


Abstract
Replication of damaged DNA is challenging because lesions in the replication template frequently interfere with an orderly progression of the replisome. In this situation, complete duplication of the genome is ensured by the action of DNA damage bypass pathways effecting either translesion synthesis by specialized, damage-tolerant DNA polymerases or a recombination-like mechanism called template switching (TS). Here we report that budding yeast Pif1, a helicase known to be involved in the resolution of complex DNA structures as well as the maturation of Okazaki fragments during replication, contributes to DNA damage bypass. We show that Pif1 expands regions of single-stranded DNA, so-called daughter-strand gaps, left behind the replication fork as a consequence of replisome re-priming. This function requires interaction with the replication clamp, proliferating cell nuclear antigen, facilitating its recruitment to damage sites, and complements the activity of an exonuclease, Exo1, in the processing of post-replicative daughter-strand gaps in preparation for TS. Our results thus reveal a novel function of a conserved DNA helicase that is known as a key player in genome maintenance.
INTRODUCTION
Proliferating cells need to replicate their DNA fully and with high fidelity in order to avoid genome instability and carcinogenesis. During DNA replication, one particularly dangerous situation arises when a replication fork encounters a lesion on the DNA template, leading to polymerase stalling. In order to prevent a permanent replication arrest, cells employ DNA damage bypass mechanisms (also termed DNA damage tolerance) that allow the complete replication of the genome in the presence of lesions (1). In eukaryotes, damage bypass is controlled by ubiquitylation of the DNA sliding clamp, proliferating cell nuclear antigen (PCNA), via components of the RAD6 pathway (2): PCNA is monoubiquitylated on a conserved lysine residue, K164, by the ubiquitin E2-E3 pair Rad6-Rad18, which promotes the recruitment of damage-tolerant DNA polymerases capable of copying damaged DNA in an often mutagenic process termed translesion synthesis (TLS); extension of this modification with a polyubiquitin chain by a different pair of enzymes, in budding yeast composed of the dimeric E2 Ubc13-Mms2 and the E3 Rad5, promotes an error-free pathway called template switching (TS), where the undamaged sister chromatid serves as a transient replication template.
How polyubiquitylated PCNA promotes TS is still an unresolved question. Although a few proteins binding to polyubiquitylated PCNA have been identified, such as Mgs1/WRNIP1 or ZRANB3 (3–6), their role in the pathway remains unclear. In recent years, various other factors have been reported to contribute directly or indirectly to TS based on genetic evidence. In addition to the enzymes promoting PCNA polyubiquitylation, these include the 9–1-1 checkpoint clamp, the Exo1 nuclease, the replicative polymerase δ, proteins mediating the strand invasion step of homologous recombination such as Rad51, Rad52, Rad55-Rad57 and the Shu complex, as well as the helicase Sgs1, implicated in the resolution of TS intermediates (7–9). Importantly, several lines of evidence indicate that DNA damage bypass is not restricted to the sites of replication stalling, but can be achieved via the filling of post-replicative daughter-strand gaps (10,11). The formation of such structures via re-priming downstream of a lesion on the leading or lagging strand has been reported in various experimental systems, including mammalian cells (12–14), and they—rather than free DNA termini—appear to serve as initiation points for TS (15). These observations strongly support the idea that damage bypass operates predominantly in the post-replicative mode and have led to the speculation that Exo1 may contribute to TS by expansion of daughter-strand gaps in order to facilitate access of recombination factors in preparation for strand invasion (7,8). In support of this model, we recently showed that damage-dependent accumulation of single-stranded regions within tracts of newly replicated DNA is strongly reduced by deletion of EXO1 in budding yeast (16).
In the context of that study, we found that daughter-strand gap expansion by Exo1 not only promotes TS, but also generates the predominant signal that leads to checkpoint activation in response to damaged replication templates. In a negative feedback involving the checkpoint kinase Rad53, the damage signal emanating from these gaps subsequently restricts their further erosion by phosphorylation-mediated inhibition of Exo1 activity, thus preventing large-scale genome instability (16). Intriguingly, we observed that the same regulatory mechanism, involving a contribution to checkpoint activation at daughter-strand gaps and subsequent inhibition by Rad53, applies to a multi-functional DNA helicase, Pif1 (16), raising the question whether Pif1—like Exo1—contributes to TS.
Saccharomyces cerevisiae Pif1 is a member of a family of 5′-3′ DNA helicases conserved from prokaryotes to humans (17). Via alternative translational start sites, the PIF1 gene expresses a mitochondrial as well as a nuclear form of the protein (18). Nuclear Pif1 is involved in numerous DNA transactions (19), including telomerase inhibition both at telomeres and DNA double-strand breaks (DSBs) (18,20), resolution of G-quadruplex structures (21), inhibition of replication fork progression at the replication fork barrier within ribosomal DNA (rDNA) (22), stimulation of DNA synthesis during break-induced replication (BIR) (23), and removal of R-loops at transfer-RNA genes (24). Furthermore, Pif1 plays a role in Okazaki fragment maturation, in combination with the helicase/nuclease Dna2, by processing flaps that escape endonuclease cleavage via Rad27/Fen1, thus constituting the so-called alternative pathway (25–27). Finally, Pif1 is known to generate single-stranded DNA (ssDNA) both at uncapped telomeres, where it stimulates checkpoint activation (28), and at stalled replication forks, where it promotes fork reversal and chromosome fragility in the absence of Rad53 (29).
By using a combination of genetic assays along with direct visualization of DNA structures, we demonstrate here that Pif1 acts downstream of PCNA polyubiquitylation and contributes to TS by promoting the efficient expansion of post-replicative daughter-strand gaps. Moreover, this function of Pif1 requires binding to PCNA, which enhances Pif1 recruitment to damage foci. We envision a model where the concerted action of Exo1 and Pif1 at daughter-strand gaps arising from the replication of damaged DNA facilitates the topological transactions associated with the TS pathway of DNA damage bypass.
