

Abstract
Background:
Facioscapulohumeral muscular dystrophy (FSHD) is characterized by asymmetric muscular deficit of facial, shoulder-girdle muscles, and descending to lower limb muscles, but it exists in several extramuscular manifestations or overlapping syndromes. Herein, we report a “complex disease plus” patient with FSHD1, accompanied by peripheral neuropathy and myoclonic epilepsy.
Methods:
Standard clinical assessments, particular auxiliary examination, histological analysis, and molecular analysis were performed through the new Comprehensive Clinical Evaluation Form, pulsed-field gel electrophoresis-based Southern blot, Multiplex Ligation-dependent Probe Amplification (MLPA), whole exome sequencing (WES), and targeted methylation sequencing.
Results:
The patient presented with mild facial weakness, humeral poly-hill sign, scapular winging, peroneal weakness, drop foot, pes cavus, and myoclonic epilepsy. Furthermore, electrophysiology revealed severely demyelinated and axonal injury. The muscle and nerve biopsy revealed broadly fiber Type II grouping atrophy and myelinated nerve fibers that significantly decreased with thin myelinated fibers and onion bulbs changes. Generalized sharp and sharp-slow wave complexes on electroencephalography support the diagnosis toward myoclonic epilepsy. In addition, molecular testing demonstrated a co-segregated 20-kb 4q35-EcoRI fragment and permissive allele A, which corresponded with D4Z4 hypomethylation status in the family. Both the patient's mother and brother only presented the typical FSHD but lacked overlapping syndromes. However, no mutations for hereditary peripheral neuropathy and myoclonic epilepsy were discovered by MLPA and WES.
Conclusions:
The present study described a “tripe trouble” with FSHD, peripheral neuropathy, and myoclonic epilepsy, adding the spectrum of overlapping syndromes and contributing to the credible diagnosis of atypical phenotype. It would provide a direct clue on medical care and genetic counseling.
Keywords: Facioscapulohumeral Muscular Dystrophy, Myoclonic Epilepsy, Overlapping Syndromes, Peripheral Neuropathy, Triple Trouble
摘要
背景:
面肩肱型肌营养不良症(FSHD)表现为不对称性肌无力和肌萎缩,主要累及面肌、肩胛带肌和上臂肌群,逐渐向下进展累及躯干肌和下肢肌群,除此还有骨骼肌系统外症状以及“合并综合征”。本研究旨在分析一例面肩肱型肌营养不良症合并周围神经病和肌阵挛性癫痫的病例。
方法:
采用标准的临床评估方法,完善临床辅助检查(生化指标、肌电图、肌肉MRI、肌肉神经活检等)。基于脉冲凝胶电泳的Southern Blot的分子检测;目标区域甲基化测序的方法检测甲基化水平;通过MLPA技术和全外显子测序方法,排除及寻找潜在的基因突变。
结果:
先证者为25岁男性表现为轻度的面肌、肩胛带肌、上臂肌群的萎缩,典型的“弓形足”、“鹤腿”,以及肌阵挛性癫痫。脑电图提示右颞叶区可见显著的尖波和尖慢波支持肌阵挛性癫痫的诊断,电生理提示严重的脱髓鞘和轴突损伤,腓骨肌肌活检病理染色提示肌纤维中-重度萎缩伴II型肌灶区同型肌群化,腓肠神经活检提示有髓神经纤维显著减少伴薄髓纤维和典型的“洋葱球”样改变。分子杂交检测到先证者存在一条缺失为20-kb的4q35-EcorI片段和许可的4qA等位序列,进一步分析PAS区域甲基化提示存在显著的低甲基化,支持面肩肱型肌营养不良症的诊断,同样先证者的母亲和弟弟仅仅表现为典型的FSHD患者。然而,进一步MLPA技术和全外显子测序未发现遗传性周围神经病和肌阵挛性癫痫相关的基因突变。
结论:
本研究描述一例FSHD合并遗传性周围神经病和肌阵挛性癫痫的家系,增加临床上非典型疾病病例,并对非典型疾病的诊断提供参考,为临床治疗和基因咨询提供一个可靠的线索。
INTRODUCTION
Facioscapulohumeral muscular dystrophy (FSHD, OMIM 158900) accounts for one of the most frequent adult-onset muscular dystrophies, which has an estimated prevalence of 1:20,000. Clinically, FSHD is primarily characterized by the asymmetric muscular deficit of the initial onset of facial, scapular muscles, and descends to the lower limb muscle.[1] Its prevalent form, FSHD1, results from the heterozygous contraction of the tandem D4Z4 repeats (DRs, the D4Z4 macrosatellite array [<10 units] located on chromosome 4q35, corresponding to the polymorphic EcoR I alleles of 9 to 38 kb).[2] However, the contraction alone is insufficient to cause FSHD. Several additional DNA sequences flanking the 4qter-DRs are essential for disease development, including the simple sequence length polymorphism proximal to D4Z4 and the distal polyadenylation signal (PAS, ATTAAA) for the DUX4 transcript.[3,4] Present pathogenic models were proposed to correlate with chromatin relaxation on D4Z4 contraction. This resulted in the epigenetic depression of the DUX4 retrogene (OMIM 606009) embedded within the last D4Z4 repeat and subsequently inducing a toxic gain of function.[5]
There is a remarkable variable disease expression that ranges from asymptomatic carriers to wheelchair dependent. Generally, the symptoms present in the first or second decade of the disease course. However, rare cases of infantile or early onset have been reported with severe effects.[6] The great clinical heterogeneity of age onset and disease severity may be correlated with the size of DRs or other regulated factors. Particularly, extramuscular manifestations or overlapping syndromes are the unusual symptoms found.[7,8] All of the above emphasize the diagnostic challenges. In the present study, a special patient with facioscapulohumeral and peroneal muscle weakness and myoclonic epilepsy, who had a co-segregated 20-kb 4qA-allele fragment and a typical peripheral neuropathy similar to Charcot–Marie–Tooth (CMT) syndrome, was described. The clinical and molecular data were consistent with the uncommon diagnosis of FSHD/peripheral neuropathy/myoclonic epilepsy “triple trouble.”
