

Abstract
Background
Acyclovir (ACV) resistant herpes simplex virus (HSV) isolates can be readily selected in animal infection models receiving suboptimal ACV treatment, however no comparative studies of the emergence of resistance following suboptimal treatment with valacyclovir (VCV) or famciclovir (FCV), the prodrugs of acyclovir and penciclovir, respectively, have been reported.
Methods
Mice (n = 30) were infected with HSV type 1 or 2 in the ear pinnae and administered oral prodrugs at one fifth a dose previously shown to be effective. To select and amplify drug-resistant HSV, a total of seven consecutive in vivo passages with suboptimal treatment were performed for each virus sample and progeny virus from each passage was characterized by the plaque reduction (PRA) and plating efficiency assays (PEA).
Results
No drug-resistant HSV-2 and only a single drug-resistant HSV-1 variant were identified. Virus recovered from the first three sequential passages of this HSV-1 sample was susceptible by PRA, although the proportion of resistant virus recovered gradually increased upon passage. The resistant HSV-1 phenotype was confirmed by PRA after four sequential passages in mice. Unexpectedly, this in vivo-selected drug-resistant HSV-1 failed to yield an infection completely refractory to treatment in subsequent passages.
Conclusions
Sub-optimal therapy of immunocompetent mice with either VCV or FCV did not readily select for HSV-mutants resistant to either ACV or PCV, suggesting that selection of resistance with either prodrug remains difficult using this system. Futhermore, this study suggests that the PEA may represent a useful adjunct to the PRA for monitoring alterations in the proportion of drug-resistant virus even when no change in IC50 is apparent.
Background
Resistance to acyclovir (ACV) or penciclovir (PCV) generally arises by a single base mutation in either the HSV thymidine kinase (TK) or DNA polymerase (Pol) gene [for a review, see reference [5]]. Unlike Pol, TK is not essential for virus replication in cell culture [7], although in vivo analyses implicate it in HSV virulence, pathogenicity and reactivation from latency [4,8,16,22]. Since ACV and PCV are dependent upon HSV TK for monophosphorylation, an event obligatory for the formation of the active triphosphate inhibitor [14], ACVr isolates often exhibit high levels of cross-resistance to PCV [2,3].
Viral populations are naturally heterogeneous; there are approximately 6 to 8 TK deficient variants per 104 plaque-forming units (PFU) of HSV-1 (laboratory strains and clinical isolates) that have never been exposed to antiviral selection [6,13,15,17]. Resistance to nucleoside inhibitors of HSV arises readily in tissue culture probably by selection of pre-existing mutants [10,13,18]. Moreover, Field et al.[9] demonstrated sequential HSV inoculation from mouse to mouse receiving sub-optimal ACV therapy led to the selection and amplification of resistant virus in vivo. After four serial passages in the immunocompetent mice, the infection became completely refractory to ACV treatment.
Acyclovir has been in part superceded by the oral prodrugs valaciclovir (VCV) and famciclovir (FCV), the latter being the oral version of PCV [20,24]. No comparative studies of the emergence of resistance following sub-optimal VCV or FCV have been reported and therefore set out to to select for HSV-1 and HSV-2 isolates resistant to ACV and PCV after using prodrug selection.
Materials and Methods
Cell lines and virus strains
MRC-5 (pass 18–25) and Vero were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS) and incubated at 37°C, 5% CO2. Vero cells were utilized for virus plaque purification and protein lysate preparations and MRC-5 cells were used for PRA and PEA. TK-negative (TK-) human osteosarcoma (143) cells were propagated in the medium described above and utilized for the 3H-thymidine kinase enzymatic assay.
Virus stocks were prepared by inoculating cells at 0.01 plaque-forming units per MRC-5 cell (PFU/cell). Laboratory virus strains utilized were HSV-1 SC16 and HSV-2 SB5. SC16 was kindly provided by Dr. Sharon Safrin (Gilead Sciences, CA). SB5 (ATCC; VR-2546) was plaque-purified from HSV-2 333 kindly provided by Dr. Priscilla Schaffer (University of Pennsylvania, PA) at passage 6.
Antiviral agents
PCV and FCV were synthesized at GlaxoSmithKline. VCV was extracted from Valtrex™ tablets and purity was confirmed by HPLC.
