


Abstract
p53 transcriptional networks are well-characterized in many organisms. However, a global understanding of requirements for in vivo p53 interactions with DNA and relationships with transcription across human biological systems in response to various p53 activating situations remains limited. Using a common analysis pipeline, we analyzed 41 data sets from genome-wide ChIP-seq studies of which 16 have associated gene expression data, including our recent primary data with normal human lymphocytes. The resulting extensive analysis, accessible at p53 BAER hub via the UCSC browser, provides a robust platform to characterize p53 binding throughout the human genome including direct influence on gene expression and underlying mechanisms. We establish the impact of spacers and mismatches from consensus on p53 binding in vivo and propose that once bound, neither significantly influences the likelihood of expression. Our rigorous approach revealed a large p53 genome-wide cistrome composed of >900 genes directly targeted by p53. Importantly, we identify a core cistrome signature composed of genes appearing in over half the data sets, and we identify signatures that are treatment- or cell-specific, demonstrating new functions for p53 in cell biology. Our analysis reveals a broad homeostatic role for human p53 that is relevant to both basic and translational studies.
INTRODUCTION
The tumor suppressor p53 is a stress-activated transcription factor (TF) that recognizes a 20-base pair (bp) degenerate motif in chromatin consisting of two decamers of the structure 5′-RRRCWWGYYY-3′, where R = [A,G], W = [A,T] and Y = [C,T] (1–3). Recently, we also identified the functional recognition of single decamer half-sites as part of the sequence repertoire that p53 binds in vivo (4). Because of its role in suppressing human cancers, the protein, its gene, and the networks it regulates have been intensively studied for nearly 40 years. Nevertheless, it is unlikely that p53 primarily evolved to be a tumor suppressor because it is present in primitive organisms, and it has many other functions (5) that may play a role long before the occasional appearance of cancer. Hundreds of thousands of potential binding sites (p53 motifs) exist in the human genome, yet any cell nucleus contains only a few thousand p53 molecules even after p53 is stabilized in response to stress (6). Despite the vast literature, a paucity of information addresses sites bound by p53 in normal and cancer human cells after p53 induction relative to its target sequences and with respect to its direct influence on transcription. More specifically, while there have been many studies on p53 responses at specific sites and genes, little is known at the genome level about binding and sequence relationships, binding versus expression as well as the relevance of various stress signals or the extent of commonality of responses. We anticipated that through an extensive, rigorous analysis of the combination of natural binding, target sequence and expression in response to different stresses across studies, we would have the opportunity to address a variety of important p53 universe issues at the mechanistic as well as the network level and to identify genes that are directly targeted by p53 for altered expression.
The advent of genome-wide chromatin immunoprecipitation of DNA fragments followed by high-throughput sequencing (ChIP-seq) coupled with gene expression provides a potential means to addressing the above issues. In the last seven years, multiple studies have been published, often with differences in results potentially due to biological variation, technical issues, or method of analysis. Here, we examined 44 data sets from human p53 ChIP-seq studies that contained activated or overexpressed p53 binding and associated gene expression. We also analyzed 17 data sets that correspond to control, non-activated (no treatment or DMSO) p53 (Table 1). To avoid variations resulting from differences in methods of analysis between studies as well as the many pitfalls that may occur in using conclusions to assemble information, the data sets were downloaded and reanalyzed with a common ChIP-seq workflow (Table 1; Supplementary Figure SF1, Supplementary Table ST1). We assessed the quality of the data and developed a uniform, unbiased approach to analysis. The common workflow for all raw data assures uniformity of analysis and, more importantly, uniformity of conclusions. We note that our approach can be applied to any sequence-specific transcription factor.
Table 1.
Matrix for analysis of cells and treatments for ChIP-seq and expression data sets. Rows give results for normal and cancer cells or cell lines as indicated. Columns correspond to treatments: 5-FU, 5-fluorouracil; ActD, actomycin D; Cisp, cisplatin; DXR, doxorubicin; Etop, etoposide; IR, ionizing radiation; nutlin, nutlin-3; RITA, reactivation of p53 and induction of tumor cell apoptosis; UV, ultraviolet radiation; p53 O/E, p53 overexpression; RA, retinoic acid; Ras O/E, Ras overexpression; NT, no treatment; DMSO, dimethyl sulfoxide. Numbers in cells give reference citations, which are also found in Supplementary Table ST1. * = this publication. Those with associated gene expression are highlighted in gray.
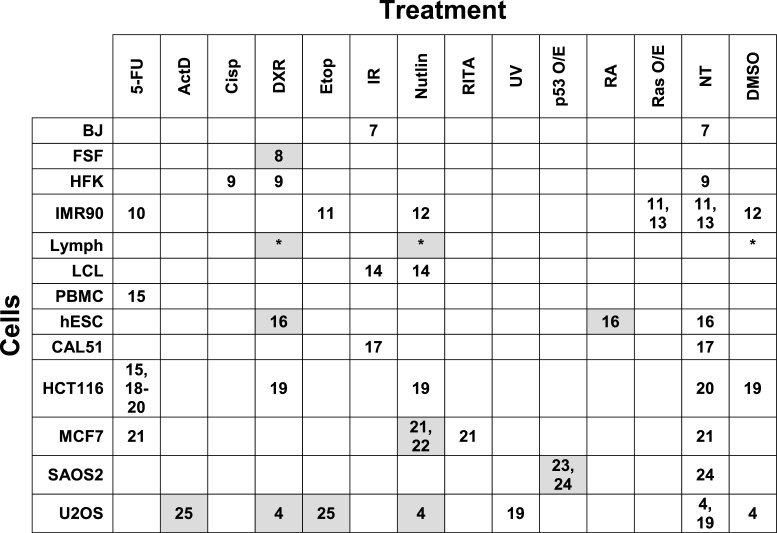
|
The ChIP-seq and gene expression data sets came from 20 publications as well as the first primary p53 ChIP-seq and expression data on normal human lymphocytes, which we report here. Included in the data sets are results from 13 cell types and 12 methods of inducing or activating p53 that span nine time points (from 1 to 48 h after treatment). Among these binding data sets in which p53 is activated or elevated, 16 also contained mRNA expression data. This enabled us to directly assess the relationship between binding and expression.
We created a human p53 Binding And Expression Resource (BAER) hub that can be accessed on the University of California Santa Cruz (UCSC) Genome Browser (26,27) alongside other publicly available annotation tracks (https://orio.niehs.nih.gov/ucscview/nguyen/p53BAER/hub.txt). The human p53 BAER hub contains tracks for read depth for each data set, identified peaks (or p53 bound sites) for each activated or control data set, and changes in gene expression (Supplementary Figure SF2).
Through our knowledge of p53 binding motifs, including half-sites, and the data in the p53 BAER hub, we a) distinguished direct vs. indirect targets, b) assessed mechanistic relationships between binding and increased vs. decreased expression changes to c) reveal a greatly increased number of p53 target genes (cistrome) and a core cistrome signature. We describe a broad homeostatic role for human p53 in diverse biological processes beyond its classical role as guardian of the genome.
MATERIALS AND METHODS
Lymphocyte ChIP-seq and microarray sample preparation and sequencing
Human lymphocytes were isolated with Histopaque-1077 (Sigma-Aldrich) from the blood of healthy volunteers per the approved NIEHS institutional review board (IRB#07-E-0023) protocol. All volunteers gave written informed consent for blood donation. Cells were grown in 1.5% (v/v) PHA for 72 h and treated with 0.3 μg/ml Doxorubicin, 10 μM Nutlin-3, or 0.1% DMSO for 24 h. 17–30 million T-lymphocytes were used per each experiment, and ChIP assays were performed as described in our previous work (4) (also see Supplementary Materials and Methods). RNA was isolated using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA), and microarrays were performed in the Affymetrix GeneChIP Instrument system using procedures as previously described (4). Sequencing and microarray data for these data sets have been deposited in GEO under accession GSE110370. ChIP-seq and microarray data were processed according to their respective analysis workflow described below.
ChIP-seq analysis workflow
Relevant ChIP-seq and associated input data sets were downloaded from publicly-available resources as listed in Supplementary Table ST1. All reads were clipped to a maximum length of 36 nucleotides (nt), then filtered to retain only sequences with a mean base quality score of at least 20. Filtered reads were aligned against the hg19 reference genome (excluding haplotype chromosomes) via Bowtie v0.12.8 (28) with parameters ‘-m1 -v2’ to accept only uniquely-mapped hits with a maximum of two mismatched bases. Multiple replicates from the same sample were merged, then duplicate reads were removed with MergeSamFiles.jar and MarkDuplicates.jar from the Picard tool suite v1.86 (http://broadinstitute.github.io/picard). For ChIP-seq data sets without an associated input sample, surrogate inputs were generated by randomly selecting 20 million uniquely-mapped, non-duplicate reads from other input data sets of the same cell type. Specifically, a surrogate U2OS input was made by downsampling the combined input data sets from DMSO, DXR, and Nutlin treatment conditions from Menendez et al. (4), and a surrogate HCT116 input was made by downsampling the combined 5FU-treated input data sets from Botcheva and McCorkle (18) and Wang et al. (15). Depth tracks were generated with BEDTools genomeCoverageBed v2.17.0 (29) and UCSC utility bedGraphToBigWig (30), after extending each uniquely-mapped, non-duplicate read to a length of 200 nt.
p53 peak calls
The SISSRs program (31) was used to identify p53 bound peaks for each p53 ChIP-seq data set using its associated input data set (or a surrogate input) as a control at default parameters (P < 0.001). The SISSRs output peaks were subsequently redefined as 200-mers centered on the called peak's midpoint. Merged peak lists were generated for the 41 activated p53 ChIP-seq data sets and for the 17 control p53 ChIP-seq data sets by BEDTools mergeBed v2.24.0 (29), where regions that had at least one nt overlap or were book-ended were merged.