METHODS
Subjects and ethical approval
The subjects including patients and available family members were recruited from academic institutions after informed consent. All clinical information and materials were collected for diagnostic purposes in the research. Clinical assessments were based on the new Comprehensive Clinical Evaluation Form.[9] This study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethics Committee for Medical Research of the First Affiliated Hospital of Fujian Medical University (No.(2016)094).
Auxiliary examination and histological analysis
The disease was further supported by laboratory examination, which included the level of serum creatine kinase (CK) and myoglobin (MYO). Significant auxiliary examinations, such as pulmonary function test (PFT), ambulatory electroencephalography (AEEG), electromyography (EMG), electrophysiology (ECG), electrocardiogram, echocardiogram, and brain magnetic resonance imaging (MRI), were accomplished. The muscle biopsy was performed on the left peroneal muscle with routine enzymatic and immune histochemical staining, including hematoxylin and eosin, modified Gomori trichrome (MGT), nicotinamide adenine dinucleotide (NADH)-tetrazolium reductase, cytochrome c oxidase (COX), adenosine triphosphates (ATPase; pH 4.5 and 9.0), three sections of dystrophin antibodies (N-terminus, rod domain, and C-terminus), and dysferlin antibody. The nerve biopsy was performed on the left sural nerve and stained with toluidine blue.
Analysis of the size of DRs and distal variation
The high-quality genomic DNA of the family numbers was extracted from peripheral lymphocytes, which were embedded in agarose plugs (InCert Agarose, USA). Then, 5 μg of DNA plugs was digested with the restriction enzyme EcoRI (or EcoRI/HindIII; New England Biolabs, USA) and EcoRI/BlnI (Takara, Japan). To determine the distal 4qA or 4qB variants, the corresponding DNA plugs were digested with HindIII. Then, the DNA digestions were separated on 1.2% agarose gel (Agarose III™, BBI, USA) for 39 h, according to the standard procedure for pulsed-field gel electrophoresis (PFGE). After PFGE, the DNA was transferred to a Nytran + membrane (GE Healthcare, USA) and hybridized with probe P13E-11, 4qA, or 4qB, as previously described.[10,11] The position of the DRs was determined using an appropriate molecular size standard (MidRange I PFG Marker, New England Biolabs, USA). The D4Z4 units were calculated, as follows: D4Z4 unit = (D4Z4 length in EcoRI digestion [kb] − 5)/3.3.
DNA methylation analysis
DNA methylation level was quantified using the MethylTarget™ technique by Genesky Biotechnologies Inc. (Shanghai, China). The gDNA from peripheral blood mononuclear cells was subjected to sodium bisulfite treatment using an EZ DNA Methylation™-GOLD Kit (Zymo Research, USA). This translates unmethylated cytosine into uracil but does not change methylated cytosine. Then, this is amplified using HotStarTaq polymerase (Takara, Japan). The methylation-sensitive regions on the distal PAS region were analyzed, which selectively amplifies PAS-positive alleles and discriminates B alleles and nonfunctional 10qA sequences. The fragment length was 247 bp with 10 CpGs.[12]
Multiplex Ligation-dependent Probe Amplification and whole exome sequencing
The gDNA was extracted from the peripheral blood of all subjects using a standard method. To explore the deletions and duplications of the peripheral myelin protein 22 (PMP22) gene, a total quantity of 50–250 ng DNA was performed in a 5 μl volume for each reaction using a Multiplex Ligation-dependent Probe Amplification (MLPA) kit (MRC-Holland P033, the Netherlands). Then, the polymerase chain reaction (PCR) products were analyzed on the ABI 3730 (Applied Biosystems, USA). To search for potential pathogenic genes, the whole exome sequencing (WES) of gDNA was performed by Novogene Bioinformatics Institute (Beijing, China). Exome capture was performed using the Agilent's SureSelect Human All Exon V5 Kit. Library sequencing was performed on an Illumina HiSeq (Illumina Inc., USA), which generated paired-end reads (100 bp ×2) and a mean target coverage of 27× with 81% of the target covered by ≥10 reads, as previously described.[13]
RESULTS
Patients
The pedigree is presented in Figure 1. The proband was a 25-year-old Chinese adult, who was observed with repeated and involuntary convulsion of the limbs or facial muscles with seizures without consciousness over the previous 24 years. The onset of the disease was associated with seizures at the limbs for a few minutes in early infant. In the following years, the symptom did not recur until the age of 6 years old. The frequency was 3–4 times a month and maintained at 10–20 s at a time. When the mood of intention, irritability, or indignation occurred, the frequency of jerks progressively increased and might have presented continuously or episodically. With the seizure frequency progressively increased, the patient drop out of school and visited a neurologist at the 3rd year of middle school. The EEG revealed generalized sharp and sharp-slow wave complexes, which was suggestive of generalized epileptic activity associated with positive myoclonus in the right temporal area [Figure 1]. Therefore, the patient was confirmed with epilepsy, and carbamazepine was initiated. The patient's mother recalled that the patient's lower extremities appeared to be thin and the arches were slightly higher in childhood. In the above therapeutic process, the weakness and atrophy of the lower limbs gradually aggravated, accompanied by thoracic invagination. Obviously, high arches and drop foot were observed [Figure 2], and orthopedic surgery was operated in the right foot. After surgery, the arches were further aggravated. The patient was recommended to our hospital for consideration of peripheral neuropathy.
Figure 1.