MATERIALS AND METHODS
Yeast strains and growth conditions
The yeast strains used in this study are derived from the DF5 background except for strains used in Figures 2A, 4, 5A, Supplementary Figures S1A and B, S3 and S4A, which were derived from W303 (Rad5+). Relevant genotypes are shown in Supplementary Table S1. Gene deletions were introduced by polymerase chain reaction (PCR)-based methods or by mating and tetrad dissection. The pif1-m1, pif1-m2 and pif1-R3E alleles, including a TRP1 cassette, were amplified from appropriate plasmids and integrated at the chromosomal PIF1 locus into pif1Δ strains. Correct integration was confirmed by sequencing. The pif1-m2 and pif1-R3E alleles were then backcrossed with their respective wild-type (WT) strains to recover mitochondrial function, confirmed by their ability to grow in non-fermentable medium (YP-glycerol). Endogenous Pif1 and Rad52 were C-terminally tagged with GFP or mRuby2, respectively, by PCR-based methods. All strains were grown at 30°C in YPD medium. Cells were synchronized in G1 by incubation with 10 μg/ml α-factor for 2 h.
Figure 2.

PIF1 participates in TS. (A) Deletion of PIF1 suppresses the MMS sensitivity of smc6–56. Serial dilutions of relevant strains were spotted onto plates containing the indicated concentrations of MMS. (B) Deletion of PIF1 rescues the cold sensitivity of pol32Δ. Serial dilutions of relevant strains were spotted onto YPD plates and incubated at the indicated temperatures. (C) Schematic representation of DNA replication intermediates detected by 2D gel electrophoresis. X-shaped molecules represent recombination structures, Ys and bubbles are replication intermediates, and the n-spot represents non-replicating DNA. The 3.9 kbp EcoRV-HindIII fragment containing the early origin ARS305 analysed by 2D gel electrophoresis is shown on the right. (D) Deletion of PIF1 reduces the amount of X-shaped molecules in early S phase. 2D gel analysis of the ARS305 region was performed with the indicated strains. Cells were synchronized in G1 with α-factor, released in the presence of 0.033% MMS and samples were taken after 60 and 120 min. The arrow indicates X-shaped molecules. Cell cycle profiles are shown at the bottom. (E) Quantification of X-shaped molecules, relative to sgs1Δ and normalized to the n-spot. Error bars indicate SD derived from three independent experiments.
Figure 4.

Pif1 and Exo1 jointly contribute to the expansion of daughter-strand gaps during replication of damaged DNA. (A) DNA fibres were isolated from cells synchronized in G1, treated with 0.04% MMS for 30 min, washed and released into S phase for 30 min in the presence of EdU (added 15 min prior release) without MMS. Fibres were stained with YOYO-1 for total DNA (blue). EdU incorporation was visualized by a click reaction with Alexa Fluor 647 (red), and ssDNA was detected by means of an antibody (green). Scale bar = 10 kbp. (B) Quantification of the fraction of ssDNA within newly replicated DNA, determined for individual EdU-stained tracts by measuring total tract length and total length of ssDNA within that tract. Number of replication tracts analysed: WT = 177; exo1Δ = 120; pif1Δ = 81; exo1Δ pif1Δ = 150. Significance was calculated by the Mann–Whitney test (ns: not significant; *: P < 0.05; ***: P < 0.001; ****: P < 0.0001). Black bar = mean.
Figure 5.

Interaction of Pif1 with PCNA is required for efficient TS and recruitment to damage foci. (A) The pif1-R3E mutation suppresses the MMS sensitivity of smc6–56. Serial dilutions of relevant strains were spotted onto plates containing the indicated concentrations of MMS. (B) The pif1-R3E mutation rescues the cold sensitivity of pol32Δ. Serial dilutions of relevant strains were spotted onto YPD plates and incubated at the indicated temperatures. (C) Interaction with PCNA promotes Pif1 recruitment to damage foci. Left: percentage of cells containing Pif1GFP foci in WT and pif1-R3E strains. Cells were synchronized in G1 with α-factor and released in the absence or presence of 0.033% MMS for 30 or 90 min, respectively, in order to quantify foci numbers in S phase cells. Error bars indicate SD derived from three independent experiments. Cell-cycle profiles are shown below the graph. Right: representative images of Pif1GFP foci. Scale bar = 5 μm. (D) Quantification of Pif1 foci intensity in WT and pif1-R3E (a.u. = arbitrary units, averaged absolute values). Error bars indicate SD derived from three independent experiments.
Viability assays
Sensitivity to methyl methanesulfonate (MMS), 4-nitroquinoline 1-oxide (4NQO) or zinc was determined by spotting 10-fold serial dilutions of exponentially growing cultures onto YPD plates containing the indicated concentrations of damaging agents or ZnCl2. Plates were incubated at 30°C for 3 days before imaging. For cold sensitivity assays, plates were incubated at 19, 17 or 15°C for 5, 6 or 7 days, respectively, before imaging.
Analysis of cell cycle profiles
Samples were taken at the indicated times and fixed in 70% ethanol. Cells were then resuspended in 50 mM sodium citrate, pH 7.0, treated with RNase A, sonicated, stained with propidium iodide and analysed by flow cytometry using a FACSVerse (BD Biosciences).
