

To the Editor: Neurofibromatosis type 1 (NF1) is one of the most common autosomal dominant inherited disorders with a prevalence of approximately 1 in 3000 individuals.[1] Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the gastrointestinal tract. Studies have suggested that NF1 associated GISTs (NF1-GISTs) manifest at younger ages than sporadic GISTs; they tend to be located in the small intestine and they affect females predominantly.[1,2] Most sporadic GISTs are driven by activating mutations of KIT or PDGFRA. However, these mutations in NF1-GISTs are very rare. Various mutation types GISTs coexisting in an NF1 patient are rarer still. Herein, we report a case of multiple GISTs in an NF1 patient. Mutation analysis of KIT and PDGFRA was performed in three tumors from different locations. Interestingly, the molecular analysis of the three tumors showed that one mutation was found in KIT exon 11 of the gastric GIST in this case, whereas the other two tumors from the duodenum and jejunum were identified as wild-type KIT and PDGFRA.
A 69-year-old woman was admitted with complaints of intermittent melena and fatigue for the past 2 years. On examination, there were several café-au-lait spots and diffuse cutaneous neurofibromas over her body. On further inquiry, the patient revealed that her mother also had the similar lesions on her body. Based on this history, a diagnosis of NF1 was made. Abdominal contrast-enhanced computed tomography (CT) showed a tumor (1.8 cm × 1.8 cm) in the posterior wall of the stomach and another mass (3.9 cm × 3.4 cm) in the descending part of the duodenum. The CT and endoscopic ultrasound images suggested that the lesions in both stomach and duodenum were likely to be GISTs.
Surgery was performed. During exploration, a 1.4 cm tumor was seen in the stomach and a 3.0 cm tumor was in the duodenum. The latter had abundant blood supply located in the descending part of duodenum. In addition, we found 12 tumors ranging from 0.2 cm to 1.4 cm in the jejunum.
The patient underwent R0 surgical excision of the stomach and small intestine tumors. The specimen from the duodenal tumor revealed that the tumor involved mucosal tissue. In addition, the “umbilical concave” sign and focal hemorrhage were visible.
The specimen from the duodenum measured 3.5 cm × 3.3 cm × 2.9 cm in size with gray-white appearance. Hematoxylin and eosin staining of the specimen revealed that spindle cell neoplasia arranged in a bundle pattern with mild atypia. Mitotic figures were visible [Figure 1a]. The immunohistochemical findings showed that CD117 was positive [Figure 1b], of DOG1 was focally positive, and desmin, SMA, CD34, S-100, SOX10, and p53 were negative, and Ki-67 was 20%+ [Figure 1c and 1d] suggesting high risk.
Figure 1.
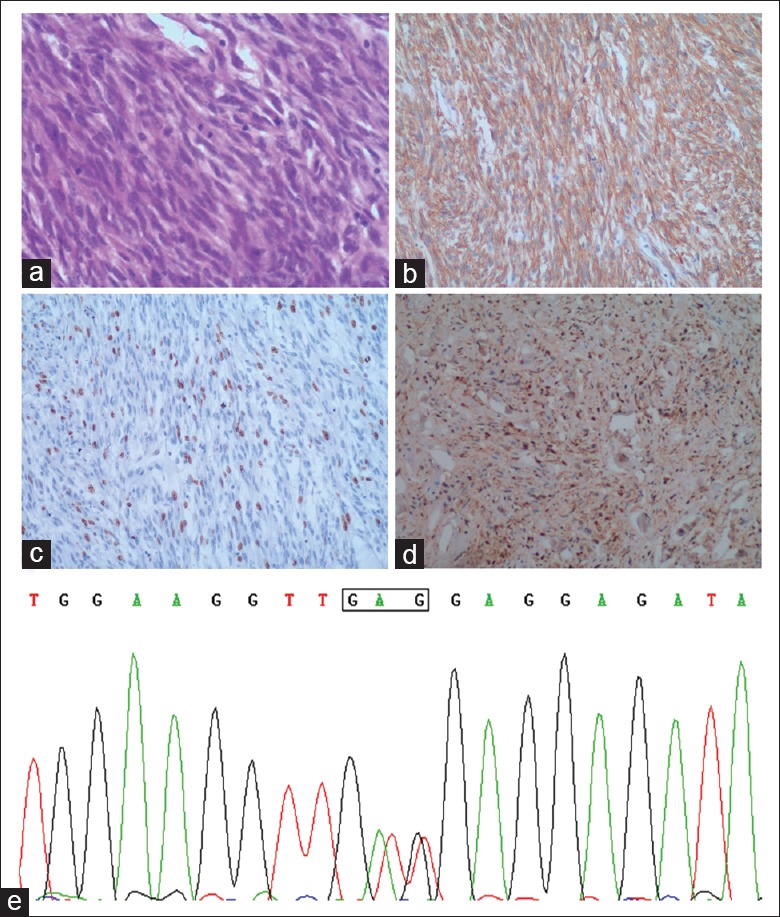
(a) Hematoxylin and eosin staining of the duodenal specimen showing spindle cells arranged in bundles with mild atypia. (b) Immunohistochemical staining was positive for CD117. (c) Immunohistochemical staining showing a medium ki-67 labeling index. (d) Immunohistochemical staining showing that S-100 was negative (a-d, ×100). (e) A constitutional GTT to GAG transition at codon 560 of exon 11 shown by the molecular analysis of gastric tumor.