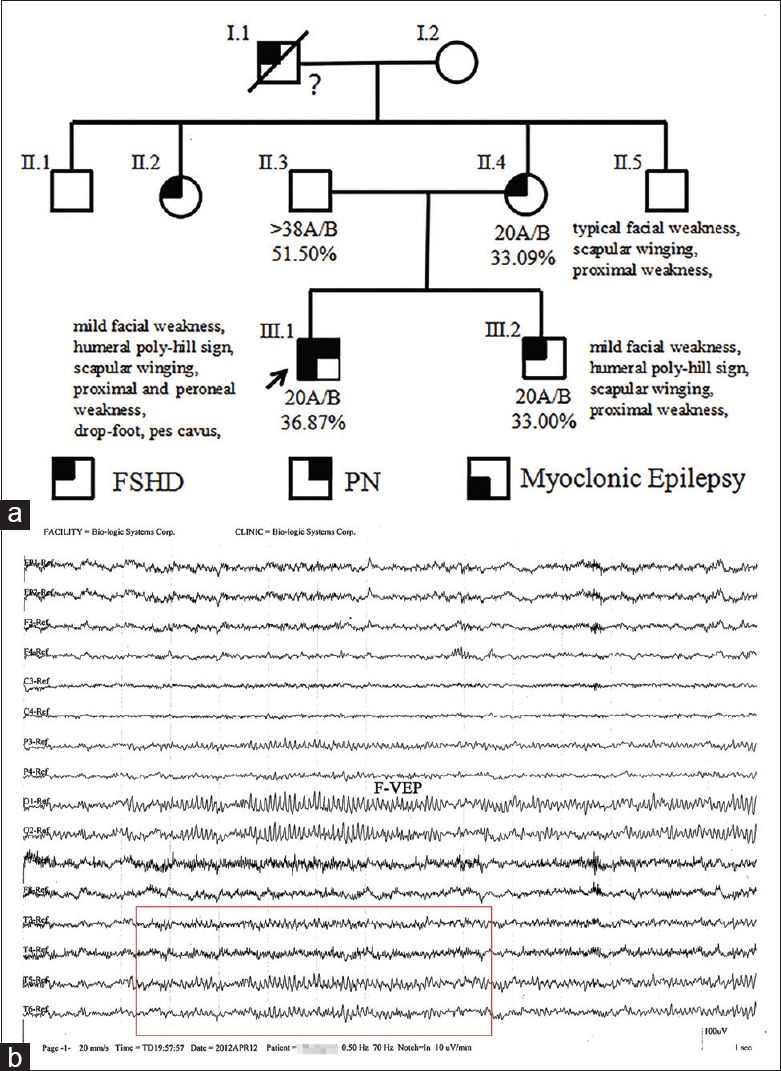
Pedigree of the family and EEG of the proband. (a) The D4Z4 fragment sizes, 4qA/B distal, PAS methylation statue, and summaries of the physical examination were listed below the pedigree symbols. (b) The EEG revealed generalized sharp and sharp-slow wave complexes in the right temporal area (red box). EEG: Electroencephalography; PAS: Polyadenylation signal; F-VEP: Flash visual evoked potentials.
Figure 2.

Clinical phenotypes of the family. (a) The proband exhibited mild facial weakness, humeral poly-hill sign, thoracic invagination, scapular winging, thoracic invagination, proximal and peroneal weakness, drop foot, and pes cavus (CT image in the bottom right). (b and c) The proband's mother and brother presented with typical FSHD symptoms. The patients have given consent for their images to be reported in the journal. CT: Computed tomography; FSHD: Facioscapulohumeral muscular dystrophy.
However, further examination revealed the following: moderate paucity of facial expression, the lips remained horizontal during smiling, and unable to completely close both eyelids or whistle or blow up a balloon. In addition, the patient was unable to lift his arms above the horizontal level with scapular winging. There was mild asymmetrical atrophy in the thorax, shoulder, erector spinae, and lower limb muscles with typical thoracic invagination. The patients' right peroneal muscle obviously atrophied, which lead to drop foot and pes cavus. Both patellar tendon and Achilles tendon reflexes were absent. Sensitive examination was normal. Fasciculation, myotonia, and pathological reflex were not present. There was no history of visual, hearing, or intellectual problems. After the review of the course of epilepsy for the proband, who experienced brief episodes of impairment of consciousness associated with “jerks” at one side of the upper limbs, the diagnosis of myoclonic epilepsy was taken into consideration, and carbamazepine was replaced with sodium valproate.
Family members
The mother of the proband, who was 47 years old, was unable to raise her arms above the horizontal level for over 20 years, which the weakness in her bilateral lower limbs progressed for 2 years. At the age of approximately 25 years old, she realized that she had trouble lifting heavy stuff and gradually had the inability to raise the upper limbs above the head. In the previous 2 years, she noticed difficulty in climbing stairs, squatting, and standing up. On neurological examination, she was unable to tightly close her eyelids, and it was easy to pry them open. Furthermore, her facial expression decreased with lips hypertrophied, and she was unable to puff up her cheeks and whistle. The shoulder and limb muscles had mild asymmetrical atrophy with scapular winging. She had atrophy in the humeral and pelvic girdle muscle with a marked lordosis and waddling gait. The brother of the proband was a 22-year-old adult who presented with weakness and myalgia of limbs. He was unable to whistle or blow since his childhood. Two years ago, he was diagnosed with mild depression by Hamilton Depression Rating Scale. The result of the neurological examination was similar to his mother, with asymmetry atrophy of the facial, shoulder, and limb muscles. Unfortunately, his grandfather died, and aunt was unavailable for further investigation, who presented with a similar phenotype.
Auxiliary examination
The proband's CK was 646 U/L (normal: 38–174 U/L), and MYO was 170.68 μg/L (normal: 1–110 μg/L). His brain MRI did not reveal any structural lesions, and his electrocardiogram, echocardiogram, and PFT were normal. His mother's CK and MYO were 244 U/L and 92.570 μg/L, respectively. His brother's CK and MYO were 267 U/L and 94.23 μg/L.