Animal inoculation and treatment
Female BALB/c mice (18–20 g; Charles River, Raleigh, NC) were infected intradermally in both ear pinnae with 20 μl of virus suspension containing 105 PFU of HSV as previously described [1]. The same viral challenge was used for each subsequent in vivo passage. VCV or FCV were administered via drinking water at either 1 mg/ml (a dose previously shown to be a highly effective [23]; and D Sutton, unpublished observations) or 0.2 mg/ml (suboptimal treatment) from the time of infection until day 4 post-infection. No significant difference in daily consumption between either of the treated groups or the placebo group (drug-free water) was observed. Daily drug intake equated to 75 mg/kg or 15 mg/kg, for the 1 mg/ml and 0.2 mg/ml doses, respectively.
To facilitate processing of blinded samples, a coding system using sample number-passage number was applied. Virus sample numbers 1 through 15 were HSV-1, and 16 through 30 were HSV-2. The ears from one mouse were pooled (passage number 1 inoculation) and used to inoculate the ear pinnae of one mouse (passage number 2). There were five replicate mice per treatment group (either suboptimally treated with 0.2 mg/ml FCV, VCV or placebo-treated) and isolates from seven serial in vivo passages examined. Five individual plaques were purified three times to homogeneity from the single drug-resistant virus mixture identified after in vivo passage, 14-P4 (sample 14, passage 4). The clonal purified isolates from this sample were designated 14-P4A-E.
The in vivo pathogenicity of selected virus preparations was assessed in mice by monitoring lesion severity for 14 days following inoculation of the left ear pinna with 5 × 105 PFU. Cumulative lesion scores (day 0–14) were then calculated from the individual daily scores.
Virus isolation and in vitro assays
Virus stocks were prepared as described previously [17], titrated and characterized by PRA and PEA. On day 4 post-infection, mice were euthanized, and both ear pinnae removed and pooled in coded tubes, one tube per mouse. The code was not broken until all tissue culture assays had been completed. The tissue was homogenized and virus stocks were prepared in MRC-5 cells from cell-free supernatants. Following virus adsorption onto the MRC-5 monolayer, two washes with phosphate-buffered saline were performed to remove any residual antiviral drug. Cell-free virus was harvested, titrated and characterized by PRA and PEA prior to use in subsequent passage experiments (Figure 1). All PRA, PEA and in vivo studies were carried out using virus that had undergone a single in vitro passage. PRAs were performed in MRC-5 cells as described previously [17]. The plating efficiency of the virus preparations generated in vivo was determined according to the method developed by Hall et al. with minor modifications [13] as reported previously [17]. Briefly, six serial 10-fold dilutions of virus were inoculated onto MRC-5 cells in the absence of antiviral drug or in the presence of either ACV or PCV at a concentration of antiviral 10 times higher than the average IC50 for wild type parental HSV-1 and HSV-2, respectively. The mutation frequency was calculated as follows: mutation frequency = (virus titer in the presence of drug)/(virus titer in the absence of drug). The frequency is often expressed as a plating efficiency, or percentage of viruses that are resistant, which is calculated as percent resistant = (mutation frequency) × 100.
Figure 1.
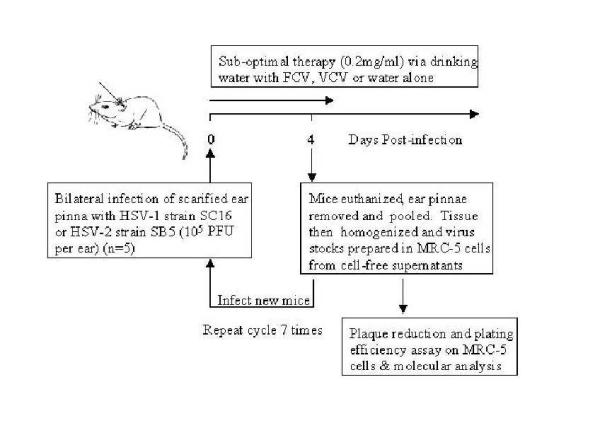
Experimental design for in vivo selection of drug-resistant HSV. This cartoon depicts the methodology used to select drug-resistant virus. Both ear pinnae of each mouse were infected with HSV. There were five replicate mice per treatment group (treated suboptimally with 0.2 mg/ml FCV or VCV, or with placebo) and isolates from seven serial in vivo passages examined. The resulting virus preparation from the pooled ears of an individual mouse (sample number 1, passage number 1) were used to inoculate both ear pinnae of one mouse for the subsequent serial passage (sample number 1, passage number 2).