The molecular analysis for activating mutations of KIT and PDGFRA of the three tumors was performed. Interestingly, the gastric tumor showed a point mutation of the KIT exon 11 [Figure 1e] and no mutation of PDGFRA. The other two tumors had wild type of KIT and PDGFRA.
The patient refused to take imatinib because of poor economic conditions. The patient remains under follow-up without any evidence of recurrence after 18 months (as of April 1, 2018).
Approximately 85% of GISTs are driven by activating mutations in the oncogenes KIT or alternatively PDGFRA, whereas this mutation of KIT and PDGFRA is very rare in NF1-GISTs.[3] Cases such as ours where various mutation types GISTs coexisting in a NF1 patient are rarer still. To the best of our knowledge, only a few similar cases have been reported.[4,5]
In summary, we presented a highly unusual case of various mutation types of GISTs in NF1. The tumorigenesis of various mutation types of GISTs remains unclear due to the insufficiency of evidence, requiring further investigation in the future.
Declaration of patient consent
We certify that we have obtained all appropriate patient consent form. In the form, the patient has given his consent for his images and other clinical information to be reported in the journal. The patient understands that his name and initial will not be published and due efforts will be made to conceal his identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
This work was supported by a grant from the Peking University People's Hospital Research and Development Funds (No. RDE2017-01).
Conflicts of interest
There are no conflicts of interest.
Footnotes
Edited by: Qiang Shi
REFERENCES
- 1.Hakozaki Y, Sameshima S, Tatsuoka T, Okuyama T, Yamagata Y, Noie T, et al. Rectal carcinoma and multiple gastrointestinal stromal tumors (GIST) of the small intestine in a patient with neurofibromatosis type 1: A case report. World J Surg Oncol. 2017;15:160. doi: 10.1186/s12957-017-1231-3. doi: 10.1186/s12957-017-1231-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pan D, Liang P, Xiao H. Neurofibromatosis type 1 associated with pheochromocytoma and gastrointestinal stromal tumors: A case report and literature review. Oncol Lett. 2016;12:637–43. doi: 10.3892/ol.2016.4670. doi: 10.3892/ol.2016.4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nishida T, Tsujimoto M, Takahashi T, Hirota S, Blay JY, Wataya-Kaneda M, et al. Gastrointestinal stromal tumors in Japanese patients with neurofibromatosis type I. J Gastroenterol. 2016;51:571–8. doi: 10.1007/s00535-015-1132-6. doi: 10.1007/s00535-015-1132-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Namgung H. Gastrointestinal stromal tumor with KIT mutation in neurofibromatosis type 1. J Korean Surg Soc. 2011;81:276–80. doi: 10.4174/jkss.2011.81.4.276. doi: 10.4174/jkss.2011.81.4.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamamoto H, Tobo T, Nakamori M, Imamura M, Kojima A, Oda Y, et al. Neurofibromatosis type 1-related gastrointestinal stromal tumors: A special reference to loss of heterozygosity at 14q and 22q. J Cancer Res Clin Oncol. 2009;135:791–8. doi: 10.1007/s00432-008-0514-z. doi: 10.1007/s00432-008-0514-z. [DOI] [PubMed] [Google Scholar]