Electrophysiological studies
The proband's nerve conduction study revealed that the motor nerve conduction velocities (NCVs) decreased to 13.6 m/s and distal motor latency (DML) was markedly prolonged, which demonstrated demyelinating motor neuropathy. Compound motor action potentials (CMAPs) in amplitude decreased to 0.039 mV, showing severe axonal injury. Sensory NCVs and the H-reflection of the tibial nerve were not educible [Table 1]. Needle EMG revealed impaired recruitment and abnormal spontaneous activities such as fibrillation potentials and positive sharp waves. In addition, increased durations and polyphasic motor unit action potentials (MUAPs) in the musculature revealed chronic neurogenic changes in the bicipital, abductor digiti minimi muscle on the right, as well as the tibialis anterior and quadriceps femoris muscles on the left. However, the result of the ECG in his mother and brother only revealed a typical myogenic damage in the proximal and distal muscles.
Table 1.
Electrophysiological data of the proband
| Nerve | Motor NCVs | Sensory NCVs | ||
|---|---|---|---|---|
| Distal latency (ms) | Distal CMAP amplitude (mV) | Conduction velocity (m/s) | ||
| Median (L) | 14.5 | 3.2 | 26.5 | – |
| Median (R) | 14.9 | 2.8 | 28.6 | – |
| Ulnar (L) | 16.0 | 3.1 | 28.4 | – |
| Ulnar (R) | 13.5 | 4.8 | 31.7 | – |
| Tibia (L) | 34.6 | 0.10 | 23.7 | NR |
| Tibia (R) | 34.7 | 0.039 | 17.4 | NR |
| Peroneal (L) | 34.6 | 0.055 | 13.6 | – |
| Peroneal (R) | 39.2 | 0.079 | 19.6 | – |
L: Left; R: Right; NCV: Nerve conduction velocity; NR: No response; –: Not examined; CMAP: Compound motor action potential.
Muscle biopsy
The neurogenic and myopathological characteristic changes appeared in small groups of atrophy predominating in the left peroneal muscle biopsy, accompanied with diffuse round muscle fiber atrophy, hypertrophic fiber, and few inflammatory cell infiltrations. The ATPase staining revealed a predominant Type I fiber and extensive fiber Type II grouping atrophy. Furthermore, MGT staining exhibited individual rimmed vacuoles without evidence of ragged red fibers. NADH, succinate dehydrogenase (SDH), and COX/SDH staining were of significant normality, as well as the immunohistochemistry of dystrophin and dysferlin. These muscle biopsy findings helped exclude rarer causes of FSHD-like phenotypes including desmin storage and mitochondrial, structural, and inflammatory myopathies [Figure 3].
Figure 3.
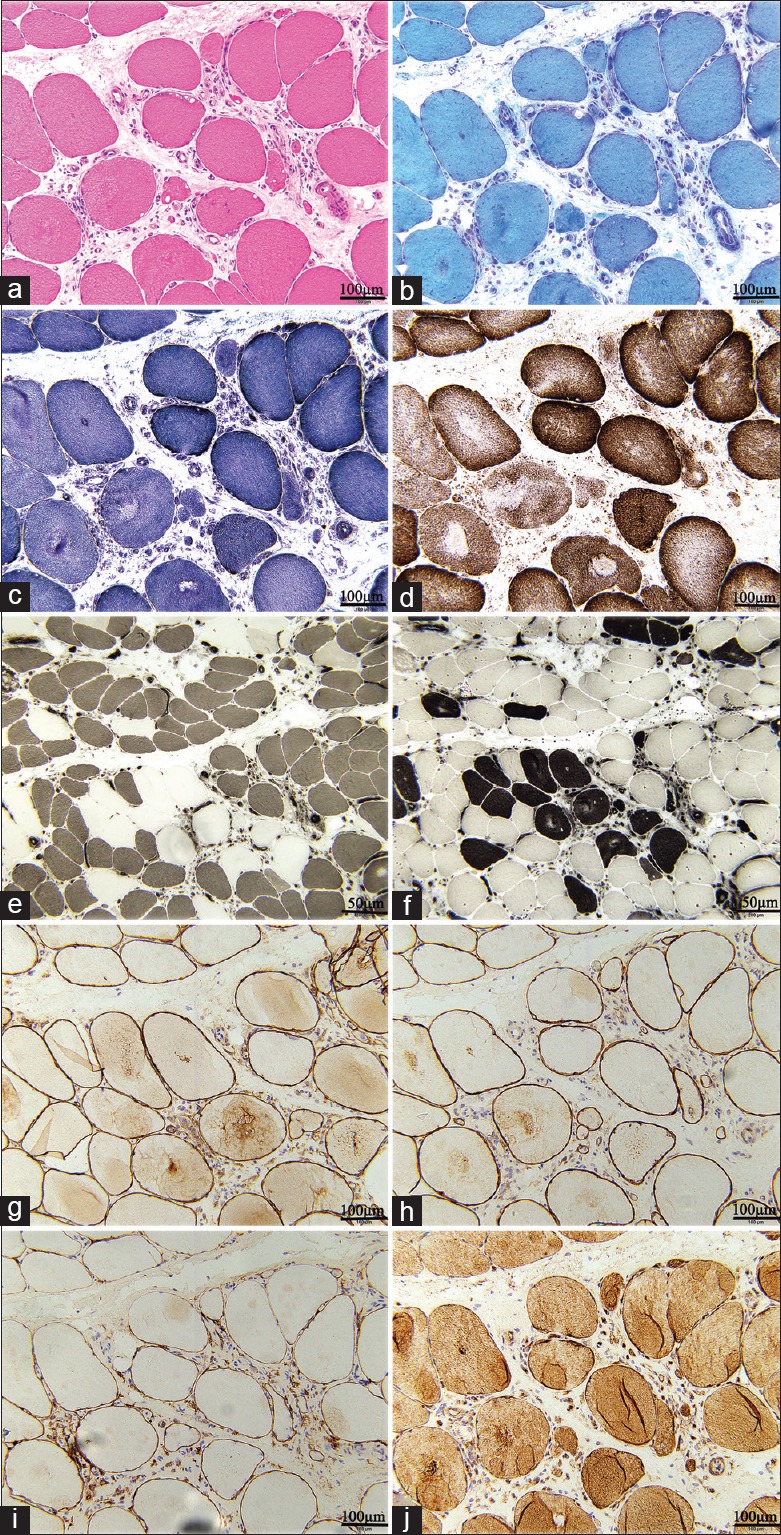
Pathological images of the muscle biopsy. (a and b) H and E and MGT staining revealed the predominance of small group atrophy, accompanied with diffuse round muscle fiber atrophy, hypertrophic fiber, and few inflammatory cell infiltrations. (Bar = 100 μm). (c and d) NADH and COX/SDH staining were of significant normality. (e and f) ATPase (pH 4.5 and 9.0) revealed a type I fiber predominance and extensive fiber type II grouping atrophy. (Bars = 50 μm). (g-j) The immunohistochemistry of dystrophy and dysferlin antibodies did not reveal any significant abnormality (Bar = 100 μm). MGT: Modified Gomori trichrome; NADH: Nicotinamide adenine dinucleotide; COX/SDH: Cytochrome c oxidase/Succinate dehydrogenase; ATPase: Adenosine triphosphates.