Analysis of DNA structures by 2D gel electrophoresis
For isolation of DNA replication or recombination intermediates, 2 × 109 cells per time point were arrested with 0.1% sodium azide. After centrifugation, cells were resuspended in 2.4 ml spheroplasting buffer (1 M sorbitol, 100 mM ethylenediaminetetraacetic acid (EDTA), 2 mM Tris–HCl pH 8.0, 0.1% β-mercaptoethanol and 15 U/ml zymolyase) and incubated for 20 min at 30°C, followed by 30 min at 37°C. Spheroplasts were collected by centrifugation and carefully resuspended in 1.125 ml of G2 solution (800 mM guanidine HCl, 30 mM Tris–HCl pH 8.0, 30 mM EDTA, 5% Tween-20, 0.5% Triton X-100), followed by addition of 25 μl of RNase A (10 mg/ml) and 75 μl of proteinase K (20 mg/ml) and incubation at 50°C for 40 min. Samples were then centrifuged at 12 000 g for 10 min, and the supernatant was transferred to a new tube and extracted with 750 μl of chloroform/isoamyl alcohol (24:1). The DNA in the aqueous phase was precipitated by addition of two volumes of Solution II (1% w/v cetyl-trimethyl-ammonium-bromide (CTAB), 50 mM Tris–HCl pH 7.6, 10 mM EDTA pH 8.0) and centrifugation at 12 000 g for 10 min. The pellet was resuspended in 0.8 ml Solution III (1.4 M NaCl, 10 mM Tris–HCl pH 7.6, and 1 mM EDTA). DNA was again precipitated with one volume isopropanol, washed with 70% ethanol, air-dried and finally resuspended in 250 μl of 2 mM Tris–HCl pH 8.0. DNA was digested with HindIII and EcoRV. First dimension electrophoresis was performed in 0.4% TBE-buffered agarose gels at 40 V for 18 h. A gel slice containing DNA fragments between 3 and 12 kbp was excised for second dimension resolution in 1.1% TBE-buffered agarose gel at 140 V for 15 h. Denatured DNA was then transferred to a Hybond XL membrane (Amersham) by standard procedures, and replication intermediates were detected with a 32P-labelled probe specific for ARS305 or a region 6 kbp upstream of ARS305. Signals were acquired using a Typhoon FLA 9500 (GE Healthcare) and quantified using Image Studio software (LI-COR). The relative intensity of X-shaped intermediates was normalized to the signal intensity of the 1n-spot (non-saturating exposure).
Detection of PCNA ubiquitylation
Strains expressing His6-tagged POL30 were synchronized in G1, treated with 0.04% MMS for 30 min and released into S phase. At the indicated times, samples were collected and processed for isolation of HisPCNA by Ni-NTA pull-downs under denaturing conditions, followed by detection of PCNA and its ubiquitylated forms by western blot as described previously (30). Each experiment was performed independently at least twice with similar results.
DNA fibre analysis
Visualization and quantification of ssDNA on DNA fibres was performed as described previously (16).
Fluorescence microscopy
In order to visualize Pif1GFP foci, live cell images were acquired with a 63× objective on a wide-field fluorescence microscope (AF7000, Leica) equipped with an ORCA-Flash 4.0 V2 digital CMOS camera (Hamamatsu) under the control of LAS AF software (Leica). Pif1GFP foci were identified with an auto-threshold from maximum intensity-projected images using Image J (https://imagej.nih.gov/ij/). Percentage of cells containing foci and foci intensity were quantified from at least 200 cells derived from three independent experiments. To visualize co-localization of Pif1GFP and Rad52mRuby2, live cell images were acquired with a 60× objective on a DeltaVision Elite system (GE Healthcare) equipped with a DV Elite sCMOS camera under the control of softWoRx software. Images were processed with Image J.
RESULTS
Pif1 participates in the TS pathway of DNA damage bypass
The notion that Pif1—like Exo1—promotes checkpoint activation during replication of damaged DNA and is inhibited by Rad53-mediated phosphorylation to prevent genome instability (16) suggested that this helicase might also contribute to the expansion of daughter-strand gaps and thus facilitate TS. In order to address this issue, we examined potential epistatic relationships between mutants of PIF1 and of genes involved in damage bypass with respect to their sensitivity to DNA-damaging agents. REV3, encoding the catalytic subunit of DNA polymerase ζ, was chosen as representative of the TLS pathway, and UBC13, encoding the E2 that mediates PCNA polyubiquitylation, as a factor involved in error-free TS (Figure 1). As the pif1Δ single mutant did not show any noticeable sensitivity to MMS or 4NQO, an epistatic relationship with ubc13Δ could neither be confirmed nor ruled out. However, mutants in the TS pathway such as rad5Δ or exo1Δ generally exhibit synergistic defects in combination with TLS mutants such as rev3Δ (31,32) (Figure 1). Similarly, the pif1Δ rev3Δ double mutant displayed a greater MMS and 4NQO sensitivity than either corresponding single mutant, while pif1Δ ubc13Δ was as sensitive as the ubc13Δ single mutant. Thus, these findings are consistent with a role of Pif1 in the TS pathway of damage bypass, possibly in a back-up function.
Figure 1.

Contribution of PIF1 and EXO1 to DNA damage bypass pathways. DNA damage sensitivities of the indicated strains were assessed by spotting serial dilutions of exponential yeast cultures on YPD plates containing the indicated concentrations of MMS or 4NQO.
In order to provide further evidence in support of this relationship, we used three previously described genetic readouts diagnostic for TS factors (7,8,11,33). First, we assessed the suppression of the MMS sensitivity associated with a mutant allele of SMC6 (Figure 2A). This sensitivity results from a defect in the resolution of recombination intermediates that emerge during TS and can therefore be rescued to some extent by abolishing the formation of such structures in TS mutants (33). Consistent with a role in TS, exo1Δ and—to a lesser extent—pif1Δ suppressed the MMS sensitivity of the smc6–56 allele (Figure 2A). The effect could be ascribed to the nuclear form of Pif1, since the pif1-m2 allele, which exclusively produces the mitochondrial form, suppressed the MMS sensitivity of smc6–56 as efficiently as the pif1Δ mutant, whereas pif1-m1, which only expresses the nuclear form, did not afford any rescue (Supplementary Figure S1A). Interestingly, the double mutant exo1Δ pif1Δ suppressed the sensitivity of smc6–56 even more efficiently than either single mutant, suggesting that Exo1 and Pif1 might fulfil parallel or redundant functions within TS (Figure 2A). As expected, complete abrogation of TS by deletion of UBC13 fully rescued viability of smc6–56 at MMS concentrations that permit the growth of the ubc13 single mutant (0.002% MMS). Importantly, deletion of both EXO1 and PIF1 did not increase the suppressive effect of ubc13Δ any further, indicating that Exo1 and Pif1 function in the same pathway as Ubc13 (Supplementary Figure S1B).