Antibodies and biochemical assays
Western blot analysis was performed as described previously [18]. Viral TK activity was determined by a modification of the method utilized by Coen and Schaffer [4] as described in [17]. Primers utilized for TK sequencing were described previously [18].
Results
Virus yield after antiviral treatment
To evaluate efficacy of prodrug treatment in this model, VCV and FCV were provided in the drinking water at 1.0 mg/ml or at 0.2 mg/ml (sub-optimal treatment) for mice infected with either HSV-1 SC16 or HSV-2 SB5. Titers (day 4 post-infection) of approximately 4.5 log10 virus per ear were recovered from placebo-treated mice, regardless of virus type (Table 1). As expected, a significant reduction in virus yield (greater than 3.5 log10 decrease) was noted for mice treated with either VCV or FCV at 1.0 mg/ml following infection with either HSV-1 or HSV-2.
Table 1.
Virus Titer Day 4 post-infection in mice inoculated with HSV-1 strain SC16 or HSV-2 strain SB5 and treated orally with Placebo, FCV or VCV
| Virus Titer (log10 PFU/ear) | |||||
| Virus inoculumb | Placebo | FCV | VCV | ||
| 0.2 mg/mlc | 1.0 mg/mlc | 0.2 mg/mlc | 1.0 mg/mlc | ||
| HSV-1 (SC16) | 4.54 ± 0.04a | 2.92 ± 0.23 (2.4)d | 1.0 ± 0.21 (0.03) | 2.45 ± 0.15 (0.80) | 0.74 ± 0.35 (0.02) |
| HSV-2 (SB5) | 4.73 ± 0.30 | 2.55 ± 0.25 (0.67) | 0.98 ± 0.13 (0.02) | 2.06 ± 0.17 (0.20) | 0.72 ± 0.44 (0.01) |
aGeometric mean titer ± standard deviation of virus obtained from the ears of five mice and titrated independently. bA total of 105 PFU was inoculated into each ear pinna. cTreatment in the drinking water from the time of virus inoculation. Mice drank approximately 1.5 ml per day. No differences in intake volume or spillage between treatment groups were evident. dPercent virus yield compared to placebo-treated mice.
In the same experiment, virus yield from suboptimally-treated mice was reduced approximately 1.6–2.6 log10 compared to placebo-treated mice, and these data are consistent with a previous report in which ACV was administered at a concentration of 0.25 mg/ml [10]. At 0.2 mg/ml, viral replication, albeit reduced, was still ongoing, a prerequisite for the in vivo selection of resistant HSV.
A reproducible difference in virus yield between the two antiviral treatment groups at the suboptimal concentration was evident, regardless of virus type. For HSV-1, the difference in titer between VCV-treated mice (2.45 log10 PFU/ml) and FCV-treated (2.92 log10 PFU/ml) was statistically significant (p = 0.049). Similarly for HSV-2, VCV at 0.2 mg/ml was significantly more effective than FCV in this mouse model. Titers from VCV- or FCV-treated mice were 2.06 log10 PFU/ml or 2.55 log10 PFU/ml, respectively (p = 0.001). Similar trends were noted when the antivirals were compared at 1.0 mg/ml for both HSV-1 and HSV-2 (Table 1). These observations can not be accounted for by differences in exposure to ACV and PCV since oral administration of VCV and FCV results in almost identical blood concentration-time curves for ACV and PCV [11]. A more probable explanation is that ACV is approximately four-fold more potent than PCV in murine cells [21].
To select and amplify drug-resistant HSV in mice, a total of seven consecutive passages were performed, and progeny virus from each passage was characterized in vitro by PRA and PEA.
PRA susceptibility
IC50s were determined for each virus sample by the PRA. The parental control virus stock (which was not passaged in mice) was tested in parallel to provide a baseline for every PRA. Consistent with previous studies [2], IC50s for PCV tended to be marginally higher than for ACV, and IC50s were higher for HSV-2 SB5 than for HSV-1 SC16. The in vitro breakpoint for defining ACV resistance (2.0 μg/ml) and the provisionally-defined PCV resistance breakpoint, an IC50 greater than 10-fold above the wild-type control virus IC50 within a particular assay [17], were utilized.