Nerve biopsy
Semithin transverse sections of the left sural nerve revealed a decreased density of large myelinated fibers, and the remaining fibers frequently presented with thin myelinated axon and onion bulb changes caused by the proliferation of Schwann cells. The transverse sections did not reveal any inflammatory change and abnormal deposits around the blood vessels. According to the pathological examination, hereditary neuropathy could not be excluded [Figure 4].
Figure 4.
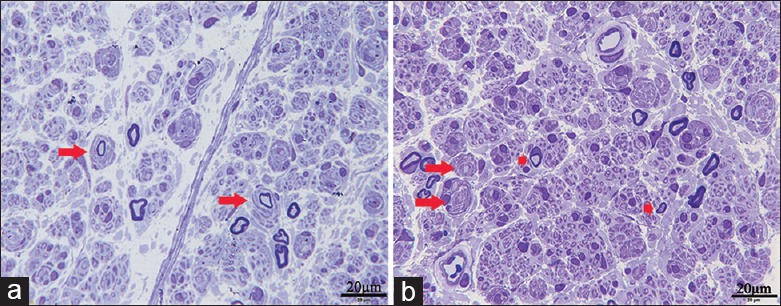
Pathological images of the nerve biopsy. (a and b) Toluidine blue staining revealed a decreased density of large myelinated fibers with frequently thin myelinated axons and onion bulb changes (Bars = 20 μm; longer arrow, onion bulbs changes; shorter arrow, thin myelinated axons).
Molecular genetics and D4Z4 methylation analysis
FSHD molecular testing demonstrated that the proband harbored a co-segregated 20 kb fragment on EcoRI/BlnI double digestion derived from chromosome 4q35 on permissive allele A. His mother and brother were identified with the same contraction fragment [Figure 5]. The methylation result revealed that a significantly lower methylation affected the patients (average: II.4 = 33.09%, III.1 = 36.87%, and III.2 = 33.00%), when compared to subject II.6 (51.50%) [Figure 1].
Figure 5.
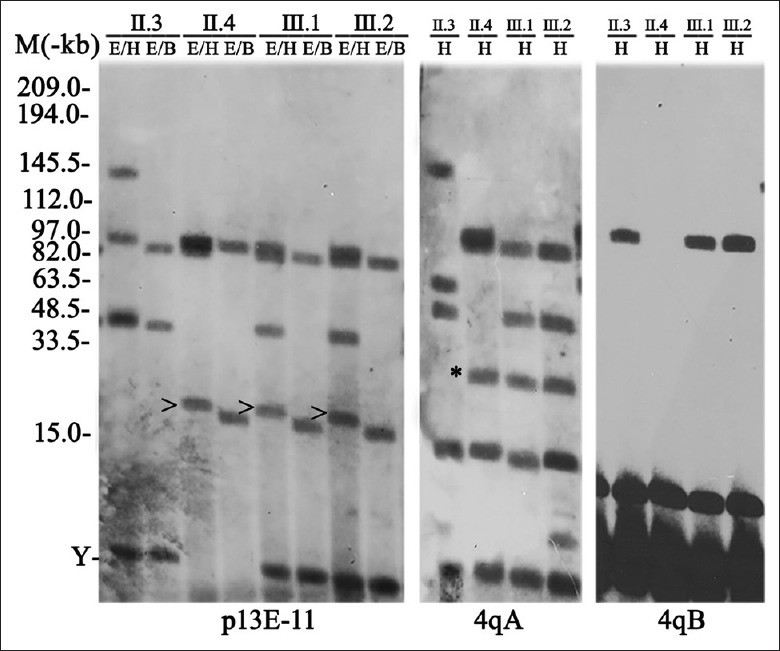
The southern blotting analysis of the family revealed that the proband, and his mother and brother carried one co-segregated 20 kb (arrowhead) with 4qA permissive allele (asterisk) (M: MidRange I PFG Marker; Y: Y chromosome).
Multiplex Ligation-dependent Probe Amplification and whole exome sequencing
MLPA technology was performed to search for deletions or duplications in the PMP22 gene. However, no deletions or duplications encompassing the PMP22 gene were found. Then, WES was operated to screen single nucleotide polymorphisms for variations or splice mutations. Nevertheless, the data did not reveal a heterozygous or homozygous mutation in any of the exons of the CMT (GJB1, MFN2, MPZ, TNF, GDAP1, etc.) or progressive myoclonic epilepsy (PME) (CSTB, KCNT1, ASAH1, GOSR2, etc.) genes.
DISCUSSION
The proband with unique overlapping phenotypes in this pedigree was characterized with concomitant involvement of FSHD, peripheral neuropathy, and myoclonic epilepsy. The clinical feature and laboratory examination may be explained by FSHD due to the contraction fragment of 20 kb on the 4qter D4Z4 region.