Next, we examined the effects of PIF1 and EXO1 on the cold sensitivity conferred by deletion of POL32, encoding a non-essential polymerase δ subunit (Figure 2B). This phenotype is efficiently suppressed by defects in the TS pathway (11), and as previously reported, deletion of EXO1 or PIF1 showed the expected effects (7,25) (Figure 2B). In contrast to the suppression of the MMS sensitivity of smc6–56, exo1Δ and pif1Δ did not exhibit any additivity in their suppressive effects. However, this was attributable to a damage-independent growth defect observed upon deletion of PIF1, likely due to mitochondrial malfunction, since the pif1-m2 allele did not display such growth defect and exhibited an moderately additive suppression in combination with exo1Δ (Supplementary Figure S1C).
Finally, we investigated the formation and resolution of TS intermediates during replication of damaged DNA using 2D gel electrophoresis. Such X-shaped structures are formed in the proximity of replication forks in the course of TS and are dissolved by the action of Sgs1-Top3 (34,35). Thus, their accumulation in an sgs1Δ background is suppressed by inactivating the TS pathway (8). In order to detect these intermediates, cells were synchronized in G1 and released into S phase in the presence of MMS. At the indicated times, genomic DNA was extracted, subjected to restriction enzyme digestion and a region containing an early replication origin, ARS305, was analysed by 2D gel electrophoresis and Southern blotting (Figure 2C). As previously reported, exo1Δ showed a significant reduction in the accumulation of X-molecules in sgs1Δ (8) (Figure 2D and E). Deletion of PIF1 also reduced the formation and/or accumulation of X-molecules after 60 min, but had no more effect after 120 min, indicating that pif1Δ might cause a delay, but not a complete abolishment of TS. Strikingly, the combination of EXO1 and PIF1 deletions further reduced the amount of X-molecules after 60 min, suggesting an additive effect at this early time point, while after 120 min exo1Δ pif1Δ recovered to exo1Δ levels (Figure 2D and E). Similar results were obtained when a passively replicated region upstream of ARS305 was analysed, thereby indicating that the effect was not replication origin-specific (Supplementary Figure S2). Taken together, our findings suggest that Pif1 is required for efficient TS, but appears to act in an auxiliary function that becomes apparent predominantly in the absence of Exo1.
PCNA ubiquitylation is prolonged in exo1Δ and pif1Δ mutants
The failure of exo1Δ as well as pif1Δ to efficiently promote TS could in principle be ascribed to defects either in PCNA ubiquitylation or in more downstream events. Deletion of EXO1 has indeed been suggested to decrease the levels of di-ubiquitylated PCNA after MMS treatment in yeast whole cell extract (31). In order to re-examine this finding by means of improved detection of modified PCNA with temporal resolution, we performed affinity purifications of His6-tagged PCNA under strongly denaturing conditions, thus facilitating the detection of PCNA modifications after MMS damage in the absence of Exo1 and/or Pif1 (Figure 3). Cells were synchronized in G1, treated with MMS for 30 min and released into the cell cycle. Contrary to previous reports based on an analysis of crude extracts (31), deletion of EXO1 did not decrease the levels of polyubiquitylated forms of PCNA (Figure 3A). Instead, we observed a significant accumulation of ubiquitylated PCNA, suggesting an enhanced persistence of the damage signal in exo1Δ. Deletion of PIF1 had a similar, albeit less dramatic effect (Figure 3B). The combination of exo1Δ and pif1Δ led to a further accumulation of ubiquitylated PCNA, especially at the later time points (Figure 3C). Taken together, our observations indicate that the effects of Exo1 and Pif1 on the TS pathway are not caused by a reduction in PCNA modifications, thus implying that both proteins act downstream of PCNA ubiquitylation. In addition, the persistence of ubiquitylated PCNA provides further evidence for the requirement of both Exo1 and Pif1 for efficient completion of damage bypass.
Figure 3.

PCNA ubiquitylation is prolonged in pif1Δ and exo1Δ mutants. Ubiquitylation of His6-tagged PCNA was analysed by Ni-NTA pull-down followed by western blotting in exo1Δ (A), pif1Δ (B) and exo1Δ pif1Δ (C). G1-synchronized cells were treated with 0.04% MMS for 30 min prior to release into S phase without MMS. At the indicated times, samples were collected for isolation of His6-PCNA under completely denaturing conditions as described in the ‘Materials and Methods’ section. Cell-cycle profiles are shown below the blots. Quantification of polyubiquitylated PCNA, relative to G1 (0 min) and normalized to unmodified PCNA, is plotted on the right. The fold increase in the mutants is indicated above the bars.
Pif1 expands post-replicative daughter-strand gaps
Regions of ssDNA accumulate along replicated tracts on DNA fibres isolated from UV- or MMS-treated cells (16). Prompted by our previous finding that Exo1 expands such daughter-strand gaps (16), we asked whether Pif1 is also implicated in this process. To this end, we visualized the distribution of ssDNA within newly replicated DNA on fibres isolated from early S-phase cells that had been treated with MMS in G1 (Figure 4A). In agreement with our previous results, exo1Δ cells significantly reduced the amount of ssDNA within replicated tracts after MMS damage (16) (Figure 4B). Although deletion of PIF1 alone did not have any significant effect, the double mutant exo1Δ pif1Δ exhibited a further decrease in the fraction of ssDNA along newly replicated DNA. Notably, this reduction cannot be explained by a general slowdown of replication fork progression upon deletion of PIF1 since both pif1Δ and exo1Δ pif1Δ showed a similar reduction in replication tract length, but only the double mutant was defective in gap expansion (Supplementary Figure S3). Taken together, our observations suggest that Pif1 contributes to the expansion of daughter-strand gaps left behind replication forks upon replication of damaged templates, most likely as an alternative to Exo1-dependent processing.