Peak annotation
Presence of a p53-like motif (with or without spacers between the two half-sites or just one half-site) was established by scanning each p53 peak via MAST v4.9.0 (32) with parameter ‘-comp"; the position weight matrix (PWM) used for this scan was the highest-scoring de novo motif within the top 1000 sequences from the DXR-treated lymphocyte peak set (GEO # GSM2988950) as identified by MEME v4.9.0 (33) with parameters ‘-mod zoops -evt 0.1 -w 20 -revcomp -minsites 25 -maxsites 1000 -maxsize 20000000’. For the differentiation of potential direct versus indirect p53 targets in the cistrome analysis, a more relaxed identification of p53-like sites was implemented, whereby individual peak sequences were evaluated via p53scan (34) allowing for spacers of size 0 through 15 bp, and a score >4.39.
Association of p53 peaks with other genomic features were assigned by a simple distance heuristic. The genomic context of individual p53 peaks were assigned based on proximity to hg19 RefSeq gene models, as downloaded from the UCSC Genome Browser 15 December 2014. Categories for genomic context are defined as ‘near TSS’ (5 kb upstream of TSS through end of first intron), ‘intragenic’ (second exon through 3′ untranslated region (UTR)), or ‘intergenic’ (everything else). Assignment of specific genes to p53 peaks are made based on this same assessment. p53 peaks defined as having ‘near TSS’ or ‘intragenic’ status are considered associated with those genes for the gene expression and cistrome analyses. miRNA potentially associated with p53 peaks were defined as miRBase v21 (http://www.mirbase.org/, 35) entries within 5 kb of a peak. lincRNA transcripts potentially associated with p53 peaks were defined as Human Body Map lincRNA (http://portals.broadinstitute.org/genome_bio/human_lincrnas/, 36) entries within 5 kb of a peak. eRNAs potentially associated with p53 peaks were defined as Andersson et al.’s active enhancers (37) within 5 kb of a peak. G-quadruplexes (G4s) potentially associated with p53 peaks were defined as Hansel-Hertsch et al.’s HaCat or NHEK G4s (38) within 1 kb of a peak.
Enriched motifs
Searches for enriched known or de novo motifs within peak sets were performed by HOMER findMotifsGenome v4.9.1 (39). Known motif searches were performed with parameters ‘-size given -nomotif’; de novo motif searches were performed with parameters ‘-size given -noknown -len 20,22 -S 10’.
Scanning genome for p53 consensus motifs
We downloaded the genomic sequences of the 24 human chromosomes from the UCSC genome browser (build hg19). For each chromosome, we counted the number of exact match of RRRCWWGYYY (10-mer), RRRCWWGYYYRRRCWWGYYY (20-mer, 0-spacer) or RRRCWWGYYYN(1–15)RRRCWWGYYY (for 20-mer, 1–15-spacer) using a custom C code. Similarly, we counted the number of exact match of the 20-mer, 0-spacer with one mismatch.
Enrichment of chromatin marks in p53 binding peaks that appear in ≥20 data sets
The ENCODE genome segmentation data for the 16 chromatin marks in nine cell lines (GM12878, H1-hESC, HEP-G2, HNEC, HSMM, HUVEC, K562, NHEK and NHLF) were downloaded from the UCSC site (http://genome.ucsc.edu/cgi-bin/hgFileUi?db=hg19&g=wgEncodeBroadHmm, 40). First, we counted the number of p53 binding peaks that appear in ≥20 data sets and p53 binding peaks that appear in only two data sets that fell into the each of the 16 chromatin states identified by hidden Markov model (HMM) (41). Chromatin state enrichment was then carried out on the resultant 2 × 2 contingency table using Fisher's exact test.
Microarray analysis
Microarray perfect match pixel intensity data from the Affymetrix raw CEL files of a given study (see Supplementary Table ST1) were preprocessed in the Partek Genomics Suite v6.13 software (Partek, St. Louis, MO, USA) using the robust multichip average (RMA) approach (42,43) that includes log2 transformation, background correction, quantile normalization, and summarization by median polish to combine data from the probes in a probe set to get a single data value. The data was then modeled with an N-way analysis of variance (ANOVA) where N denotes the number of factors in the given study design. Fisher's least significant difference contrasts (44) between the mean of treated replicates and the mean of the respective control replicates (not treated [NT], mock/vehicle, DMSO, etc.) were performed to identify statistically significant differentially expressed genes (DEGs) using a Benjamini & Hochberg multiple testing (45) false discovery rate (FDR) threshold <0.01 based on two-sided nominal P-values and an absolute fold-change (FC) >1.5.
RNA-seq analysis of downloaded data sets
RNA-seq data from previous studies (GSE55727, GSE47042, GSE15780) (Supplementary Table ST1) were downloaded from the Gene Expression Omnibus (GEO) (46,47). Data quality was assessed with FastQC, and the adapter sequences were removed (if detected) prior to alignment. Preprocessed RNA-seq short reads were aligned with TopHat (48) to human genome hg19 guided with refseq-based gene model (time stamp as of Dec. 15th, 2014). The alignment .bam files were directly processed with HTseq modules (49), and count level measurement was produced based on refseq-based gene model. In the end, count level data was used for further statistical analysis. To test for biological hypotheses, pair-wise tests with replicates were conducted with DEseq (50). Differentially expressed genes at each comparison condition were obtained based on the negative binomial test at a FC >|2| and P-value <0.01.
RESULTS
Nature and distribution of p53 peaks
For each activated p53 data set included in the meta-analysis, we asked how all identified peaks are distributed with respect to transcriptional start sites (TSS) (regions from 5 kb upstream through the first intron), intragenic (second exon through the 3′-UTR), or intergenic regions. The TSS region comprises about 10% of the total genome. However, nearly 35% of the peaks found in at least one data set appear in the TSS region, while a smaller amount (25%) were in the intragenic region, and more were intergenic (41%) (Figure 1A). (No differences could be detected in terms of TSS or intergenic binding when data sets are divided into normal cells, cancer cells or p53 overexpressing cells (see Supplementary Figure SF3)). Thus, p53 presents a strong bias to bind near a TSS, which is consistent with its role as a TF.
Figure 1.

p53 binding distribution across the genome. (A) Genomic distribution of peaks relative to genic regions of the 44 activated p53 ChIP-seq data sets: nearTSS (5 kb upstream through the first intron); intragenic (second exon through 3′UTR); and intergenic (everything else). Each point is a ChIP-seq data set. Tukey box plot: box = 25th–75th percentile, black line = median, and whiskers = 1.5 * interquartile range (IQR). Open symbols are outliers for each genomic region. (B) Distribution of called peaks on chromosome 5 in some normal (GM00011, GM6170, HFK) and cancer (MCF-7, U2OS) cells after DXR or Nutlin-3 treatment. Data set reference citations are in () following the peak descriptions.
We addressed the distribution of p53 binding across chromosomes. As shown in Supplementary Figure SF4, peaks are distributed across the genome in a non-random manner, which may simply reflect the non-randomness of genome structure. All chromosomes show a clustering of peaks in several chromosomal regions, and the clustering was similar for all cell types examined. Importantly, as shown in the example for Chromosome 5 (Figure 1B), peak regions were distributed similarly in both normal and cancer cells (the average number of peaks for cancer and normal cells was 2923 and 3499, respectively, excluding the two p53 overexpression data sets (see Supplementary Figure SF5)), suggesting a commonality for the potential for binding. This similarity between cell types was observed for all chromosomes (see p53 BAER hub).
Visualization of the p53 data sets with the UCSC genome browser provides opportunities to identify major genetic and epigenomic annotated elements, including gene density, repeats, CpG islands, chromatin markers and promoters, as well as p53 peaks across chromosomes. For example, as presented in Figure 1B and Supplementary Figure SF4, a substantial correlation exists among the p53 ChIP-seq peaks and the appearance across the genome of CpG islands regardless of the experimental conditions.
We examined peaks for the presence of a p53-like motif, including half-sites and motifs with spacers between half-sites, using the methods we described in Menendez et al. (4). We previously established that p53 can bind half-sites to modulate transcription. The range of peaks containing a p53-like motif in the 44 data sets with activated p53 was from 21 to 95%, except for three data sets (Figure 2A). (For a summary of the distribution of p53-like motifs including half-sites and spacers, see Supplementary Figure SF6.) These three data sets were excluded from further assessment due to an abnormally low frequency of peaks with a p53 motif (3%) or low number of total peaks (only 17) (identified as outliers in Figure 1A, Supplementary Figure SF6, and Supplementary Table ST1). The remaining 41 data sets associated with activated p53 are the focus of this meta-analysis.
Figure 2.

p53 motif analysis. (A) Tukey boxplot of the peaks from the 44 activated p53 ChIP-seq data sets based on the type of sequence bound: p53-like motif (includes p53 consensus motif containing two decamers with no spacer sequence, with spacers of 1–15 bp between the decamers, or half-site/one decamer) or no motif (no evidence of a half site in the 200 bp peak). (B) Tukey boxplot of the percent of p53-like motifs in peaks that are frequently bound in multiple data sets as indicated. (C) Tukey boxplot of p53 ‘binding strength’ as measured by ChIP-over-Input fold enrichment of 54 947 peaks called in the data sets. Symbols beyond the whiskers are outliers. (D) de novo consensus motif identified from peaks in ≥2 data sets; (E) the % sites bound in vivo when there is a perfect motif with 0–15-spacer or just a half-site (HS); (F) the % sites bound when there is a perfect motif with 0-spacer with no mismatches (no MM) or with single mismatches at each position of the p53 motif positions.