The initial sign of the proband was considered as epilepsy, and the treatment of carbamazepine brought about some improvement on the myoclonia. However, in the review of the proband's course of epilepsy, he experienced myoclonic episodes with impairment of consciousness only at one side of the limbs. Hence, he was re-diagnosed as myoclonic epilepsy. Generalized sharp and sharp-slow wave complexes on EEG in the right temporal area supported the epileptic activity with myoclonus pointing toward myoclonic epilepsy. The unilateral myoclonus was the main performance, which was distinguished from epileptic encephalopathies, and started with polymorphic seizures in early infancy.[14] The result of the brain MRI excluded secondary epilepsy caused by brain structural changes. To avoid the risks of exacerbations with carbamazepine, the medicine was regulated with sodium valproate. The proband's condition of myoclonic epilepsy was stable during the recent follow-up.
The notable peroneal muscle atrophy, drop foot, and pes cavus of the proband might be explained by the hereditary peripheral neuropathy (HPN), which was consistent with the electrophysiological results. The reduction of NCVs to 13.6 m/s and the markedly prolonged DMLs support the demyelinating motor neuropathy. The CMAPs in amplitude decreased to 0.039 mV which barely could not generate an action potential with severe neuromuscular damage for a long period and obvious pontaneous activities, increased durations in needle EMG demonstrated severe axonal injury. Therefore, the patient's symptoms were between the HPN1 (demyelinating sensory–motor polyneuropathy [PN] with reduced NCVs) and HPN2 (axonal injury with decreased in amplitude motor responses). The EMG revealed impaired recruitment, abnormal spontaneous activities, and a marked change in MUAPs, which identified the chronic neurogenic changes in the proximal and distal muscles. Furthermore, the muscle and nerve biopsy revealed typical fiber Type II grouping atrophy, significantly decreased myelinated nerve fibers with abundantly thin myelinated fibers and onion bulbs changes. These typical clinical signs and the EMG examination supported the diagnosis of HPN. The CMT syndrome is the most common form of HPN, and most of which are caused by a 1.5 Mb deletion encompassing the PMP22.[15] The deletions and duplications of PMP22 were first excluded using the MLPA. Unfortunately, none of the point mutations and splices of various HPN genes were discovered during the further WES. With the development of high-throughput technologies, an increasing number of new CMT-associated genes have been identified. However, summaries from recent studies with relatively large cohorts of CMT patients indicated that molecular diagnosis could only be established in 60–70% of them.[16,17]
Classical FSHD is primarily characterized by the asymmetric muscular deficit of the initial onset of facial and shoulder-girdle muscles, which descends to the lower limb muscle. However, D4Z4 contractions have also been reported in patients with atypical or overlapping symptoms, including SHD,[11] Duchenne muscular dystrophy[18]/Becker's muscular dystrophy (BMD),[19] limb-girdle muscular dystrophy (LGMD),[20] and caveolinopathy.[21] Interestingly, HPN was easily accompanied by muscular dystrophy or other syndromes, such as BMD[22] and dopamine beta-hydroxylase gene-related dysautonomia.[23] To date, the overlapping symptoms of HPN and FSHD have been reported in only four cases: only one patient had been genetically confirmed overlapping diagnoses of CMT1A and FSHD due to a PMP22 gene duplication and a 19 kb deletion of the D4Z4 locus,[8] while three cases did not proceed with the sequencing techniques, and the pathogenic mutation was unknown.[24,25,26] Although none of the mutations for CMT and PME were discovered by MLPA and WES in the present patient, it is possible that the present sequencing technology was restricted or had novel genes. To the best of our knowledge, epilepsy has been described in patients with several muscular dystrophies such as Fukuyama congenital muscular dystrophy[27] and LGMD2A or calpainopathy.[28] FSHD together with epilepsy might be observed in the early onset in patients carrying large D4Z4 deletions and might be accompanied by mental retardation, speech delay, or hearing loss.[7,29] Furthermore, there was no cognitive decline or other concomitant symptoms in the present patient who carried the common size of 20 kb with moderate phenotype. It was speculated that myoclonic epilepsy was the patient's own complication, rather than being due to FSHD.
The present molecular epigenetic model of FSHD proposes that the contraction of the 4q35 DRs is accompanied by DNA hypomethylation,[30,31] which leads to the relaxed heterochromatinization of the region. In turn, these resulted in the de-repression and transcription of the DUX4 gene.[32] One of the most intriguing clinical observations in FSHD is the apparent inter- and intra-familial variability in disease onset and progression. More recent studies have shown a correlation between the D4Z4 repeat size and D4Z4 methylation and suggest that the susceptibility to somatic DUX4 expression and disease presentation is dependent on a combination of genetic and epigenetic risk factors.[33] Furthermore, the data of the PAS region methylation revealed that all contracted carriers had significant hypomethylation. In addition to the epigenetic effect, the environmental influences, allelic variation and somatic mosaicism, might play significant roles in the special “disease plus.”
In conclusion, a “complex disease plus” patient with FSHD, PN, and myoclonic epilepsy was described and added the disease spectrum of overlapping phenotypes. Consequently, more attention should be given to atypical phenotypes in clinical assessment, which contributes to credible diagnosis, providing a direct clue on medical care and genetic counseling.
Financial support and sponsorship
This work was supported by grants from National Natural Science Foundation of China (No. 81671237 and No. U1505222), Joint Fund for Program of Science innovation of Fujian Province, China (No. 2016Y9010), and National Natural Science Foundation of Fujian Province, China (No. 2017J01196).
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
We sincerely thank Dr. Silvère Van der Maarel of Leiden University for his kindly providing the p13E11, 4qA/4qB, and B31 plasmids. We also thank the participants for their help and willingness to participate in this study.