Pif1 binding to PCNA is required for TS and efficient recruitment of Pif1 to damage foci
Pif1 interacts with PCNA via a non-conventional epitope located at the C-terminus of the protein (23,36). This interaction has been recently shown to facilitate Pol δ-mediated DNA synthesis during BIR (36). In order to investigate whether Pif1 binding to PCNA is required for TS, we used a previously characterized allele, pif1-R3E, which harbours the point mutations I817R, M820R, L821R and R823E, to abolish interaction with PCNA (36). Mutant pif1-R3E was expressed at near WT levels (Supplementary Figure S4A). Moreover, pif1-R3E did not exhibit increased tolerance to zinc, a phenotype previously linked to the loss of the mitochondrial form of Pif1 (37) (Supplementary Figure S4B), indicating that the mutations did not compromise the mitochondrial form of Pif1. Remarkably, pif1-R3E suppressed the MMS sensitivity of smc6–56 to a similar extent as the full deletion of PIF1 (Figure 5A) and also suppressed the cold sensitivity associated with pol32Δ (Figure 5B). Thus, our observations suggest that an interaction of Pif1 with PCNA is required for efficient TS.
Pif1 is recruited to DNA repair foci after induction of DSBs by ionizing irradiation and co-localizes with the recombination protein Rad52 (38). Importantly, the formation of Pif1 foci is apparently not restricted to the induction of DSBs, since Pif1 foci also appeared upon release of G1-synchronized cells into medium under moderately damaging conditions (0.033% MMS) that do not promote DSBs (39) (Figure 5C). These Pif1 foci corresponded to DNA damage or repair foci since they almost exclusively co-localized with Rad52 (Supplementary Figure S5). Interestingly, the mutant pif1-R3E exhibited a significant decrease in the percentage of cells containing foci after 90 min in MMS, and such foci were overall less intense (Figure 5C and D). Thus, our results indicate that Pif1 binding to PCNA is required for the efficient recruitment of Pif1 to damage sites.
DISCUSSION
The helicase Pif1 acts at telomeres, rDNA, G-quadruplexes, R-loops and during break-induced replication and Okazaki fragment maturation (18,20–24,26), thus playing important roles in several aspects of genome maintenance. In this work, we have uncovered a novel function of Pif1 in the TS pathway of DNA damage bypass. As a mechanistic basis for our data, we propose a model where replication through damaged DNA generates daughter-strand gaps behind replication forks due to re-priming events. These gaps are then expanded, on one hand by the 5′-3′ exonuclease activity of Exo1 and on the other hand by the action of Pif1 (Figure 6).
Figure 6.
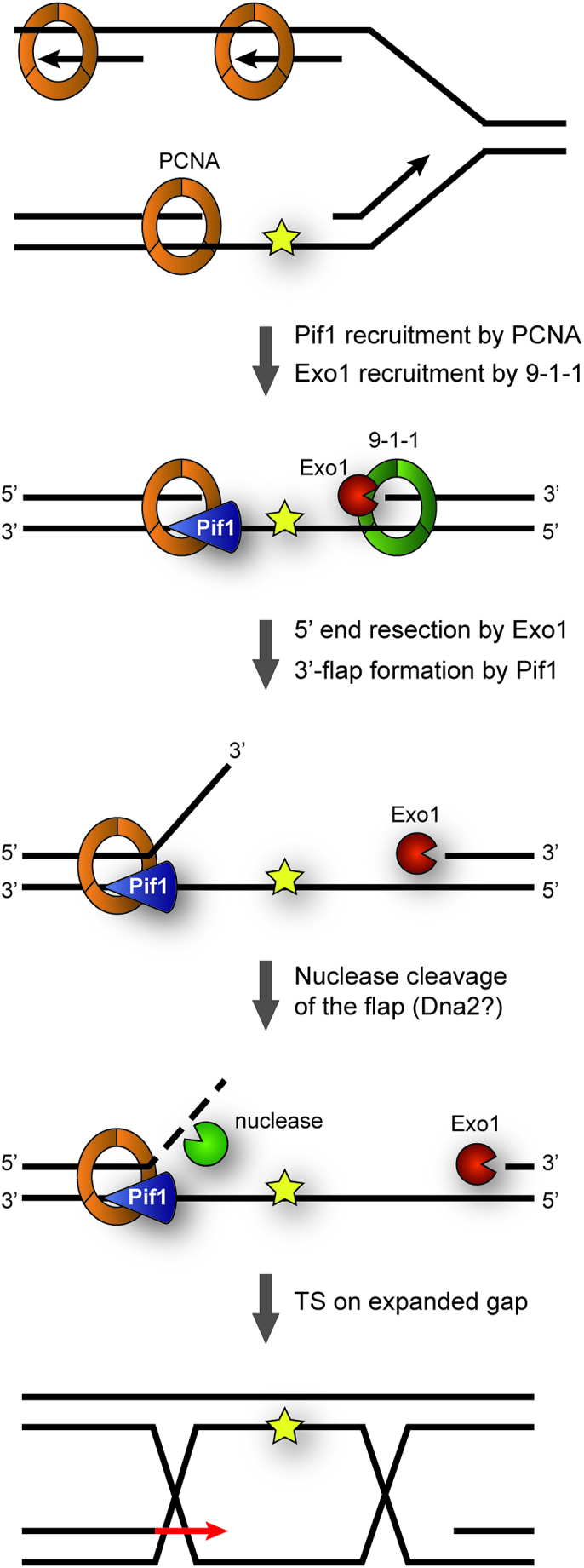
Model for contributions of Pif1 and Exo1 to TS. Replication through damaged DNA generates daughter-strand gaps behind replication forks due to re-priming events. These gaps are expanded at their 5′-junction via Exo1’s 5′-3′-exonucleolytic activity in cooperation with the 9–1-1 checkpoint clamp. Pif1, recruited via interaction with PCNA, contributes to gap expansion at the 3′-junction by generating ssDNA 3′-flaps that subsequently undergo nuclease cleavage, possibly via Dna2. In this manner, gap expansion facilitates invasion of the damaged strand into the newly synthesized sister chromatid, thus initiating TS.