The peaks from the 41 activated p53 data sets were merged to generate one master list of 54 947 peaks for further analysis (we considered only peaks from canonical chromosomes 1–22 and XY) (Supplementary Table ST2). Of these, 23% (12 885 peaks) contain a p53-like motif consisting of the commonly described p53 consensus 20-mer with a 0–15 bp spacer or with only a half-site. We identified ∼770 000 corresponding sequences that were not bound (see below). Thus, it appears that in this extensive sampling of potential sites only ∼1.6% of the p53-like sequences in the genome, at most, have the potential to bind activated p53, which suggests multiple factors are required to enable binding.
We then asked about peaks that appeared in at least two (≥2) independent data sets. This analysis resulted in 19 088 peaks, of which 48% (9126) contained a p53-like motif (Supplementary Table ST2). As the incidence of a given peak increases across data sets, the likelihood that it contains a p53-like motif also increases, implying less influence of other factors on binding at sites bound in multiple data sets. For example, increasing the minimum number of data sets that identified a given peak to 5 or 11 increased the percent of peaks with p53-like motifs to 82% (4923/6001) or 96% (2324/2409), respectively (Figure 2B). Increasing the incidence to at least 20 data sets resulted in 99% (1000/1011) with a p53-like motif. The 1011 p53 peaks that appear in ≥20 data sets bind with a similar distribution across the genome regardless of cell type, p53 activating agent, or time after activation (Supplementary Table ST2). However, hierarchical clustering of peaks in ≥20 data sets show clustering by cell type and not by treatment or time (Supplementary Figure SF7).
In the parallel 17 control p53 data sets, we found 16 209 p53 peaks, of which 2532 are peaks that appear in ≥2 data sets (Supplementary Table ST3). Of these, 68% (1727 peaks) contain a p53-like motif and are distributed disproportionately in genic regions: 43% TSS, 20% intragenic and 37% intergenic. Interestingly, 11% (111 peaks) of the activated-p53 peaks in ≥20 data sets are also pre-bound in at least eight control p53 data sets. Although the numbers are much smaller, the distribution of the 111 peaks is similar to that for the 2532 peaks: 34% TSS; 24% intragenic and 49% intergenic.
While many studies have investigated p53 binding in vitro and individual studies have addressed in vivo binding, our study provides opportunities to evaluate common features of human p53 binding in a variety of cell types and conditions. For example, the more frequently a peak appears across data sets the greater the ratio of ChIP reads to input reads at that peak. Thus, the fold-change of ChIP reads over input reads of peaks that appear in 75% or more of data sets is higher than that which appears in only 25 or 50% of data sets (Figure 2C) and correlates with the likelihood of containing a p53-like motif (Figure 2B). We generated a single p53 logo from our list of peaks in ≥2 data sets (Figure 2D) that is comparable to other published logos, which include a variety of conditions and cells, and displays a strong bias for binding to motifs with no spacer.
We also examined the specific impact of sequence variation, spacers between decamer motifs, as well as half-sites on p53 binding in vivo, by looking at the relative frequencies of bound sites. Assuming an equal distribution of ATs and GCs, we estimate that the 3 × 109 bp human genome should contain ∼732 400 perfect p53 half-sites (10 bp) and 179 perfect full-sites (defined here as an exact match to the 20 bp consensus sequence RRRCWWGYYYRRRCWWGYYY). A scan of the genome sequence gave a value for perfect half-sites similar to that expected (784 101), but the number of perfect sites with no spacer, or a one-base spacer between the two decamers was 550 and 373, respectively, i.e. two–three-fold higher than expected, suggesting possible selection. The 550 perfect 20-mer sites with zero spacers (20-mer, 0-spacer) were distributed across the genome in a manner similar to all sites: 15% were near a TSS, 26% were intragenic, while 59% were intergenic. There was a strong preference for binding to perfect 20-mer, 0-spacer sites: 75% (410/550) were bound in at least one data set (Figure 2E), and 70% (386 sites) were bound in at least two data sets. Importantly, among these perfect 20-mer, 0-spacer sites, nearly all (53/54) of those with CATG cores in both half-sites were bound in at least one data set.
For in vitro binding studies, the effects of increased spacer length on binding are variable (51,52). Here, we found that only 36%, or 133 of 20-mer (two perfect decamers) sites with a one-base spacer were bound in at least one data set; the fraction binding in at least two data sets was 31% (115 sites). The greatly reduced binding observed with a one-base spacer suggests an intimate relationship between sequence organization and in vivo binding. Increasing the spacer to two or more bp reduces the frequency of in vivo binding to a range of just ∼2–11% (Figure 2E), suggesting that the sites are detected similarly to half-sites (2%), consistent with our previous report (4). Thus, presence of a spacer strongly influences the probability of p53 binding. Our meta-analysis establishes p53 binding preferences in vivo that can be related to in vitro structural analyses of p53 binding (52–54).
The observation that perfect sequences with no spacers have a high likelihood of binding led us to explore the consequences of a single departure (i.e., mismatch) from the perfect consensus. The data presented in Supplementary Table ST4 provides a genomic approach to evaluating the contribution of each base to the potential for in vivo p53 binding. In essence, the frequency of bound sites for a given target sequence provides information on the relative impact of individual base changes on the direct binding of p53 to targets across the genome under a variety of conditions in many cell types. As described above, ∼75% of the perfect sites in the genome are bound in at least one data set. A mismatch (i.e. R to Y) in position 1 and 2 or in the mirror image direction 20 and 19 (1→RRRCWWGYYY← 10 11→RRRCWWGYYYRRR←20) reduced the binding of these singly mismatched sites to ∼55–60% (as compared to ∼75%), whereas R to Y changes at 3 and 18 reduced the sites bound to ∼35% (Figure 2F). As expected, changes in the core C and G positions (4 and 7, or 17 and 14), which are key contact positions for p53, reduced binding much more, ∼11% and ∼3%, respectively. Overall this unique approach to assessing the influence of a mismatch on binding in vivo in natural chromosomal contexts provides insights into the relationship between the p53 protein and its preferred target sequences.
Over 52% (9962/19 088) of p53 peaks in ≥2 data sets do not contain a p53-like motif within the peak region. The binding of p53 to sites that lack a p53 motif could result from a variety of factors, including p53 interactions with other proteins bound to DNA such as other TFs (55) as well as to specific DNA structures (56). A search for known motifs in peaks that lack a p53 motif using HOMER identified sequences commonly bound by CTCF, and the bZIP, ETS, and Runx family of TFs (Supplementary Figure SF8). p53 has been shown to interact with some members of each family (e.g. ATF3, ETS2 and Runx1, respectively) (57–59). HOMER also identified 1350 peaks that contained a p53 motif, which we did not call, suggesting our method for identifying p53 motifs is conservative. In vitro, p53 binds various DNA structures in a non-sequence-specific manner. By way of example, we chose to examine p53 interactions with G-quadruplexes (G4s), which have been investigated in vitro (38,60). G4s form when single-stranded guanine-rich DNA sequences fold into stable four-stranded helical (non-B DNA) structures. G4s have been detected in the human genome in vitro and recently more than 10 000 have been mapped in vivo; these are enriched in gene regulatory regions and open chromatin (38,60). We found that 20% of all p53 peaks that appear in ≥2 data sets are within 1 kb of a G4 structure that was identified in vivo (P-value < 0.0001; Supplementary Table ST5). Of the p53 peaks that do not contain a p53 motif, ∼28% are near G4s, which is consistent with the view that p53 may be able to bind G4 structures based on in vitro studies (61,62).
p53 binding and potential for transcription
Using ingenuity pathway analysis (IPA), we addressed pathways that might be regulated by p53 based on p53 bound peaks in ≥2 data sets that contained a p53-like motif near a TSS. As expected, we found 93 genes known to be involved in p53 signaling, molecular mechanisms of cancer, and cell cycle regulation that could be directly targeted by p53. In addition to these well-known pathways, we uncovered additional pathways that include G-protein coupled receptor (GPCR) signaling, neurotransmitters and other nervous system signaling, growth factor signaling, cellular immune response, and cardiovascular signaling (Supplementary Figure SF9). Thus, there is the potential for stress-responsive p53 to broadly influence cellular biology through direct targeting of a wide range of genes. This finding supports current views that p53 functions as more than just a tumor suppressor (5).
We also examined opportunities for DNA-bound p53 to influence RNA expression by means other than direct transcription. Recently, p53 transcriptional regulation has been expanded to include microRNAs (miRNA) (63–65) and other non-coding RNAs (ncRNA), such as long intergenic non-coding RNAs (lncRNAs) and enhancer RNAs (eRNAs) (8,20,66,67), which together are five times more numerous than protein coding genes. miRNAs regulate gene expression post-transcriptionally by RNA interference. The lncRNAs, while less well understood, are emerging as key regulators of diverse cellular processes. A p53 peak in ≥2 data sets was found within 5 kb of ∼12% of all annotated ncRNA regions (see Materials and Methods), such that 256 miRNA and 973 lncRNA coding sequences were near a p53 peak in ≥2 data sets (Supplementary Table ST5). In addition, we identify the following putative p53 targets near p53 peaks that appear in ≥20 data sets: miR-2116, -4774, -6512, -6867 and lnc-ADSS-2, -HMP19.1-1, -C9orf69-2, -ATAD1-1. While the extent to which p53 may transcriptionally regulate these ncRNAs and downstream gene expression remains to be determined, our findings with bound p53 suggest a broad potential impact of p53 on expression dynamics across the genome.