Footnotes
Edited by: Yuan-Yuan Ji
REFERENCES
- 1.Deenen JC, Arnts H, van der Maarel SM, Padberg GW, Verschuuren JJ, Bakker E, et al. Population-based incidence and prevalence of facioscapulohumeral dystrophy. Neurology. 2014;83:1056–9. doi: 10.1212/WNL.0000000000000797. doi: 10.1212/WNL.0000000000000797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Upadhyaya M, Maynard J, Rogers MT, Lunt PW, Jardine P, Ravine D, et al. Improved molecular diagnosis of facioscapulohumeral muscular dystrophy (FSHD): Validation of the differential double digestion for FSHD. J Med Genet. 1997;34:476–9. doi: 10.1136/jmg.34.6.476. doi: 10.1136/jmg.34.6.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang ZQ, Wang N, van der Maarel S, Murong SX, Wu ZY. Distinguishing the 4qA and 4qB variants is essential for the diagnosis of facioscapulohumeral muscular dystrophy in the Chinese population. Eur J Hum Genet. 2011;19:64–9. doi: 10.1038/ejhg.2010.143. doi: 10.1038/ejhg.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin F, He JJ, Lin XD, Wang DN, Lin HX, Liu XY, et al. A large cohort study confirming that specific haplotype 4A161PAS is exclusively associated with the Chinese FSHD1. Clin Genet. 2016;90:558–9. doi: 10.1111/cge.12858. doi: 10.1111/cge.12858. [DOI] [PubMed] [Google Scholar]
- 5.Geng LN, Yao Z, Snider L, Fong AP, Cech JN, Young JM, et al. DUX4 activates germline genes, retroelements, and immune mediators: Implications for facioscapulohumeral dystrophy. Dev Cell. 2012;22:38–51. doi: 10.1016/j.devcel.2011.11.013. doi: 10.1016/j.devcel.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lunt PW, Jardine PE, Koch MC, Maynard J, Osborn M, Williams M, et al. Correlation between fragment size at D4F104S1 and age at onset or at wheelchair use, with a possible generational effect, accounts for much phenotypic variation in 4q35-facioscapulohumeral muscular dystrophy (FSHD) Hum Mol Genet. 1995;4:951–8. doi: 10.1093/hmg/4.5.951. doi: 10.1093/hmg/4.5.951. [DOI] [PubMed] [Google Scholar]
- 7.Grosso S, Mostardini R, Di Bartolo RM, Balestri P, Verrotti A. Epilepsy, speech delay, and mental retardation in facioscapulohumeral muscular dystrophy. Eur J Paediatr Neurol. 2011;15:456–60. doi: 10.1016/j.ejpn.2011.04.003. doi: 10.1016/j.ejpn.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 8.Schreiber O, Schneiderat P, Kress W, Rautenstrauss B, Senderek J, Schoser B, et al. Facioscapulohumeral muscular dystrophy and charcot-marie-tooth neuropathy 1A – Evidence for “double trouble” overlapping syndromes. BMC Med Genet. 2013;14:92. doi: 10.1186/1471-2350-14-92. doi: 10.1186/1471-2350-14- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ricci G, Ruggiero L, Vercelli L, Sera F, Nikolic A, Govi M, et al. A novel clinical tool to classify facioscapulohumeral muscular dystrophy phenotypes. J Neurol. 2016;263:1204–14. doi: 10.1007/s00415-016-8123-2. doi: 10.1007/s00415-016-8123-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu ZY, Wang ZQ, Murong SX, Wang N. FSHD in chinese population: Characteristics of translocation and genotype-phenotype correlation. Neurology. 2004;63:581–3. doi: 10.1212/01.wnl.0000133210.93075.81. [DOI] [PubMed] [Google Scholar]
- 11.He JJ, Lin XD, Lin F, Xu GR, Xu LQ, Hu W, et al. Clinical and genetic features of patients with facial-sparing facioscapulohumeral muscular dystrophy. Eur J Neurol. 2018;25:356–64. doi: 10.1111/ene.13509. doi: 10.1111/ene.13509. [DOI] [PubMed] [Google Scholar]
- 12.Calandra P, Cascino I, Lemmers RJ, Galluzzi G, Teveroni E, Monforte M, et al. Allele-specific DNA hypomethylation characterises FSHD1 and FSHD2. J Med Genet. 2016;53:348–55. doi: 10.1136/jmedgenet-2015-103436. doi: 10.1136/jmedgenet-2015-103436. [DOI] [PubMed] [Google Scholar]
- 13.Basit S, Albalawi AM, Alharby E, Khoshhal KI. Exome sequencing identified rare variants in genes HSPG2 and ATP2B4 in a family segregating developmental dysplasia of the hip. BMC Med Genet. 2017;18:34. doi: 10.1186/s12881-017-0393-8. doi: 10.1186/s12881-017-0393-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Classification of progressive myoclonus epilepsies and related disorders. Marseille consensus group. Ann Neurol. 1990;28:113–6. doi: 10.1002/ana.410280129. doi: 10.1002/ana.410280129. [DOI] [PubMed] [Google Scholar]
- 15.Chance PF. Overview of hereditary neuropathy with liability to pressure palsies. Ann N Y Acad Sci. 1999;883:14–21. doi: 10.1111/j.1749-6632.1999.tb08562.x. [PubMed] [Google Scholar]
