases work for us. Curr. Opin. Microbiol. 2017; 38:130–136. [DOI] [PubMed] [Google Scholar]
- 37. Thyagarajan B., Guimaraes M.J., Groth A.C., Calos M.P.. Mammalian genomes contain active recombinase recognition sites. Gene. 2000; 244:47–54. [DOI] [PubMed] [Google Scholar]
- 38. Chalberg T.W., Genise H.L., Vollrath D., Calos M.P.. phiC31 integrase confers genomic integration and long-term transgene expression in rat retina. Invest. Ophthalmol. Vis. Sci. 2005; 46:2140–2146. [DOI] [PubMed] [Google Scholar]
- 39. Ehrhardt A., Engler J.A., Xu H., Cherry A.M., Kay M.A.. Molecular analysis of chromosomal rearrangements in mammalian cells after phiC31-mediated integration. Hum. Gene Ther. 2006; 17:1077–1094. [DOI] [PubMed] [Google Scholar]
- 40. Liu J., Skjorringe T., Gjetting T., Jensen T.G.. PhiC31 integrase induces a DNA damage response and chromosomal rearrangements in human adult fibroblasts. BMC Biotechnol. 2009; 9:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chalberg T.W., Portlock J.L., Olivares E.C., Thyagarajan B., Kirby P.J., Hillman R.T., Hoelters J., Calos M.P.. Integration specificity of phage phiC31 integrase in the human genome. J. Mol. Biol. 2006; 357:28–48. [DOI] [PubMed] [Google Scholar]
- 42. Vijaya Chandra S.H., Makhija H., Peter S., Myint Wai C.M., Li J., Zhu J., Ren Z., D’Alcontres M.S., Siau J.W., Chee S. et al. Conservative site-specific and single-copy transgenesis in human LINE-1 elements. Nucleic Acids Res. 2016; 44:e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lorbach E., Christ N., Schwikardi M., Droge P.. Site-specific recombination in human cells catalyzed by phage lambda integrase mutants. J. Mol. Biol. 2000; 296:1175–1181. [DOI] [PubMed] [Google Scholar]
- 44. Siau J.W., Chee S., Makhija H., Wai C.M., Chandra S.H., Peter S., Droge P., Ghadessy F.J.. Directed evolution of lambda integrase activity and specificity by genetic derepression. Protein Eng. Des. Select.: PEDS. 2015; 28:211–220. [DOI] [PubMed] [Google Scholar]
- 45. Jessop L., Bankhead T., Wong D., Segall A.M.. The amino terminus of bacteriophage lambda integrase is involved in protein-protein interactions during recombination. J. Bacteriol. 2000; 182:1024–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Corona T., Bao Q., Christ N., Schwartz T., Li J., Droge P.. Activation of site-specific DNA integration in human cells by a single chain integration host factor. Nucleic Acids Res. 2003; 31:5140–5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Narsinh K.H., Jia F., Robbins R.C., Kay M.A., Longaker M.T., Wu J.C.. Generation of adult human induced pluripotent stem cells using nonviral minicircle DNA vectors. Nat. Protoc. 2011; 6:78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Munye M.M., Tagalakis A.D., Barnes J.L., Brown R.E., McAnulty R.J., Howe S.J., Hart S.L.. Minicircle DNA provides enhanced and prolonged transgene expression following airway gene transfer. Scientific Rep. 2016; 6:23125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Imai C., Mihara K., Andreansky M., Nicholson I.C., Pui C.H., Geiger T.L., Campana D.. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004; 18:676–684. [DOI] [PubMed] [Google Scholar]
- 50. Wang J., Song L., Grover D., Azrak S., Batzer M.A., Liang P.. dbRIP: a highly integrated database of retrotransposon insertion polymorphisms in humans. Hum. Mutat. 2006; 27:323–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Alexander R.P., Fang G., Rozowsky J., Snyder M., Gerstein M.B.. Annotating non-coding regions of the genome. Nat. Rev. Genet. 2010; 11:559–571. [DOI] [PubMed] [Google Scholar]