We extended our p53 binding analysis to sites that might associate with enhancers or enhancer RNAs. p53 peaks in ≥20 data sets were significantly enriched in transcriptional enhancers based on chromatin state data from ENCODE (41) (Supplementary Figure SF10). In addition, 6820 (∼16%) of the ∼43 000 annotated active enhancers in the human genome (37) are within 5 kb of a p53 peak in ≥2 data sets (Supplementary Table ST5). Thus, p53 also may bind preferentially to enhancers with the consequent activation of transcription of distant genes through chromatin looping, supporting selection for its broad role in transcription. Indeed, a recent study showed that p53 is stably bound to hundreds of enhancers including many that are within inaccessible chromatin regions in normal human cells (68).
The transcriptome within p53 ChIP-seq gene expression data sets
We addressed changes in expression within the p53 ChIP-seq gene expression data sets. Of the 41 data sets that passed all filters in our p53 ChIP-seq meta-analysis, only 22 had associated gene expression data (microarray or RNA-seq). However, of these only 16 contained replicate samples and met our criteria for inclusion in our meta-analysis (see Materials and Methods). Among these 16 data sets, 7880 genes were differentially expressed in at least one data set as compared to controls following p53 activation (Supplementary Table ST6). This information also has been incorporated into the human p53 BAER hub. Of these differentially expressed genes (DEGs), 3613 appear in ≥2 independent data sets. Among these p53-associated DEGs in ≥2 data sets, 47% are solely upregulated, 38% are solely downregulated, and 15% are either upregulated or downregulated across multiple data sets.
p53 cistrome–p53 binding targets with associated changes in gene expression
While independent binding and transcriptome analyses can identify genes potentially subject to transcriptional regulation by p53, we wanted to identify the universe of gene-specific p53 binding targets with a p53 motif that had associated changes in gene expression. Combining the information above for p53 bound peaks with the activated p53 transcriptome, we extended our meta-analysis to genes at which p53 bound at a p53 motif near the TSS and for which associated changes in expression were described in the same study. We refer to these DEGs, which are potential direct p53 targets, as p53 cistrome genes. Alternatively, they could be indirect p53 targets (possibly p53 associated with another TF or structure, as described above).
Consistent with the previous analysis of Riley et al., which identified p53 binding sites that might directly affect gene expression (3), we identified p53 binding peaks near a TSS. In the 16 data sets, a total of 16 882 peaks were found near a TSS, and 7880 genes were differentially expressed as noted above (Supplementary Table ST7). Only 11%, or 1840 peaks, were associated with differential gene expression in at least one data set. This number corresponds to 1291 unique p53-associated DEGs, since some genes were associated with several peaks. We then identified the p53-associated DEGs that contained a p53-like motif based on a modified p53 scan algorithm (4). This analysis gave 943 unique p53 cistrome genes corresponding to ∼73% of the DEGs. HOMER analysis within peaks that did not contain a p53 motif but also resulted in differential gene expression found sequences commonly bound by the bZIP family of TFs (P-value 1 × 10−13). However, manual evaluation of each of the remaining 27% of peaks (348 genes) indicated that most (307 genes) contained a p53-like motif, such as a half- or three-quarter-site or sites with multiple mismatches from the perfect consensus. We also analyzed within +/- 1kb of the TSS or within the p53 peak region (∼200 bp) of the cistrome genes for other TF binding sites. Only the p53 motif was found in both analyses. Additionally, the bZIP family of TFs (P-value 1 × 10−31), such as Fra1, JunB, BATF, Fra2, Atf3, AP-1, Fosl2, was also found when the analysis is done within the p53 binding peak regions. Among the 943 p53 cistrome genes, 70% are upregulated, 27% are downregulated, and 3% were either upregulated or downregulated (Figure 3A and Supplementary Table ST8). The association between p53 peaks, DEGs, and cistrome genes for each of the 16 data sets are shown in Supplementary Figure SF11. To make this cistrome analysis more accessible, we have developed an interactive R Shiny application of the data presented in Supplementary Table ST8a.
Figure 3.

The p53 cistrome. (A) Distribution of the 943 p53 cistrome genes with respect to direction of expression regulation. (B) Distance of p53 motifs from TSS of cistrome genes. The ‘-0’ category corresponds to -1 to -999 bp and the ‘0’ corresponds to 1–999 bp from the TSS. The distribution for p53 motifs beyond 10 kb downstream are not shown. The inset summarizes the distribution of p53 motifs for the 943 cistrome targets and for the 2375 genes, with a p53 motif containing peak in ≥2 data sets near the TSS, that had no change in expression. (C) Overlap of p53 cistrome genes with previously identified p53 targets. (D) IPA signaling pathways of the 713 potential new p53 cistrome target genes (with a Fisher's Exact test P-value < 1 × 10−3).
We also examined whether p53 cistrome genes were more likely to contain a perfect p53 consensus motif. Even though 54 perfect motifs located near a TSS were frequently bound across data sets, only 25 are associated with changes in expression, corresponding to 24 cistrome genes (the perfect motif with one spacer has a similar ratio (11/22)) (Supplementary Table ST8). This result establishes that even with an ideal sequence that is bound by p53, the likelihood of changes in expression is similar regardless of the p53 sequence motif, suggesting the importance of factors in addition to binding that influence expression. Expression characteristics of the cistrome genes with a perfect motif were similar to the expression characteristics of the rest of the cistrome genes: 14 were upregulated, 8 were downregulated and 2 were upregulated or downregulated in different data sets.
We then asked how p53 bound motifs for the 943 cistrome genes were distributed with respect to distance from the TSS compared to the 2375 genes, with a p53-motif-containing peak in ≥2 data sets, in the same region (from -5 kb through the 1st intron) that did not exhibit a change in expression (Figure 3B). For the cistromes, similar numbers of p53 motifs were found within 5 kb upstream or downstream of the TSS. Among all cistromes, 73% have a motif within this region; the remainder were further downstream of the TSS. This distribution is similar to that reported by Riley et al. (3). Moreover, the majority of bound motifs fell within 1 kb upstream or downstream of the TSS. However, bound motifs near the TSS for genes that did not exhibit a change in expression upon p53 activation exhibited a very similar distribution, again emphasizing the conclusion that binding is not sufficient for the induction (or possible repression) of transcription.
No p53 cistrome genes were found across all 16 data sets. While p53 was found to bind 30 sites near a TSS in all 16 data sets, none of these sites resulted in changes in expression in all data sets. Yet for three of these sites, a change in expression was found in 14 of the 16 data sets. Thus, although p53 binding and associated expression is frequent across data sets, binding does not always result in changes in expression.
Among the 943 cistrome genes, 713 are newly identified p53 targets (Figure 3C). The remaining 230 (22%) correspond to most of the previously identified p53 target genes (69). Of the 713 new genes, 441 exhibited enhanced expression, 252 showed repression and 20 had enhanced or repressed expression in different data sets (Supplementary Table ST8). These 713 genes fall into the following broad biological categories based on IPA analysis: neurotransmitters and other nervous system signaling, cellular immune response, GPCR signaling, cardiovascular signaling, cellular stress and injury, and cancer and cell junction signaling (Figure 3D). We saw no apparent differences in pathways between the upregulated and downregulated cistromes. As examples, upregulated p53 targets include the RhoA inhibitor gene RIPOR2 (or FAM65B) that is involved in neutrophil polarization (70) and PDLIM1, a negative regulator of actin cytoskeleton organization (71) and more recently of NF-κB-mediated inflammatory responses (72). Downregulated p53 cistrome genes include the adaptor protein gene APBB2 that enhances the generation of the amyloidogenic peptide Aβ associated with Alzheimer's disease (73) and the GPHN gene coding for a scaffold protein (Gephyrin) in the postsynaptic protein network of inhibitory synapses (74).
We asked if there is a core cistrome signature that identifies common, directly responsive p53 target genes. As shown in Figure 4A, 28 p53 core cistrome signature genes were identified as appearing in at least eight of the 16 data sets. Unlike for the bulk of the p53 cistrome genes where a third are downregulated, the core cistrome signature genes only exhibited upregulation. Eight of these genes (CDKN1A, DCP1B, DDB2, MDM2, PLK2, RPS27L, TNFRSF10B and TRIAP1) are bound in p53 control cells as well. We suggest that these genes may be pre-bound for rapid upregulation after p53-induction regardless of mechanism. Included in the 28 core cistrome signature genes responsive to p53 activation are genes involved in cell cycle, apoptosis, DNA repair, p53 feedback, metabolism and survival/proliferation. Additionally, we identify mRNA decay/protein degradation, immune, and motility genes.
Figure 4.

p53 core cistrome signature. (A) The p53 core cistrome signature (genes that appeared in eight or more of the 16 data sets that had both p53 binding and associated expression) is composed of genes functioning in multiple effector networks. The core cistrome signature genes were separated into functional categories based on their GeneCards summary. (B) A word cloud representation of 124 cistrome genes that appear frequently (common in at least four data sets). The size of each word indicates its frequency of overlap across data sets.
DISCUSSION
Although p53 may be the most extensively studied TF, much about its function and its targets remains unknown. Because of its importance as a human tumor suppressor and its other recently discovered functions (e.g. Figure 3D and 4A), identifying the universe of p53 direct transcriptional targets is clinically relevant, especially since many of these may have therapeutic value. Over the past seven years, several studies have used genome-wide, next-generation sequencing approaches to identify p53 binding sites in the human genome, and in some studies genome expression also was analyzed. The individual studies have had limited success in identifying new, direct p53 transcriptional targets. Subsequent meta-analyses that have compared conclusions between studies (reviewed in (69)) have resulted in little increase in p53 cistromes or underlying mechanisms. We have taken a different, more rigorous approach by reanalyzing all the raw data from the individual ChIP-seq studies using a single analysis workflow, then combining information using a common set of criteria. The 16 studies in which both binding and associated expression could be compared were similarly analyzed. Our approach provides a uniform analysis of the large body of p53 binding and expression information.