- 16.Saporta AS, Sottile SL, Miller LJ, Feely SM, Siskind CE, Shy ME, et al. Charcot-marie-tooth disease subtypes and genetic testing strategies. Ann Neurol. 2011;69:22–33. doi: 10.1002/ana.22166. doi: 10.1002/ana.22166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murphy SM, Laura M, Fawcett K, Pandraud A, Liu YT, Davidson GL, et al. Charcot-marie-tooth disease: Frequency of genetic subtypes and guidelines for genetic testing. J Neurol Neurosurg Psychiatry. 2012;83:706–10. doi: 10.1136/jnnp-2012-302451. doi: 10.1136/jnnp-2012-302451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Korngut L, Siu VM, Venance SL, Levin S, Ray P, Lemmers RJ, et al. Phenotype of combined duchenne and facioscapulohumeral muscular dystrophy. Neuromuscul Disord. 2008;18:579–82. doi: 10.1016/j.nmd.2008.03.011. doi: 10.1016/j.nmd.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 19.Rudnik-Schöneborn S, Weis J, Kress W, Häusler M, Zerres K. Becker's muscular dystrophy aggravating facioscapulohumeral muscular dystrophy – Double trouble as an explanation for an atypical phenotype. Neuromuscul Disord. 2008;18:881–5. doi: 10.1016/j.nmd.2008.06.387. doi: 10.1016/j.nmd.2008.06.387. [DOI] [PubMed] [Google Scholar]
- 20.Simeoni S, Russo V, Gigli GL, Scalise A. Facioscapulohumeral muscular dystrophy and limb-girdle muscular dystrophy: “double trouble” overlapping syndrome? J Neurol Sci. 2015;348:292–3. doi: 10.1016/j.jns.2014.12.009. doi: 10.1016/j.jns.2014.12.009. [DOI] [PubMed] [Google Scholar]
- 21.Ricci G, Scionti I, Alì G, Volpi L, Zampa V, Fanin M, et al. Rippling muscle disease and facioscapulohumeral dystrophy-like phenotype in a patient carrying a heterozygous CAV3 T78M mutation and a D4Z4 partial deletion: Further evidence for “double trouble” overlapping syndromes. Neuromuscul Disord. 2012;22:534–40. doi: 10.1016/j.nmd.2011.12.001. doi: 10.1016/j.nmd.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bergmann C, Senderek J, Hermanns B, Jauch A, Janssen B, Schröder JM, et al. Becker muscular dystrophy combined with X-linked charcot-marie-tooth neuropathy. Muscle Nerve. 2000;23:818–23. doi: 10.1002/(sici)1097-4598(200005)23:5<818::aid-mus23>3.0.co;2-o. doi: 10.1002/(SICI)1097-4598(200005)23:5%3C818::AID-MUS23%3E3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 23.Bartoletti-Stella A, Chiaro G, Calandra-Buonaura G, Contin M, Scaglione C, Barletta G, et al. A patient with PMP22-related hereditary neuropathy and DBH-gene-related dysautonomia. J Neurol. 2015;262:2373–81. doi: 10.1007/s00415-015-7896-z. doi: 10.1007/s00415-015-7896-z. [DOI] [PubMed] [Google Scholar]
- 24.Bütefisch CM, Lang DF, Gutmann L. The devastating combination of charcot-marie-tooth disease and facioscapulohumeral muscular dystrophy. Muscle Nerve. 1998;21:788–91. doi: 10.1002/(sici)1097-4598(199806)21:6<788::aid-mus11>3.0.co;2-p. doi: 10.1002/(SICI)1097-4598(199806)21:6<788::AID-MUS11>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 25.Auer-Grumbach M, Wagner K, Strasser-Fuchs S, Löscher WN, Fazekas F, Millner M, et al. Clinical predominance of proximal upper limb weakness in CMT1A syndrome. Muscle Nerve. 2000;23:1243–9. doi: 10.1002/1097-4598(200008)23:8<1243::aid-mus13>3.0.co;2-z. doi: 10.1002/1097-4598(200008)23:8%3C1243::AID-MUS13%3E3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 26.Lecky BR, MacKenzie JM, Read AP, Wilcox DE. X-linked and FSH dystrophies in one family. Neuromuscul Disord. 1991;1:275–8. doi: 10.1016/0960-8966(91)90101-w. doi: 10.1016/0960-8966(91)90101-W. [DOI] [PubMed] [Google Scholar]
- 27.Segawa M, Nomura Y, Hachimori K, Shinoyama N, Hosaka A, Mizuno Y, et al. Fukuyama type congenital muscular dystrophy as a natural model of childhood epilepsy. Brain Dev. 1979;1:113–9. doi: 10.1016/s0387-7604(79)80019-4. doi: 10.1016/S0387-7604(79)80019-4. [DOI] [PubMed] [Google Scholar]
- 28.Tsao CY, Mendell JR. Partial epilepsy in an adolescent male with limb-girdle muscular dystrophy 1B. J Child Neurol. 2009;24:346–8. doi: 10.1177/0883073808323525. doi: 10.1177/0883073808323525. [DOI] [PubMed] [Google Scholar]
- 29.Trevisan CP, Pastorello E, Tomelleri G, Vercelli L, Bruno C, Scapolan S, et al. Facioscapulohumeral muscular dystrophy: Hearing loss and other atypical features of patients with large 4q35 deletions. Eur J Neurol. 2008;15:1353–8. doi: 10.1111/j.1468-1331.2008.02314.x. doi: 10.1111/j.1468-1331.2008.02314.x. [DOI] [PubMed] [Google Scholar]
- 30.van Overveld PG, Lemmers RJ, Sandkuijl LA, Enthoven L, Winokur ST, Bakels F, et al. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat Genet. 2003;35:315–7. doi: 10.1038/ng1262. doi: 10.1038/ng1262. [DOI] [PubMed] [Google Scholar]
- 31.Lemmers RJ, Goeman JJ, van der Vliet PJ, van Nieuwenhuizen MP, Balog J, Vos-Versteeg M, et al. Inter-individual differences in cpG methylation at D4Z4 correlate with clinical variability in FSHD1 and FSHD2. Hum Mol Genet. 2015;24:659–69. doi: 10.1093/hmg/ddu486. doi: 10.1093/hmg/ddu486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lemmers RJ, van der Vliet PJ, Klooster R, Sacconi S, Camaño P, Dauwerse JG, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329:1650–3. doi: 10.1126/science.1189044. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gaillard MC, Roche S, Dion C, Tasmadjian A, Bouget G, Salort-Campana E, et al. Differential DNA methylation of the D4Z4 repeat in patients with FSHD and asymptomatic carriers. Neurology. 2014;83:733–42. doi: 10.1212/WNL.0000000000000708. doi: 10.1212/WNL.0000000000000708. [DOI] [PubMed] [Google Scholar]