Drug-selected HSV-1 SC16
Mean PCV susceptiblities of the HSV-1 isolates (n = 105) from passages 1 through 7 are listed in Table 2. All isolates from mice treated with either placebo or FCV remained sensitive to PCV (IC50s ranging from 0.28–1.50 μg/ml) and to ACV (results not shown) according to the standard resistance breakpoint criteria, whereas isolates from mice treated with VCV had PCV IC50s ranging from 0.31–8.3 μg/ml. One isolate (4th passage; 14-P4) in this treatment group was confirmed as resistant to both PCV (IC50 = 8.3 μg/ml) and ACV (IC50 = 9.5 μg/ml). Generally, test sample IC50s varied within two-fold of the non-passaged control virus IC50, within each particular assay (data not shown); however, isolate 14-P4 was clearly different, with an IC50 greater than 11-fold over the control virus IC50 for both PCV and ACV.
Table 2.
Susceptibility of serial passaged HSV-1 SC16 isolates to PCV as measured by the PRA.
| Parental SC16a | Passage No. | PCV IC50 (μg/ml) | ||
| Placebo | FCV | VCV | ||
| 0.39 | 1 | 0.36 ± 0.04b | 0.42 ± 0.07 | 0.42 ± 0.05 |
| 0.57 | 2 | 0.48 ± 0.16 | 0.76 ± 0.23 | 0.74 ± 0.35 |
| 0.90 | 3 | 0.33 ± 0.08 | 0.50 ± 0.24 | 0.49 ± 0.36 |
| 0.73 | 4 | 1.13+0.46 | 1.31 ± 0.42 | 2.73 ± 3.11c |
| 0.77 | 5 | 1.15 ± 0.24 | 0.68 ± 0.15 | 0.68 ± 0.07 |
| 0.70 | 6 | 0.79 ± 0.13 | 0.74 ± 0.09 | 0.80+0.12 |
| 0.70 | 7 | 0.81 ± 0.38 | 0.57 ± 0.27 | 0.66 ± 0.24 |
aIC50 against PCV for the original parental control virus preparation (not passaged in mice). bIC50 against PCV ± standard deviation for test isolates for n = 5 samples. cIC50 (μgml) PCV for samples 26–30 ranged from 1.09–8.3.
Interestingly, although 14-P4 was resistant, the third pass isolate for this sample was completely sensitive to both antiviral agents (ACV IC50 = 0.16 μg/ml; PCV IC50 = 0.53 μg/ml). Therefore, the PRA provided no early indication at pass 3 of potentially increasing resistant variants upon subsequent passage. Furthermore, the resulting virus preparation from subsequent passaging of 14-P4 reverted to a sensitive phenotype (ACV IC50 = 0.74 μg/ml; PCV IC50 = 0.74 μg/ml), although suboptimal treatment was still utilized.
Drug-selected HSV-2 SB5
Mean PCV susceptiblities of the HSV-2 isolates (n = 105) from passages 1 through 7 are listed in Table 3. No resistant HSV-2 emerged among the total of 105 HSV-2 isolates tested from the three treatment groups (PCV IC50s ranged from 0.66–1.85 μg/ml across treatment groups). These results were confirmed when the same isolates were tested for susceptibility to ACV (results not shown) and all the IC50s were sensitive over a similar range as PCV. As with HSV-1, the HSV-2 test samples were generally within two-fold of the control virus IC50 (data not shown).
Table 3.
Susceptibility of serial passaged HSV-2 SB5 isolates to PCV as measured by the PRA
| Parental SB5a | Passage No. | PCV IC50 (μg/ml) | ||
| Placebo | FCV | VCV | ||
| 0.79 | 1 | 0.76 ± 0.08b | 0.78 ± 0.05 | 0.87 ± 0.09 |
| 1.02 | 2 | 0.98 ± 0.08 | 0.82 ± 0.23 | 0.96 ± 0.27 |
| 1.06 | 3 | 1.10 ± 0.23 | 1.04 ± 0.22 | 0.87 ± 0.04 |
| 1.43 | 4 | 1.55 ± 0.15 | 1.02 ± 0.51 | 1.14 ± 0.58 |
| 1.11 | 5 | 1.32 ± 0.38 | 1.47 ± 0.48 | 1.14 ± 0.18 |
| 0.80 | 6 | 0.96 ± 0.18 | 0.81 ± 0.05 | 0.85 ± 0.16 |
| 0.90 | 7 | 0.81 ± 0.08 | 0.77 ± 0.05 | 0.82 ± 0.12 |
aIC50 against PCV for the original parental control virus preparation (not passaged in mice). bIC50 against PCV ± standard deviation for test isolates for n = 5 samples.