Factors influencing p53 binding
Our meta-analysis information provides opportunities to address factors that influence in vivo p53 binding at endogenous sites and to compare with previously described in vitro interactions between p53 and various target sequences as determined kinetically (52,75,76) or by structural interactions (53,54). We found that the more frequently a site is bound across data sets, the more likely it contains a p53-like motif. Most sites bound by p53 contain at least one mismatch with respect to the perfect consensus sequence as suggested from previous studies (77). For instance, only 15% of our p53 binding sites that are seen in ≥20 p53 ChIP-seq data sets contain a perfect consensus, while the remaining 85% have at least one mismatch. However, even if a site has a perfect consensus sequence, p53 binding to the site is not guaranteed as 25% of the perfect sites that exist in the human genome do not show enriched ChIP-seq signal in any of the 41 data sets.
We addressed the influence of specific mismatches from consensus (see Supplementary Table ST4), role of core CWWG sequences, as well as proposed p53/DNA contacts. Nucleotide changes in the C and G core dramatically affect binding across data sets unlike mismatches at other positions. Interestingly, the in vivo differences in binding to a target with a single mismatch at the C (4 and 14) position vs. the G (7 and 17) position of the core CWWG in Figure 2F can be explained in terms of structural relations. Based on the findings of Kitayner et al. (78), mismatches at the core C and G positions highly impede DNA binding as these base pairs anchor p53 to the DNA via two strong interactions between the G base and Arg280. The differential effects (4,14 versus 7,17) could be related to the different DNA environments of the G/C bases (Shakked, personal communication). The four p53 core domains interacting with DNA have the shape of a parallelogram with two internal and two external p53 monomers across the DNA (referred to as A, C and B, D, respectively, in Kitayner et al. (53)). The internal monomers closer to the center of the DNA interact with the G/C base pairs at positions 7 and 14 and play a greater role in stabilizing the tetramer on the DNA relative to the external monomers that interact with the C/G base pairs at positions 4 and 17, which explains the greater reduction in binding of mismatches at 7,14 relative to 4,17.
We also explored possible reasons for binding to sites without a p53-like motif. Many could be due to crosslinking artifacts (especially if they do not appear in ≥2 data sets (79,80)), interaction with other chromatin-associated proteins, or binding to DNA structures such as G4s. As described in the clutch-like model of TF binding (81), molecules that undergo rapid turnover at non-motif sites that may be non-functional (called ‘treadmilling’) may become fixated during the ChIP-seq protocol. Quantitation of p53 after activation or stabilization (6) indicates that cell nuclei contain roughly a twentieth as many p53 dimers as there are half-sites (780 000) in the human genome. p53 likely spends the vast majority of its time in the nucleus closely associated with DNA, but only a fraction of this time is it bound specifically to its motif (82,83). As noted above, 52% of the p53 peaks in ≥2 data sets do not contain a p53 motif, and half of these are near a TSS. p53 is known to bind in vitro to several DNA structures, and it may well be that it spends longer in association with certain non-specific sequences or possibly is confined within chromatin structures during its search for specific binding sites. Thus, the existence of p53 peaks in ≥2 data sets without a motif but with similar ChIP-seq signal as peaks with a motif may not be surprising. Analysis of how p53 is bound at commonly bound sites should be possible using current mass spectrometry techniques to determine the bases and amino acid residues crosslinked.
p53 binding and transcription
Many questions about p53 and its transcriptional network are addressable through our meta-analysis and p53 BAER hub. For example, we investigated the influence of p53 motif and non-motif binding sites on expression as well as the potential effect of other p53 targets (such as lncRNAs and miRNAs) on gene expression, whether p53 is a direct transcriptional repressor, as well as the significance of the extensive p53 whole genome binding revealed by multiple ChIP-seq studies that, so far, has not been associated with changes in gene expression.
In addition to determining the role of sequence and spacers in binding (Figure 2E), our analysis of p53 binding sites allowed us to address a possible role for p53 bound sequences in expression across the genome. While the ability to bind would obviously influence the level of expression, there remains the question of whether the sequence itself, including spacers, might influence expression. Although the number of perfect sites (with no spacer) bound and near a TSS is not large, nearly half of them (25/54) are associated with a change of expression. As noted in Figure 2E, the likelihood of binding is greatly reduced by a spacer. However, associated expression for bound sites near a TSS does not appear to change. For a one base spacer 11/22 had associated expression changes. Summing up all the bound perfect motifs with 2–15 bp spacers near a TSS, we found ∼45% (17/38) are associated with expression changes. From this we propose that while spacer greatly affects binding, the likelihood of expression is independent of spacer (or spacer sequence). The results were similar when this approach was extended to the 943 cistrome genes where the p53 motifs contain multiple mismatches to the consensus. Among these, 449 had no spacer, 72 had a single spacer and for the remaining 422 cistrome genes they were approximately evenly distributed between p53 motifs containing 2–15 bp spacers (average 30 cistrome genes).
Based on our summary results, binding of p53 to a motif in a promoter region is not sufficient for transcription, as only 11% of all p53 bound sites near the TSS of genes result in differential expression. Additionally, the bound motifs near the TSS of genes that did not exhibit a change in expression upon p53 activation were distributed similarly to those of the cistrome genes, with respect to distance from the TSS. Overall, the incidence of bound motifs is greatly enhanced in the region within 1 kb of the TSS. Thus, the lack of correspondence between binding and expression suggests that additional context-dependent signals are required for a transcriptional response for many p53 regulated genes.
An example of the differences in binding versus associated transcriptional program is demonstrated by comparing data sets from doxorubicin (DXR)- versus nutlin-3-treated cells. For DXR treatment, our meta-analysis contains 10 binding data sets, of which six included gene expression data sets, compared with 10 and 5, respectively, for nutlin treatment. While there is a 70% overlap between the DXR- and nutlin-3-treated peaks in ≥2 data sets, only 36% of the transcriptome DEGs in ≥2 data sets and 22% of cistrome changes were common between these two treatments (Supplementary Table ST9). Thus, each condition/cell type can be considered as having its own group of p53 cistrome targets.
Our approach also provides the opportunity to examine DEGs that do not have an associated p53 motif target sequence. Gene expression analysis following p53 activation from 16 data sets revealed 3613 p53-associated DEGs that appear in ≥2 independent data sets. Only 25% of these genes have a p53 binding site near the TSS leaving ∼2700 genes with no direct connection to p53 regulation, as defined by binding near the TSS of the gene. It is possible that p53 may regulate some of these 2700 genes, but via mechanisms other than binding near their TSS. One such mechanism may involve regulation of other expression regulatory nodes or ncRNAs. p53 is able to actively influence transcription from distal sites containing p53 binding sequences that are located in regions classified as closed chromatin based on chromatin histone modifications profiles (12) as well as at active enhancers (84). Our meta-analysis reveals that p53 binds and may regulate 12–15% of all ncRNAs, namely miRNAs, lncRNAs, and eRNAs or enhancers. Therefore, future studies to determine whether p53 regulates the expression of the ncRNAs that p53 binds and the subsequent gene targets of these ncRNAs will greatly expand the p53-influenced network/universe.
Since p53 peaks that appear in ≥20 data sets are enriched in enhancer marks and p53 peaks in ≥2 data sets are near 15% of active enhancers, understanding the 3D architecture of the genome and how chromosomal folding and looping bring distant enhancers in close contact with promoters of target genes may also increase the p53 responsive universe. Thus, chromosome conformation capture studies to map topologically associating domains (TADs) in various cell types and conditions to determine the neighborhood of target genes that p53 enhancers could target would greatly benefit the p53 field. While enhancers usually regulate expression within TADs and not between neighboring TADs (85), it would be interesting to examine whether p53 gain-of-function cancer mutants could ignore boundary regions to regulate across neighboring TADs (86). Therefore, mapping the spatial configuration of the genome may reveal that some of these 2700 genes are additional p53 direct targets.
Recent studies suggest p53 binding may play a role in opening chromatin (12,87). p53 recognition and interaction with genome regions associated with epigenetic changes such as CpG islands may induce global redistribution of the p53 binding and reprogram its transcriptional network depending on the context of the stress detected. In our meta-analysis, we found that approximately a third of the p53 cistrome targets have a peak within or near CpG islands. Future studies could determine if accessible chromatin is required for p53 binding or p53-mediated transcriptional regulation and whether this affects target gene transcription in a cell- or treatment-dependent manner. Such studies will facilitate interpretations of the p53-dependent tumor suppression program as well as other emerging functions in the context of dynamic chromatin and potentially would reveal novel TF-epigenomic global patterns.
p53 cistrome genes
In this study, we establish that the direct, targeted influence of p53 on gene expression is much larger than previously indicated. We identified 943 human p53 cistrome genes, which corresponds to a nearly three-fold increase in the number of p53 target genes (69). The increase is due in part to our method of p53 motif analysis that goes beyond the more traditional approaches that primarily employ 20 base position weight matrix methods to identify p53 binding. We also include half-sites as well as half-sites separated by up to 15 bases (4). Recalling that all 16 expression studies were rigorously analyzed using the same criteria (see Materials and Methods), the DEGs in each study represent potential valid p53 regulated genes regardless of the number of studies in which they appear.