Pif1 has been shown in vitro to exhibit ssDNA-stimulated 5′-3′ helicase activity on DNA substrates containing single-stranded 5′-overhangs (40). Thus, based on its biochemical properties, we propose that Pif1 binds to ssDNA within daughter-strand gaps and unwinds the duplex DNA at their 3′-junction, thus generating a 3′-flap that is likely cleaved by a nuclease in order to extend the size of the gap (Figure 6). Given the moderate TS defect of pif1 mutants and its prominent role in the processing of G-quadruplexes and other complex structures, the helicase could also act in an auxiliary function with other helicases and/or nucleases, possibly by resolving elements of secondary structure. Alternatively, Pif1 itself might act as a 3′-5′ exonuclease. In fact, Candida albicans Pif1 was recently found to harbour such activity within its helicase domain, and this activity appears to be conserved in other yeasts (41). However, in contrast to C. albicans Pif1, the S. cerevisiae protein exhibited very weak exonuclease activity, arguing against such possibility. The notion that Pif1 requires interaction with PCNA for its function in TS is consistent with an action at the 3′-junction of the gap, as the replication clamp is known to be loaded specifically at such structures (42). Thus, interaction with PCNA helps to recruit Pif1 to damage sites and could also serve to enhance its processivity during gap extension. Association between Pif1 and PCNA has also been shown to enhance Pol δ-mediated DNA synthesis during BIR (36). It is therefore tempting to speculate that such interaction might also assist a Pol δ-mediated DNA synthesis step during TS.
Notably, this model is analogous to a scenario that might apply at the 5′-junction of the gap, the loading site of the 9–1-1 checkpoint clamp (43,44). In addition to acting as a damage sensor for checkpoint signalling, the 9–1-1 complex has been reported to promote TS (7,43,45). Intriguingly, the complex has been shown to stimulate DNA resection at uncapped telomeres by Exo1, and physical interactions with the nuclease have been detected in the two-hybrid system (7,46). Thus, we propose that the two clamps, bound to opposite ends of the daughter-strand gap, may jointly coordinate the recruitment of factors involved in error-free TS, such as Pif1 and Exo1. Whether polyubiquitylation of PCNA further enhances interaction with Pif1 remains to be explored.
Given that Exo1 can largely compensate for the lack of Pif1 and the TS defect of pif1 mutants becomes apparent mostly in an exo1 background, Pif1-mediated expansion of daughter-strand gaps might serve predominantly as a back-up pathway for the Exo1-dependent mechanism. A similar relationship between the two proteins has in fact been observed during resection and checkpoint activation at uncapped telomeres (28). At DSBs, the situation appears to be different here, budding yeast Pif1 has no effect on resection, while Exo1 plays a prominent role (47). However, recent observations have implicated human PIF1 in DNA resection at DSBs (Pablo Huertas, personal communication), suggesting that a cooperative action may apply more generally.
During the alternative pathway of Okazaki fragment maturation, Pif1 cooperates with the nuclease Dna2 by further unwinding the short 5′-flaps created by Pol δ-dependent strand displacement, thus providing the substrate for the nuclease (25–27). Based on the notion that recombinant Dna2 harbours both 5′-3′ and 3′-5′ ssDNA endonuclease activities with a preference for free single-stranded ends over ssDNA gaps (48,49), it is attractive to hypothesize that Dna2 may also be responsible for flap cleavage during Pif1-mediated daughter-strand gap processing. Unfortunately, the inherent sensitivity of dna2–1 and dna2–2 mutants (mutated in the nuclease and helicase domains, respectively) to cold stress and DNA-damaging agents prevented us from assessing a potential contribution to TS genetically.
Saccharomyces cerevisiae expresses a second Pif1 family helicase, Rrm3, which facilitates progression of replication forks past protein–DNA complexes (50). Both helicases suppress genome instability at G-quadruplexes (51), promote the collapse of stalled replication forks in the absence of checkpoint signalling (29) and interact with PCNA (23,52). Yet, our previous observation that checkpoint-dependent inhibition of Rrm3 was not required to prevent daughter-strand gap instability (16) argues against a role of Rrm3 in gap processing. Furthermore, deletion of RRM3 is lethal in combination with smc6 or sgs1 mutants (53,54), thus not only making it impossible to perform some of the genetic assays used here to diagnose TS factors, but also implying a different genetic relationship to the TS pathway. Indeed, deletion of RRM3 did not afford any rescue of the cold sensitivity of pol32Δ mutants. Instead, the combination of rrm3Δ and pol32Δ resulted in an additive effect (Supplementary Figure S6). Thus, in the context of DNA damage bypass the two helicases, Pif1 and Rrm3, clearly act in different ways.
In summary, our study uncovers a novel function of the Pif1 helicase in DNA damage tolerance and provides insight into the DNA transactions at a particularly vulnerable replication intermediate, the daughter-strand gap. Unravelling the mechanisms by which Pif1 family helicases maintain genomic integrity is relevant to human disease, as mutations of human PIF1 have been associated with cancer (55). Given the complexity of the interplay between helicases and nucleases in DNA replication and repair, future studies will be required to provide more detailed insights into the regulation of their processing activities at different substrates, such as DSBs, uncapped telomeres and internal stretches of ssDNA.
Supplementary Material
ACKNOWLEDGEMENTS
We thank IMB’s media lab for supplies, and Sonja Braun for technical support.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Research Council, Advanced Grant [ERC AdG 323179: DAMAGE BYPASS to H.D.U.]. Funding for open access charge: Institutional Core Funding.
Conflict of interest statement. None declared.