PEA profiling
All isolates were tested by the PEA, a test which may provide a more sensitive indicator of emergence of drug resistance than the PRA, since it measures the proportion of resistant isolates in the population. The parental control virus stock was tested in parallel in order to provide a baseline for every PEA.
Drug-selected HSV-1 SC16
The HSV-1 control virus preparation contained between 0.002–0.02% ACVr virus and 0.004–0.009% PCVr, which equates to an average of 6.6 ACVr and 6.0 PCVr resistant viruses per 105 PFU. Generally, the proportion of PCVr virus varied only two-fold between tests when compared to the control virus, ranging from 0.004 to 0.009% (data not shown); however, three serial samples in the VCV-treatment group, 14-P2, 14-P3 and 14-P4, clearly showed a substantial increase in the proportion of resistant virus relative to the wild type control, specifically an 11-, 300- and 8800-fold increase, respectively (14-P2, 0.002%; 14-P3, 2%; and 14-P4, 44% PCVr). Although passage 3 preparation contains a two log increase in resistant virus compared to passage 2, this level is insufficient to significantly increase the IC50. Consistent with the failure to establish an infection refractory to treatment, the proportion of resistant virus decreased to 0.08% PCVr and 0.05% ACVr by passage 5.
Drug-selected HSV-2 SB5
The HSV-2 control virus preparation contained between 0.24–0.40% ACVr and 0.13–0.40% PCVr virus, which equates to an average of 3.1 ACVr and PCVr viruses per 103 PFU. Generally, the proportion of resistant virus varied only three-fold between tests when compared to the control virus (data not shown), however a continual increase in the proportion of resistant virus was seen with the sample 30 passages (VCV treatment group). The proportion of PCVr virus in sample 30 increased from 0.40% to 2.8% to 12.2% throughout serial passages 1 through 3 and declined to 0.2% by passage 4. All of these isolates were susceptible to PCV by the PRA (IC50s for sample 30, passage 2 through 4, were 0.79, 0.90 and 1.2 μg/ml, respectively).
Analysis of drug-resistant isolate
Of 210 HSV isolates from mice treated sub-optimally with FCV or VCV, or with placebo, only one HSV-1 isolate, 14-P4 was identified in the PRA as an antiviral-resistant isolate. Five single plaques from 14-P4 were isolated after three rounds of plaque purification and characterized. The clonal isolates were tested for susceptibility to using the PRA and were found to be highly resistant to ACV (ACV IC50s > 25 μg/ml) and PCV (Table 4; PCV IC50s > 25 μg/ml), although susceptible to foscarnet.
Table 4.
Biochemial Characterization of 14-P4 clonal plaque isolates.
| Clone | 14-P4A | 14-P4B | 14-P4C | 14-P4D | 14-P4E | WT SC16 |
| TK activity | 0.3% | 0.4% | 0.5% | 0.3% | 0.3% | 100% |
| PCV IC50 | >25 μg/ml | >25 μg/ml | >25 μg/ml | >25 μg/ml | >25 μg/ml | 0.3 μg/ml |
| PFA IC50 | 48 μg/ml | 62 μg/ml | 55 μg/ml | 80 μg/ml | 55 μg/ml | 64 μg/ml |
Western blot analysis showed that each clonal isolate produced a truncated TK polypeptide compared to the wild type TK protein product of approximately 43 kD (Figure 2). Moreover, all five clones were defective for TK activity compared with HSV-1 SC16 in a 3H-thymidine kinase assay (Table 4). Sequence analysis of the TK coding region from all five plaque isolates identified a G nucleotide insertion in the homopolymeric hot-spot G7 tract at nucleotide 435, a common mutation present in other ACVr HSV isolates [18,19]. This base insertion produced a frame shift in the coding sequence, resulting in a loss of the ATP / nucleoside-binding site and the creation of a premature stop codon at amino acid 228 to encode a truncated 28 kD TK polypeptide.