Among the cistrome genes, 659 (70%) are upregulated, 256 (27%) are downregulated and 28 can be either upregulated or downregulated. Downregulation has been proposed to be through an indirect mechanism. Recently, directly bound p53 was proposed to function only as an activator (88,89). Repression by activated p53 was suggested to occur through the DREAM repression complex (DP, RB-like, E2F4, and MuvB) in a manner that requires p21 induction (90) or through the miRNA-mediated pathway (65) rather than by direct binding of p53 to motif targets. Of the p53 transcriptome genes in ≥2 data sets in our study, 533 (15%) correspond to DREAM targets (Supplementary Figure SF12A) identified by Fischer et al., (90). Consistent with that study, 88% of the 533 genes were downregulated. Thus, repression related to p53 activation can be indirect through the DREAM complex. Among the 943 cistrome genes, about one-fourth (256 genes) were only downregulated. However, only 37 of the p53 downregulated cistrome genes are DREAM targets (Supplementary Figure SF12B). In addition, HOMER analysis for sites within +/- 1 kb of the TSS or within the p53 peak region of the 256 repressed cistrome genes did not identify a DREAM-associated motif.
The mechanism(s) of downregulation of repressed cistrome genes requires further investigation, since all of these genes contain p53 binding at a p53 motif. We note that there is a difference in the frequency of downregulated vs. upregulated cistromes. While none of the 256 repressed cistrome genes appeared in more than three studies, 122 upregulated p53 cistrome genes were found in four or more studies. Furthermore, 3% of the p53 cistrome targets were found to be activated in some experimental conditions while repressed in others, suggesting repression may be cell or treatment specific. Contrary to what has been proposed, from our analysis we cannot exclude that p53 mediates direct gene repression at sites with or without the p53 motif. Repression could also be due, for example, to recruitment of co-repressors, or by displacement of specific activators from promoters due to the presence of overlapping binding sites.
The p53 cistrome is likely modulated by the nature and duration of the stress signal, the cell type, other TFs and cofactors, chromatin, etc. Consequently, other factors may be required to coordinate and cooperate with p53 to turn on the transcriptional program. For example, the cistrome compilation from our meta-analysis contains several data sets employing various exposure times to a common agent, such as DXR (six data sets) or nutlin-3 (five data sets), or collected at the same time point (24 hr) but treated with a variety of agents (ten data sets). Identification of those p53 cistrome genes appearing in at least 50% of the data sets identified 84 cistrome targets after DXR treatment, 38 targets after nutlin-3 treatment, and 21 cistrome targets 24 hr after treatment with various agents (Supplementary Figure SF13 and Supplementary Table ST8). A similar approach can be used to compare normal vs. cancer cells. Thus, there appear to be cistrome genes that are common (Figure 4B) as well as specific to each treatment condition (Supplementary Figure SF14).
From the many cistrome genes, we identified a p53 core cistrome signature of 28 genes regardless of the experimental conditions, suggesting a more universal functional importance of the corresponding genes (Figure 4A) in the p53 transcriptional network. The observation that all of them are upregulated in response to activated p53 may indicate a special biological role, possibly a more rapid response for upregulation versus downregulation, or a need to coordinate pathways. Our meta-analysis also allowed us to confirm that p53 binds cistrome genes in the absence of external stress stimuli (no treatment or DMSO), suggesting that these sites are pre-bound or ‘poised’ for rapid regulation after p53-induction to stress. p53 binding at some of these sites also may influence the basal expression of nearby genes (91). Included among the 28 core cistrome signature genes eight were also poised, including CDKN1A (92,93). This would be consistent with the idea of a rapid response by core signature genes.
Many factors in p53 biology have been evolutionarily selected that are expected to influence the p53 network beyond just tumor suppression (94). As summarized in this meta-analysis, p53 can induce qualitatively different programs that produce various biological outcomes depending on cell type and stimulus. For fully successful biological responses to p53, crosstalk is required between input and output pathways. An emerging number of pathways modulated by p53 also may modulate p53, for example, the Hippo pathway, DREAM repression, and NF-κB signaling (90,95–97). Thus, a core set of genes that touch the various pathways as a p53 super-hub (94) could provide a coordinated and integrated response system across biological processes. Knowledge of the 28 core cistrome signature genes, the overall p53 cistromes, as well as the p53 network has several utilities including assessing p53 determined responses across tissues and exposures in clinical studies and the identification of therapeutic targets.
Our approach has led to the identification of new potential p53 transcriptional targets involved in processes not immediately related to classical p53 outcomes, for example, the immune response. Furthermore, in this work we presented the first p53 binding and associated transcription in human T lymphocytes, a central component of immune responses. We identify 50 potential cistrome genes directly involved in cellular immune processes, including CD14, IL12A, IL33, and TRAF5. Our recent studies have emphasized the influence of p53 in modulating the immune system, which defends against external and internal threats as well as tumorigenesis. DNA damage can trigger p53 dependent inflammatory responses that contribute and help orchestrate the clearance of tumor cells triggering a senescence program and influencing tumor suppression. Activation of p53 leads to the transcriptional regulation of two of the major innate immune gene families: APOBEC3 and Toll-like receptor (98,99). We also show that p53 and the immune master regulator NF-κB coregulate proinflammatory gene responses in human macrophages (100). Others have reported that p53 transcriptional targets include several interferon stimulated immune genes such as IRF5, IRF9 and ISG15, as reviewed in (101). Therefore, the existence of p53 immune cistrome genes reveals p53 as a central mediator and amplifier of the global innate immune responses and highlights its important physiological role in the immune system, providing a new dimension to the broad role that p53 plays in human biology.
Overall, our meta-analysis provides the opportunity to identify signatures for p53 binding and for associated expression, thereby enhancing our understanding of similarities and differences in directly driven changes, as well as potential changes, in the p53 universe. The resulting human p53 BAER hub we developed, which can be merged with any UCSC data, provides ready access to many aspects of the human p53 universe including p53 binding, changes in expression as well as cistrome genes. Additionally, we created a p53 analysis page on the ORIO web application (102) and a p53 cistrome R Shiny application to provide a resource for users to interactively analyze and compare p53 ChIP-seq and cistrome data, respectively. Applying this information to biological networks, we identified new roles for p53 in several pathways that go beyond the traditional view as guardian of the genome. There is a commonality of bound sites and cistromes across normal and cancer cells as well as p53 activating agent or time after activation. Our analysis has created a basal and activated p53 binding map of the human genome. The information can be used to test concepts developed from in vitro studies. Questions that address the influence of sequence and chromosome structure on p53 binding at endogenous sites are now accessible.
DATA AVAILABILITY
The human p53 Binding And Expression Resource (BAER) hub (https://orio.niehs.nih.gov/ucscview/nguyen/p53BAER/hub.txt) can be accessed on the UCSC Genome Browser. ORIO web application of p53 ChIP-seq data and the p53 cistrome R Shiny application: https://www.niehs.nih.gov/research/resources/databases/p53/index.cfm. Sequencing and microarray data for the lymphocyte data sets have been deposited in GEO under accession GSE110370.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Martin Fischer, Douglas Bell and Xuting Wang for providing insightful comments and suggestions on the manuscript and Zippi Shakked for insights on p53 binding and sequence preferences. We also thank Raja Jothi and Johannes Freudenberg for initial assistance and discussions with the lymphocyte ChIP-seq analysis, and Jian-Liang Li for help in the development and implementation of the R Shiny app. We thank Kevin Gerrish, the Molecular Genomics Core and the Integrative Bioinformatics core at NIEHS for assistance, and the Clinical Research Unit for the recruitment and processing of blood from healthy volunteers.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Research Program of the National Institutes of Health/National Institute of Environmental Health Sciences (NIH/NIEHS) [Z01-ES065079 to M.A.R., Z01-ES101765 to L.L., Z01-ES102345 to P.R.B., Z01-ES103312 to D.C.F.]. Funding for open access charge: Intramural Research Program of the National Institutes of Health/National Institute of Environmental Health Sciences.
Conflict of interest statement. None declared.