REFERENCES
- 1. Friedberg E.C. Suffering in silence: the tolerance of DNA damage. Nat. Rev. Mol. Cell Biol. 2005; 6:943–953. [DOI] [PubMed] [Google Scholar]
- 2. Ulrich H.D. Regulating post-translational modifications of the eukaryotic replication clamp PCNA. DNA Repair (Amst). 2009; 8:461–469. [DOI] [PubMed] [Google Scholar]
- 3. Ciccia A., Nimonkar A.V., Hu Y., Hajdu I., Achar Y.J., Izhar L., Petit S.A., Adamson B., Yoon J.C., Kowalczykowski S.C. et al. Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic integrity after replication stress. Mol. Cell. 2012; 47:396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Saugar I., Parker J.L., Zhao S., Ulrich H.D.. The genome maintenance factor Mgs1 is targeted to sites of replication stress by ubiquitylated PCNA. Nucleic Acids Res. 2012; 40:245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yuan J., Ghosal G., Chen J.. The HARP-like domain-containing protein AH2/ZRANB3 binds to PCNA and participates in cellular response to replication stress. Mol. Cell. 2012; 47:410–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Crosetto N., Bienko M., Hibbert R.G., Perica T., Ambrogio C., Kensche T., Hofmann K., Sixma T.K., Dikic I.. Human Wrnip1 is localized in replication factories in a ubiquitin-binding zinc finger-dependent manner. J. Biol. Chem. 2008; 283:35173–35185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Karras G.I., Fumasoni M., Sienski G., Vanoli F., Branzei D., Jentsch S.. Noncanonical role of the 9-1-1 clamp in the error-free DNA damage tolerance pathway. Mol. Cell. 2013; 49:536–546. [DOI] [PubMed] [Google Scholar]
- 8. Vanoli F., Fumasoni M., Szakal B., Maloisel L., Branzei D.. Replication and recombination factors contributing to recombination-dependent bypass of DNA lesions by template switch. PLoS Genet. 2010; 6:e1001205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ball L.G., Zhang K., Cobb J.A., Boone C., Xiao W.. The yeast Shu complex couples error-free post-replication repair to homologous recombination. Mol. Microbiol. 2009; 73:89–102. [DOI] [PubMed] [Google Scholar]
- 10. Daigaku Y., Davies A.A., Ulrich H.D.. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature. 2010; 465:951–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Karras G.I., Jentsch S.. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell. 2010; 141:255–267. [DOI] [PubMed] [Google Scholar]
- 12. Elvers I., Johansson F., Groth P., Erixon K., Helleday T.. UV stalled replication forks restart by re-priming in human fibroblasts. Nucleic Acids Res. 2011; 39:7049–7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heller R.C., Marians K.J.. Replication fork reactivation downstream of a blocked nascent leading strand. Nature. 2006; 439:557–562. [DOI] [PubMed] [Google Scholar]
- 14. Lopes M., Foiani M., Sogo J.M.. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell. 2006; 21:15–27. [DOI] [PubMed] [Google Scholar]
- 15. Giannattasio M., Zwicky K., Follonier C., Foiani M., Lopes M., Branzei D.. Visualization of recombination-mediated damage bypass by template switching. Nat. Struct. Mol. Biol. 2014; 21:884–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garcia-Rodriguez N., Morawska M., Wong R.P., Daigaku Y., Ulrich H.D.. Spatial separation between replisome- and template-induced replication stress signaling. EMBO J. 2018; 37:e98369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bochman M.L., Sabouri N., Zakian V.A.. Unwinding the functions of the Pif1 family helicases. DNA Repair (Amst). 2010; 9:237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schulz V.P., Zakian V.A.. The saccharomyces PIF1 DNA helicase inhibits telomere elongation and de novo telomere formation. Cell. 1994; 76:145–155. [DOI] [PubMed] [Google Scholar]
- 19. Boule J.B., Zakian V.A.. Roles of Pif1-like helicases in the maintenance of genomic stability. Nucleic Acids Res. 2006; 34:4147–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou J., Monson E.K., Teng S.C., Schulz V.P., Zakian V.A.. Pif1p helicase, a catalytic inhibitor of telomerase in yeast. Science. 2000; 289:771–774. [DOI] [PubMed] [Google Scholar]
- 21. Paeschke K., Capra J.A., Zakian V.A.. DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell. 2011; 145:678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ivessa A.S., Zhou J.Q., Zakian V.A.. The Saccharomyces Pif1p DNA helicase and the highly related Rrm3p have opposite effects on replication fork progression in ribosomal DNA. Cell. 2000; 100:479–489. [DOI] [PubMed] [Google Scholar]
- 23. Wilson M.A., Kwon Y., Xu Y., Chung W.H., Chi P., Niu H., Mayle R., Chen X., Malkova A., Sung P. et al. Pif1 helicase and Poldelta promote recombination-coupled DNA synthesis via bubble migration. Nature. 2013; 502:393–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tran P.L.T., Pohl T.J., Chen C.F., Chan A., Pott S., Zakian V.A.. PIF1 family DNA helicases suppress R-loop mediated genome instability at tRNA genes. Nat. Commun. 2017; 8:15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Budd M.E., Reis C.C., Smith S., Myung K., Campbell J.L.. Evidence suggesting that Pif1 helicase functions in DNA replication with the Dna2 helicase/nuclease and DNA polymerase delta. Mol. Cell. Biol. 2006; 26:2490–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pike J.E., Burgers P.M., Campbell J.L., Bambara R.A.. Pif1 helicase lengthens some Okazaki fragment flaps necessitating Dna2 nuclease/helicase action in the two-nuclease processing pathway. J. Biol. Chem. 2009; 284:25170–25180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pike J.E., Henry R.A., Burgers P.M., Campbell J.L., Bambara R.A.. An alternative pathway for Okazaki fragment processing: resolution of fold-back flaps by Pif1 helicase. J. Biol. Chem. 2010; 285:41712–41723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dewar J.M., Lydall D.. Pif1- and Exo1-dependent nucleases coordinate checkpoint activation following telomere uncapping. EMBO J. 2010; 29:4020–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rossi S.E., Ajazi A., Carotenuto W., Foiani M., Giannattasio M.. Rad53-Mediated