Figure 2.
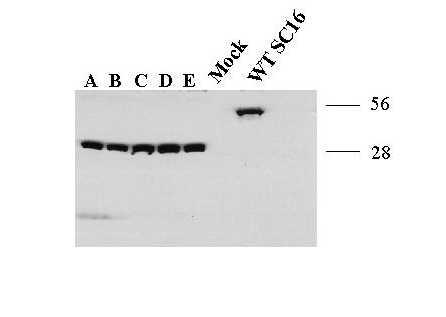
Biochemical characterization of clonal plaque isolates derived from 14-P4 in MRC-5 cells. Western blot analysis of TK protein products from five clonal isolates of virus sample 14-P4. Lanes A through E represent infected cell protein extract from isolates 14-P4A, 14-P4B, 14-P4C, 14-P4D and 14-P4E. Mock represents uninfected MRC-5 cell protein extract. Wild type TK, indicated as WT SC16, is a 43 kD polypeptide expressed from an HSV-1 SC16 infected cell extract.
Lastly, the in vivo pathogenicities of 14-P4 and one of its clonal isolates (14-P4E) were compared with the control virus, HSV-1 SC16. Isolate 14-P4 containing 45% resistant virus caused significant clinical disease in mice, only marginally less severe than that observed with HSV-1 SC16. In marked contrast, mice inoculated with the plaque-purified isolate, 14-P4E, developed a mild infection. The cumulative disease scores for parental virus control HSV-1 SC16, 14-P4 and 14-P4E were 136, 96 and 15, respectively.
Discussion
HSV-1 or HSV-2 isolates were passaged serially seven times in mice inoculated in the ear pinnae and treated with suboptimal (15 mg/kg per day) concentrations of FCV or VCV to select resistant virus. Despite ongoing, albeit reduced, viral replication in the animals, selection of resistant HSV with the prodrugs proved difficult. Interestingly, the single drug-resistant isolate identified from this study reverted to a sensitive phenotype upon reinfection, even with suboptimal antiviral treatment.
Although virus recovered at passage 4 from one of five mice treated with VCV (14-P4) was clearly resistant in the PRA (IC50s 8.3 and 9.5 for PCV and ACV, respectively), the preceding and the subsequent isolates were sensitive according to the criteria for defining resistance. The PEA identified a gradual increase in the proportion of resistant virus from 14-P2 (0.002%) to 14-P3 (~2%) to 14-P4 (44%). However, for the most part, the PEA data did not correlate with the eventual appearance of drug-resistant virus as defined by the PRA. Consistent with this, an earlier studied showed that the proportion of drug-resistant virus in a preparation needs to be approximately 50% before resulting in a signficant change in IC50[9].
Biochemical characterization of the 14-P4 isolate confirmed that impaired TK activity was most likely responsible for the resistance phenotype. Sequence analysis of one of these clonal isolates (14-P4E) revealed insertion of an extra G within the G7 homopolymeric region of the TK coding region. This mutation has also been detected in clinical isolates from patients immunocompromised [12] or with AIDS [19] as well as resistant HSV selected in tissue culture [15,18]. Interestingly, this mutation was present in ACVr clinical isolates obtained during reactivation in the absence of drug pressure, as well as in ACVr strains which evolved during ACV therapy and were followed by ACV-susceptible isolates at the next reactivation [19].
It was surprising to find that the ACV-resistant virus selected in vivo was not retained or amplified in subsequent passages despite continued suboptimal treatment with VCV. This result appears to be in direct contrast to that of Field [9] who found that following selection of ACV-resistant virus in vivo, such viruses remained refractory to treatment upon subsequent passage using suboptimal treatment with ACV. The reason for this difference is unclear. However, the failure to maintain the resistant virus on subsequent passage suggests that the pathogenicity of 14-P4 may have been only partially attenuated relative to the sensitive virus and indeed the pathogenicity of the clonal virus isolate, 14-P4E, was severely attenuated. Further studies on the 14-P4 clonal isolates and the clinical significance of their attentuation should be explored.