REFERENCES
- 1. el-Deiry W.S., Kern S.E., Pietenpol J.A., Kinzler K.W., Vogelstein B.. Definition of a consensus binding site for p53. Nat. Genet. 1992; 1:45–49. [DOI] [PubMed] [Google Scholar]
- 2. Funk W.D., Pak D.T., Karas R.H., Wright W.E., Shay J.W.. A transcriptionally active DNA-binding site for human p53 protein complexes. Mol. Cell. Biol. 1992; 12:2866–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Riley T., Sontag E., Chen P., Levine A.. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008; 9:402–412. [DOI] [PubMed] [Google Scholar]
- 4. Menendez D., Nguyen T.A., Freudenberg J.M., Mathew V.J., Anderson C.W., Jothi R., Resnick M.A.. Diverse stresses dramatically alter genome-wide p53 binding and transactivation landscape in human cancer cells. Nucleic Acids Res. 2013; 41:7286–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kastenhuber E.R., Lowe S.W.. Putting p53 in context. Cell. 2017; 170:1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang Y.V., Wade M., Wong E., Li Y.-C., Rodewald L.W., Wahl G.M.. Quantitative analyses reveal the importance of regulated Hdmx degradation for p53 activation. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:12365–12370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Williams K., Christensen J., Rappsilber J., Nielsen A.L., Johansen J.V., Helin K.. The histone lysine demethylase JMJD3/KDM6B is recruited to p53 bound promoters and enhancer elements in a p53 dependent manner. PLoS One. 2014; 9:e96545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Younger S.T., Kenzelmann-Broz D., Jung H., Attardi L.D., Rinn J.L.. Integrative genomic analysis reveals widespread enhancer regulation by p53 in response to DNA damage. Nucleic Acids Res. 2015; 43:4447–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McDade S.S., Patel D., Moran M., Campbell J., Fenwick K., Kozarewa I., Orr N.J., Lord C.J., Ashworth A.A., McCance D.J.. Genome-wide characterization reveals complex interplay between TP53 and TP63 in response to genotoxic stress. Nucleic Acids Res. 2014; 42:6270–6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Botcheva K., McCorkle S.R., McCombie W.R., Dunn J.J., Anderson C.W.. Distinct p53 genomic binding patterns in normal and cancer-derived human cells. Cell Cycle. 2011; 10:4237–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kirschner K., Samarajiwa S.A., Cairns J.M., Menon S., Pérez-Mancera P.A., Tomimatsu K., Bermejo-Rodriguez C., Ito Y., Chandra T., Narita M. et al. Phenotype specific analyses reveal distinct regulatory mechanism for chronically activated p53. PLoS Genet. 2015; 11:e1005053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sammons M.A., Zhu J., Drake A.M., Berger S.L.. TP53 engagement with the genome occurs in distinct local chromatin environments via pioneer factor activity. Genome Res. 2015; 25:179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Aksoy O., Chicas A., Zeng T., Zhao Z., McCurrach M., Wang X., Lowe S.W.. The atypical E2F family member E2F7 couples the p53 and RB pathways during cellular senescence. Genes Dev. 2012; 26:1546–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zeron-Medina J., Wang X., Repapi E., Campbell M.R., Su D., Castro-Giner F., Davies B., Peterse E.F.P., Sacilotto N., Walker G.J. et al. A polymorphic p53 response element in KIT ligand influences cancer risk and has undergone natural selection. Cell. 2013; 155:410–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang B., Niu D., Lam T.H., Xiao Z., Ren E.C.. Mapping the p53 transcriptome universe using p53 natural polymorphs. Cell Death Differ. 2013; 21:521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Akdemir K.C., Jain A.K., Allton K., Aronow B., Xu X., Cooney A.J., Li W., Barton M.C.. Genome-wide profiling reveals stimulus-specific functions of p53 during differentiation and DNA damage of human embryonic stem cells. Nucleic Acids Res. 2014; 42:205–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rashi-Elkeles S., Warnatz H.-J., Elkon R., Kupershtein A., Chobod Y., Paz A., Amstislavskiy V., Sultan M., Safer H., Nietfeld W. et al. Parallel profiling of the transcriptome, cistrome, and epigenome in the cellular response to ionizing radiation. Sci. Signal. 2014; 7:rs3. [DOI] [PubMed] [Google Scholar]
- 18. Botcheva K., McCorkle S.R.. Cell context dependent p53 genome-wide binding patterns and enrichment at repeats. PLoS One. 2014; 9:e113492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chang G.S., Chen X.A., Park B., Rhee H.S., Li P., Han K.H., Mishra T., Chan-Salis K.Y., Li Y., Hardison R.C. et al. A comprehensive and high-resolution genome-wide response of p53 to stress. Cell Rep. 2014; 8:514–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sánchez Y., Segura V., Marín-Béjar O., Athie A., Marchese F.P., González J., Bujanda L., Guo S., Matheu A., Huarte M.. Genome-wide analysis of the human p53 transcriptional network unveils a lncRNA tumour suppressor signature. Nat. Commun. 2014; 5:5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nikulenkov F., Spinnler C., Li H., Tonelli C., Shi Y., Turunen M., Kivioja T., Ignatiev I., Kel A., Taipale J. et al. Insights into p53 transcriptional function via genome-wide chromatin occupancy and gene expression analysis. Cell Death Differ. 2012; 19:1992–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Janky R., Verfaillie A., Imrichová H., Van de Sande B., Standaert L., Christiaens V., Hulselmans G., Herten K., Naval Sanchez M., Potier D. et al. iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput. Biol. 2014; 10:e1003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Koeppel M., van Heeringen S.J., Kramer D., Smeenk L., Janssen-Megens E., Hartmann M., Stunnenberg H.G., Lohrum M.. Crosstalk between c-Jun and TAp73α/β contributes to the apoptosis-survival balance. Nucleic Acids Res. 2011; 39:6069–6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schlereth K., Heyl C., Krampitz A.M., Mernberger M., Finkernagel F., Scharfe M., Jarek M., Leich E., Rosenwald A., Stiewe T.. Characterization of the p53 cistrome–DNA binding cooperativity dissects p53’s tumor suppressor functions. PLoS Genet. 2013; 9:e1003726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smeenk L., van Heeringen S.J., Koeppel M., Gilbert B., Janssen-Megens E., Stunnenberg H.G., Lohrum M.. Role of p53 serine 46 in p53 target gene regulation. PLoS One. 2011; 6:e17574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D.. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Raney B.J., Dreszer T.R., Barber G.P., Clawson H., Fujita P.A., Wang T., Nguyen N., Paten B., Zweig A.S., Karolchik D. et al. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics. 2014; 30:1003–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Langmead B., Trapnell C., Pop M., Salzberg S.L.. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kent W.J., Zweig A.S., Barber G., Hinrichs A.S., Karolchik D.. BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics. 2010; 26:2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jothi R., Cuddapah S., Barski A., Cui K., Zhao K.. Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 2008; 36:5221–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bailey T.L., Gribskov M.. Combining evidence using P-values: application to sequence homology searches. Bioinformatics. 1998; 14:48–54. [DOI] [PubMed] [Google Scholar]
- 33. Bailey T.L., Elkan C.. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994; 2:28–36. [PubMed] [Google Scholar]
- 34. Smeenk L., van Heeringen S.J., Koeppel M., van Driel M.A., Bartels S.J., Akkers R.C., Denissov S., Stunnenberg H.G., Lohrum M.. Characterization of genome-wide p53-binding sites upon stress response. Nucleic Acids Res. 2008; 36:3639–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kozomara A., Griffiths-Jones S.. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014; 42:D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cabili M.N., Trapnell C., Goff L., Koziol M., Tazon-Vega B., Regev A., Rinn J.L.. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011; 25:1915–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Andersson R., Gebhard C., Miguel-Escalada I., Hoof I., Bornholdt J., Boyd M., Chen Y., Zhao X., Schmidl C., Suzuki T. et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014; 507:455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hänsel-Hertsch R., Beraldi D., Lensing S.V., Marsico G., Zyner K., Parry A., Di Antonio M., Pike J., Kimura H., Narita M. et al. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016; 48:1267–1272. [DOI] [PubMed] [Google Scholar]
- 39. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B., Zhang X., Wang L., Issner R., Coyne M. et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011; 473:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ernst J., Kellis M.. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 2010; 28:817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Irizarry R.A., Bolstad B.M., Collin F., Cope L.M., Hobbs B., Speed T.P.. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003; 31:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Irizarry R.A., Hobbs B., Collin F., Beazer-Barclay Y.D., Antonellis K.J., Scherf U., Speed T.P.. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003; 4:249–264. [DOI] [PubMed] [Google Scholar]
- 44. Rao P.V. Single Factor Studies: Comparing Means and Determining Sample Sizes. Statistical Research Methods in the Life Sciences. 1997; 1:North Scituate: Duxbury Press; 352–360. [Google Scholar]
- 45. Benjamini Y., Hochberg Y.. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Statist. Soc. B. 1995; 57:289–300. [Google Scholar]
- 46. Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M. et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013; 41:D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Edgar R., Domrachev M., Lash A.E.. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002; 30:207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Trapnell C., Pachter L., Salzberg S.L.. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009; 25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Anders S., Pyl P.T., Huber W.. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Anders S., Huber W.. Differential expression analysis for sequence count data. Genome Biol. 2010; 11:R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Noureddine M.A., Menendez D., Campbell M.R., Bandele O.J., Horvath M.M., Wang X., Pittman G.S., Chorley B.N., Resnick M.A., Bell D.A.. Probing the functional impact of sequence variation on p53-DNA interactions using a novel microsphere assay for protein-DNA binding with human cell extracts. PLoS Genet. 2009; 5:e1000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vyas P., Beno I., Xi Z., Stein Y., Golovenko D., Kessler N., Rotter V., Shakked Z., Haran T.E.. Diverse p53/DNA binding modes expand the repertoire of p53 response elements. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:10624–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kitayner M., Rozenberg H., Rohs R., Suad O., Rabinovich D., Honig B., Shakked Z.. Diversity in DNA recognition by p53 revealed by crystal structures with Hoogsteen base pairs. Nat. Struct. Mol. Biol. 2010; 17:423–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Petty T.J., Emamzadah S., Costantino L., Petkova I., Stavridi E.S., Saven J.G., Vauthey E., Halazonetis T.D.. An induced fit mechanism regulates p53 DNA binding kinetics to confer sequence specificity. EMBO J. 2011; 30:2167–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stiewe T., Haran T.E.. How mutations shape p53 interactions with the genome to promote tumorigenesis and drug resistance. Drug Resistance Updates. 2018; 38:27–43. [DOI] [PubMed] [Google Scholar]