- 52. Penzkofer T., Jager M., Figlerowicz M., Badge R., Mundlos S., Robinson P.N., Zemojtel T.. L1Base 2: more retrotransposition-active LINE-1s, more mammalian genomes. Nucleic Acids Res. 2017; 45:D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Penzkofer T., Dandekar T., Zemojtel T.. L1Base: from functional annotation to prediction of active LINE-1 elements. Nucleic Acids Res. 2005; 33:D498–D500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Beck C.R., Collier P., Macfarlane C., Malig M., Kidd J.M., Eichler E.E., Badge R.M., Moran J.V.. LINE-1 retrotransposition activity in human genomes. Cell. 2010; 141:1159–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hardee C.L., Arevalo-Soliz L.M., Hornstein B.D., Zechiedrich L.. Advances in non-viral DNA vectors for gene therapy. Genes. 2017; 8:E65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bigger B.W., Tolmachov O., Collombet J.M., Fragkos M., Palaszewski I., Coutelle C.. An araC-controlled bacterial cre expression system to produce DNA minicircle vectors for nuclear and mitochondrial gene therapy. J. Biol. Chem. 2001; 276:23018–23027. [DOI] [PubMed] [Google Scholar]
- 57. Kreiss P., Cameron B., Darquet A.M., Scherman D., Crouzet J.. Production of a new DNA vehicle for gene transfer using site-specific recombination. Appl. Microbiol. Biotechnol. 1998; 49:560–567. [DOI] [PubMed] [Google Scholar]
- 58. Thorpe H.M., Smith M.C.. In vitro site-specific integration of bacteriophage DNA catalyzed by a recombinase of the resolvase/invertase family. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:5505–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Thorpe H.M., Wilson S.E., Smith M.C.. Control of directionality in the site-specific recombination system of the Streptomyces phage phiC31. Mol. Microbiol. 2000; 38:232–241. [DOI] [PubMed] [Google Scholar]
- 60. Chen Z.Y., He C.Y., Kay M.A.. Improved production and purification of minicircle DNA vector free of plasmid bacterial sequences and capable of persistent transgene expression in vivo. Hum. Gene Ther. 2005; 16:126–131. [DOI] [PubMed] [Google Scholar]
- 61. Jechlinger W., Azimpour Tabrizi C., Lubitz W., Mayrhofer P.. Minicircle DNA immobilized in bacterial ghosts: in vivo production of safe non-viral DNA delivery vehicles. J. Mol. Microbiol. Biotechnol. 2004; 8:222–231. [DOI] [PubMed] [Google Scholar]
- 62. Catanese D.J. Jr, Fogg J.M., Schrock D.E. 2nd, Gilbert B.E., Zechiedrich L.. Supercoiled Minivector DNA resists shear forces associated with gene therapy delivery. Gene Ther. 2012; 19:94–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Huang M., Chen Z., Hu S., Jia F., Li Z., Hoyt G., Robbins R.C., Kay M.A., Wu J.C.. Novel minicircle vector for gene therapy in murine myocardial infarction. Circulation. 2009; 120:S230–S237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hudecek M., Gogishvili T., Monjezi R., Wegner J., Shankar R., Kruesemann C., Miskey C., Ivics Z., Schmeer M., Schleef M.. Minicircle-based engineering of chimeric antigen receptor (CAR) T cells. Rec. Results Cancer Res. 2016; 209:37–50. [DOI] [PubMed] [Google Scholar]
- 65. Kay M.A., He C.Y., Chen Z.Y.. A robust system for production of minicircle DNA vectors. Nat. Biotechnol. 2010; 28:1287–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mayrhofer P., Blaesen M., Schleef M., Jechlinger W.. Minicircle-DNA production by site specific recombination and protein-DNA interaction chromatography. J. Gene Med. 2008; 10:1253–1269. [DOI] [PubMed] [Google Scholar]
- 67. Sadelain M., Riviere I., Riddell S.. Therapeutic T cell engineering. Nature. 2017; 545:423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Themeli M., Kloss C.C., Ciriello G., Fedorov V.D., Perna F., Gonen M., Sadelain M.. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013; 31:928–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Smith T.T., Stephan S.B., Moffett H.F., McKnight L.E., Ji W., Reiman D., Bonagofski E., Wohlfahrt M.E., Pillai S.P.S., Stephan M.T.. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat. Nanotechnol. 2017; 2:813–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tay Y., Ho C., Droge P., Ghadessy F.J.. Selection of bacteriophage lambda integrases with altered recombination specificity by in vitro compartmentalization. Nucleic Acids Res. 2010; 38:e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE99883.