regulation of Rrm3 and Pif1 DNA helicases contributes to prevention of aberrant fork transitions under replication stress. Cell Rep. 2015; 13:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Davies A.A., Huttner D., Daigaku Y., Chen S., Ulrich H.D.. Activation of ubiquitin-dependent DNA damage bypass is mediated by replication protein a. Mol. Cell. 2008; 29:625–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ball L.G., Hanna M.D., Lambrecht A.D., Mitchell B.A., Ziola B., Cobb J.A., Xiao W.. The Mre11-Rad50-Xrs2 complex is required for yeast DNA postreplication repair. PLoS One. 2014; 9:e109292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Johnson R.E., Henderson S.T., Petes T.D., Prakash S., Bankmann M., Prakash L.. Saccharomyces cerevisiae RAD5-encoded DNA repair protein contains DNA helicase and zinc-binding sequence motifs and affects the stability of simple repetitive sequences in the genome. Mol. Cell. Biol. 1992; 12:3807–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Choi K., Batke S., Szakal B., Lowther J., Hao F., Sarangi P., Branzei D., Ulrich H.D., Zhao X.. Concerted and differential actions of two enzymatic domains underlie Rad5 contributions to DNA damage tolerance. Nucleic Acids Res. 2015; 43:2666–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Branzei D., Vanoli F., Foiani M.. SUMOylation regulates Rad18-mediated template switch. Nature. 2008; 456:915–920. [DOI] [PubMed] [Google Scholar]
- 35. Liberi G., Maffioletti G., Lucca C., Chiolo I., Baryshnikova A., Cotta-Ramusino C., Lopes M., Pellicioli A., Haber J.E., Foiani M.. Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev. 2005; 19:339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Buzovetsky O., Kwon Y., Pham N.T., Kim C., Ira G., Sung P., Xiong Y.. Role of the Pif1-PCNA complex in Pol delta-dependent strand displacement DNA synthesis and break-induced replication. Cell Rep. 2017; 21:1707–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Guirola M., Barreto L., Pagani A., Romagosa M., Casamayor A., Atrian S., Arino J.. Lack of DNA helicase Pif1 disrupts zinc and iron homoeostasis in yeast. Biochem. J. 2010; 432:595–605. [DOI] [PubMed] [Google Scholar]
- 38. Wagner M., Price G., Rothstein R.. The absence of Top3 reveals an interaction between the Sgs1 and Pif1 DNA helicases in Saccharomyces cerevisiae. Genetics. 2006; 174:555–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lundin C., North M., Erixon K., Walters K., Jenssen D., Goldman A.S., Helleday T.. Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Res. 2005; 33:3799–3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lahaye A., Leterme S., Foury F.. PIF1 DNA helicase from Saccharomyces cerevisiae. Biochemical characterization of the enzyme. J. Biol. Chem. 1993; 268:26155–26161. [PubMed] [Google Scholar]
- 41. Wei X.B., Zhang B., Bazeille N., Yu Y., Liu N.N., Rene B., Mauffret O., Xi X.G.. A 3′-5′ exonuclease activity embedded in the helicase core domain of Candida albicans Pif1 helicase. Sci. Rep. 2017; 7:42865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moldovan G.L., Pfander B., Jentsch S.. PCNA, the maestro of the replication fork. Cell. 2007; 129:665–679. [DOI] [PubMed] [Google Scholar]
- 43. Ellison V., Stillman B.. Biochemical characterization of DNA damage checkpoint complexes: clamp loader and clamp complexes with specificity for 5′ recessed DNA. PLoS Biol. 2003; 1:E33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Majka J., Binz S.K., Wold M.S., Burgers P.M.. Replication protein A directs loading of the DNA damage checkpoint clamp to 5′-DNA junctions. J. Biol. Chem. 2006; 281:27855–27861. [DOI] [PubMed] [Google Scholar]
- 45. Majka J., Burgers P.M.. Yeast Rad17/Mec3/Ddc1: a sliding clamp for the DNA damage checkpoint. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:2249–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ngo G.H., Balakrishnan L., Dubarry M., Campbell J.L., Lydall D.. The 9-1-1 checkpoint clamp stimulates DNA resection by Dna2-Sgs1 and Exo1. Nucleic Acids Res. 2014; 42:10516–10528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhu Z., Chung W.H., Shim E.Y., Lee S.E., Ira G.. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008; 134:981–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bae S.H., Seo Y.S.. Characterization of the enzymatic properties of the yeast dna2 Helicase/endonuclease suggests a new model for Okazaki fragment processing. J. Biol. Chem. 2000; 275:38022–38031. [DOI] [PubMed] [Google Scholar]
- 49. Masuda-Sasa T., Imamura O., Campbell J.L.. Biochemical analysis of human Dna2. Nucleic Acids Res. 2006; 34:1865–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ivessa A.S., Lenzmeier B.A., Bessler J.B., Goudsouzian L.K., Schnakenberg S.L., Zakian V.A.. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol. Cell. 2003; 12:1525–1536. [DOI] [PubMed] [Google Scholar]
- 51. Paeschke K., Bochman M.L., Garcia P.D., Cejka P., Friedman K.L., Kowalczykowski S.C., Zakian V.A.. Pif1 family helicases suppress genome instability at G-quadruplex motifs. Nature. 2013; 497:458–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schmidt K.H., Derry K.L., Kolodner R.D.. Saccharomyces cerevisiae RRM3, a 5′ to 3′ DNA helicase, physically interacts with proliferating cell nuclear antigen. J. Biol. Chem. 2002; 277:45331–45337. [DOI] [PubMed] [Google Scholar]
- 53. Menolfi D., Delamarre A., Lengronne A., Pasero P., Branzei D.. Essential roles of the Smc5/6 complex in replication through natural pausing sites and endogenous DNA damage tolerance. Mol. Cell. 2015; 60:835–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schmidt K.H., Kolodner R.D.. Requirement of Rrm3 helicase for repair of spontaneous DNA lesions in cells lacking Srs2 or Sgs1 helicase. Mol. Cell. Biol. 2004; 24:3213–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chisholm K.M., Aubert S.D., Freese K.P., Zakian V.A., King M.C., Welcsh P.L.. A genomewide screen for suppressors of Alu-mediated rearrangements reveals a role for PIF1. PLoS One. 2012; 7:e30748. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.