- 56. Brázda V., Coufal J.. Recognition of local DNA structures by p53 protein. Int. J. Mol. Sci. 2017; 18:E375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Do P.M., Varanasi L., Fan S., Li C., Kubacka I., Newman V., Chauhan K., Daniels S.R., Boccetta M., Garrett M.R. et al. Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev. 2012; 26:830–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wu D., Ozaki T., Yoshihara Y., Kubo N., Nakagawara A.. Runt-related transcription factor 1 (RUNX1) stimulates tumor suppressor p53 protein in response to DNA damage through complex formation and acetylation. J. Biol. Chem. 2013; 288:1353–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yan C., Lu D., Hai T., Boyd D.D.. Activating transcription factor 3, a stress sensor, activates p53 by blocking its ubiquitination. EMBO J. 2005; 24:2425–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hänsel-Hertsch R., Di Antonio M., Balasubramanian S.. DNA G-quadruplexes in the human genome: detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol. 2017; 18:279–284. [DOI] [PubMed] [Google Scholar]
- 61. Adámik M., Kejnovská I., Bažantová P., Petr M., Renčiuk D., Vorlíčková M., Brázdová M.. p53 binds human telomeric G-quadruplex in vitro. Biochimie. 2016; 128–129:83–91. [DOI] [PubMed] [Google Scholar]
- 62. Petr M., Helma R., Polaskova A., Krejci A., Dvorakova Z., Kejnovska I., Navratilova L., Adamik M., Vorlickova M., Brazdova M.. Wild-type p53 binds to MYC promoter G-quadruplex. Biosci. Rep. 2016; 36:e00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Goeman F., Strano S., Blandino G.. MicroRNAs as key effectors in the p53 network. Int. Rev. Cell Mol. Biol. 2017; 333:51–90. [DOI] [PubMed] [Google Scholar]
- 64. Hattori H., Janky R., Nietfeld W., Aerts S., Madan Babu M., Venkitaraman A.R.. p53 shapes genome-wide and cell type-specific changes in microRNA expression during the human DNA damage response. Cell Cycle. 2014; 13:2572–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hermeking H. MicroRNAs in the p53 network: micromanagement of tumour suppression. Nat. Rev. Cancer. 2012; 12:613–626. [DOI] [PubMed] [Google Scholar]
- 66. Grossi E., Sánchez Y., Huarte M.. Expanding the p53 regulatory network: LncRNAs take up the challenge. Biochim. Biophys. Acta. 2016; 1859:200–208. [DOI] [PubMed] [Google Scholar]
- 67. Idogawa M., Ohashi T., Sasaki Y., Maruyama R., Kashima L., Suzuki H., Tokino T.. Identification and analysis of large intergenic non-coding RNAs regulated by p53 family members through a genome-wide analysis of p53-binding sites. Hum. Mol. Genet. 2014; 23:2847–2857. [DOI] [PubMed] [Google Scholar]
- 68. Younger S.T., Rinn J.L.. p53 regulates enhancer accessibility and activity in response to DNA damage. Nucleic Acids Res. 2017; 45:9889–9900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Fischer M. Census and evaluation of p53 target genes. Oncogene. 2017; 36:3943–3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gao K., Tang W., Li Y., Zhang P., Wang D., Yu L., Wang C., Wu D.. Front-signal-dependent accumulation of the RHOA inhibitor FAM65B at leading edges polarizes neutrophils. J. Cell Sci. 2015; 128:992–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Tamura N., Ohno K., Katayama T., Kanayama N., Sato K.. The PDZ-LIM protein CLP36 is required for actin stress fiber formation and focal adhesion assembly in BeWo cells. Biochem. Biophys. Res. Commun. 2007; 364:589–594. [DOI] [PubMed] [Google Scholar]
- 72. Ono R., Kaisho T., Tanaka T.. PDLIM1 inhibits NF-кB-mediated inflammatory signaling by sequestering the p65 subunit of NF-кB in the cytoplasm. Sci. Rep. 2015; 5:18327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chang Y., Tesco G., Jeong W.J., Lindsley L., Eckman E.A., Eckman C.B., Tanzi R.E., Guénette S.Y.. Generation of the β-amyloid peptide and the amyloid precursor protein C-terminal fragment γ are potentiated by FE65L1. J. Biol. Chem. 2003; 278:51100–51107. [DOI] [PubMed] [Google Scholar]
- 74. Tretter V., Mukherjee J., Maric H.-M., Schindelin H., Sieghart W., Moss S.J.. Gephyrin, the enigmatic organizer at GABAergic synapses. Front. Cell. Neurosci. 2012; 6:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Veprintsev D.B., Fersht A.R.. Algorithm for prediction of tumour suppressor p53 affinity for binding sites in DNA. Nucleic Acids Res. 2008; 36:1589–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Weinberg R.L., Veprintsev D.B., Bycroft M., Fersht A.R.. Comparative binding of p53 to its promoter and DNA recognition elements. J. Mol. Biol. 2005; 348:589–596. [DOI] [PubMed] [Google Scholar]
- 77. Menendez D., Inga A., Jordan J.J., Resnick M.A.. Changing the p53 master regulatory network: ELEMENTary, my dear Mr Watson. Oncogene. 2007; 26:2191–2201. [DOI] [PubMed] [Google Scholar]
- 78. Kitayner M., Rozenberg H., Kessler N., Rabinovich D., Shaulov L., Haran T.E., Shakked Z.. Structural basis of DNA recognition by p53 tetramers. Mol. Cell. 2006; 22:741–753. [DOI] [PubMed] [Google Scholar]
- 79. Baranello L., Kouzine F., Sanford S., Levens D.. ChIP bias as a function of cross-linking time. Chromosome Res. 2016; 24:175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Verfaillie A., Svetlichnyy D., Imrichova H., Davie K., Fiers M., Kalender Atak Z., Hulselmans G., Christiaens V., Aerts S.. Multiplex enhancer-reporter assays uncover unsophisticated TP53 enhancer logic. Genome Res. 2016; 26:882–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lickwar C.R., Mueller F., Hanlon S.E., McNally J.G., Lieb J.D.. Genome-wide protein-DNA binding dynamics suggest a molecular clutch for transcription factor function. Nature. 2012; 484:251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tafvizi A., Huang F., Fersht A.R., Mirny L.A., van Oijen A.M.. A single-molecule characterization of p53 search on DNA. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Tafvizi A., Mirny L.A., van Oijen A.M.. Dancing on DNA: kinetic aspects of search processes on DNA. Chemphyschem. 2011; 12:1481–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Léveillé N., Melo C.A., Rooijers K., Díaz-Lagares A., Melo S.A., Korkmaz G., Lopes R., Akbari Moqadam F., Maia A.R., Wijchers P.J. et al. Genome-wide profiling of p53-regulated enhancer RNAs uncovers a subset of enhancers controlled by a lncRNA. Nat. Commun. 2015; 6:6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Razin S.V., Ulianov S.V.. Gene functioning and storage within a folded genome. Cell. Mol. Biol. Lett. 2017; 22:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Valton A.-L., Dekker J.. TAD disruption as oncogenic driver. Curr. Opin. Genet. Dev. 2016; 36:34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bao F., LoVerso P.R., Fisk J.N., Zhurkin V.B., Cui F.. p53 binding sites in normal and cancer cells are characterized by distinct chromatin context. Cell Cycle. 2017; 16:2073–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Fischer M., Steiner L., Engeland K.. The transcription factor p53: not a repressor, solely an activator. Cell Cycle. 2014; 13:3037–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sullivan K.D., Galbraith M.D., Andrysik Z., Espinosa J.M.. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018; 25:133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Fischer M., Quaas M., Steiner L., Engeland K.. The p53-p21-DREAM-CDE/CHR pathway regulates G2/M cell cycle genes. Nucleic Acids Res. 2016; 44:164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zheltukhin A.O., Chumakov P.M.. Constitutive and induced functions of the p53 gene. Biochemistry. 2010; 75:1692–1721. [DOI] [PubMed] [Google Scholar]
- 92. Espinosa J.M., Verdun R.E., Emerson B.M.. p53 functions through stress- and promoter-specific recruitment of transcription initiation components before and after DNA damage. Mol. Cell. 2003; 12:1015–1027. [DOI] [PubMed] [Google Scholar]
- 93. Kaeser M.D., Iggo R.D.. Chromatin immunoprecipitation analysis fails to support the latency model for regulation of p53 DNA binding activity in vivo. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:95–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Aylon Y., Oren M.. The paradox of p53: what, how, and why. Cold Spring Harb. Perspect. Med. 2016; 6:a026328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Engeland K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018; 25:114–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Furth N., Aylon Y., Oren M.. p53 shades of Hippo. Cell Death Differ. 2018; 25:81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Menendez D., Shatz M., Resnick M.A.. Interactions between the tumor suppressor p53 and immune responses. Curr. Opin. Oncol. 2013; 25:85–92. [DOI] [PubMed] [Google Scholar]
- 98. Menendez D., Nguyen T.A., Snipe J., Resnick M.A.. The cytidine deaminase APOBEC3 family is subject to transcriptional regulation by p53. Mol. Cancer Res. 2017; 15:735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Menendez D., Shatz M., Azzam K., Garantziotis S., Fessler M.B., Resnick M.A.. The Toll-like receptor gene family is integrated into human DNA damage and p53 networks. PLoS Genet. 2011; 7:e1001360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lowe J.M., Menendez D., Bushel P.R., Shatz M., Kirk E.L., Troester M.A., Garantziotis S., Fessler M.B., Resnick M.A.. p53 and NF-kappaB coregulate proinflammatory gene responses in human macrophages. Cancer Res. 2014; 74:2182–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Munoz-Fontela C., Mandinova A., Aaronson S.A., Lee S.W.. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat. Rev. Immunol. 2016; 16:741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lavender C.A., Shapiro A.J., Burkholder A.B., Bennett B.D., Adelman K., Fargo D.C.. ORIO (Online Resource for Integrative Omics): a web-based platform for rapid integration of next generation sequencing data. Nucleic Acids Res. 2017; 45:5678–5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The human p53 Binding And Expression Resource (BAER) hub (https://orio.niehs.nih.gov/ucscview/nguyen/p53BAER/hub.txt) can be accessed on the UCSC Genome Browser. ORIO web application of p53 ChIP-seq data and the p53 cistrome R Shiny application: https://www.niehs.nih.gov/research/resources/databases/p53/index.cfm. Sequencing and microarray data for the lymphocyte data sets have been deposited in GEO under accession GSE110370.