Conclusions
Sub-optimal therapy of immunocompetent mice with either VCV or FCV did not readily select for HSV-mutants resistant to either ACV or PCV, suggesting that selection of resistance with either prodrug remains difficult using this system. All the virus isolates from each serial passage were examined by both PRA and PEA. The former is widely accepted as the gold standard for susceptibility testing of HSV, and for ACV, a correlation between PRA in vitro IC50 and clinical outcome has been established. In spite of this, the PEA appears to have potential as an adjunct to the PRA by providing earlier detection of the emergence of resistant HSV. However, it is important to note that although an increase in the number resistant variants within isolates was readily detected by the PEA, clinically-resistant virus did not develop in the immunocompetent mice. Therefore, the presence of resistant virus within an isolate, even at levels substantially above background, does not in itself indicate that increased resistance will follow, nor does it necessarily predict clinical resistance. It would be of interest to apply the PEA to an extensive series of clinical isolates, particularly isolates obtained from severely immunocompromised patients, in order to evaluate the proportions of resistant virus that occur within different clinical isolates.
Competing Interests
None declared.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgements
We thank S. Albelda, S. Safrin and P. Schaffer for generous gifts of reagents; P. Patel and N. Simon for technical assistance; B. Gagnon for statistical analyses; R. Boon for financial support of the Consumer Healthcare division; SB Laboratory Animal Science for expert handling and care of animals in accordance with the guidelines of the SB Animal Care and Use Committee; and K. Esser for support.
Contributor Information
Robert T Sarisky, Email: robert_t_sarisky@gsk.com.
H Ron Bartus, Email: henry_ron_bartus@gsk.com.
Shelley A Dennis, Email: shelley_a_dennis@gsk.com.
Matthew R Quail, Email: mquail@tektagen.com.
Tammy T Nguyen, Email: tammy_t_nguyen@gsk.com.
Robert J Wittrock, Email: robert_j_wittrock@gsk.com.
Wendy S Halsey, Email: wendy_s_halsey@gsk.com.
Teresa H Bacon, Email: teresa_h_bacon@gsk.com.
Jeffry J Leary, Email: jjl27773@gsk.com.
David Sutton, Email: david_sutton@gsk.com.
References
- Boyd MR, Bacon TH, Sutton D. Antiherpesvirus activity of 9-(4-hydroxy-3-hydroxymethylbut-1-yl) guanine (BRL 39123) in animals. Antimicrob Agents Chemother. 1988;32:358–363. doi: 10.1128/aac.32.3.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd MR, Bacon TH, Sutton D, Cole M. Antiherpesvirus activity of 9-(4-hydroxy-3hydroxymethylbut-1-yl)guanine (BRL 39123) in cell culture. Antimicrob Agents Chemother. 1987;31:1238–1242. doi: 10.1128/aac.31.8.1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd MR, Safrin S, Kern ER. Penciclovir – a review of its spectrum of activity, selectivity, and cross-resistance pattern. Antiviral Chem Chemother. 1993;4:3–11. [Google Scholar]
- Coen DM, Kosz-Vnenchak M, Jacobson JG, Leib DA, Bogard CL, Schaffer PA, Tyler KL, Knipe DM. Thymidine kinase-negative herpes simplex virus mutants establish latency in mouse trigeminal ganglia but do not reactivate. Proc Natl Acad Sci USA. 1989;86:4736–4740. doi: 10.1073/pnas.86.12.4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins P, Darby G. Laboratory studies of herpes simplex virus strains resistant to acyclovir. Rev Med Virol. 1991;1:19–28. [Google Scholar]
- Dasgupta UB, Summers WC. Ultraviolet reactivation of herpes simplex virus is mutagenic and inducible in mammalian cells. Proc Natl Acad Sci USA. 1978;75:2378–2381. doi: 10.1073/pnas.75.5.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubbs DR, Kit S. Mutant strains of herpes simplex deficient in thymidine kinase-inducing activity. Virology. 1964;22:493–502. doi: 10.1016/0042-6822(64)90070-4. [DOI] [PubMed] [Google Scholar]
- Efstathiou S, Kemp S, Darby G, Minson AC. The role of herpes simplex virus type 1 thymidine kinase in pathogenesis. J Gen Virol. 1989;70:869–879. doi: 10.1099/0022-1317-70-4-869. [DOI] [PubMed] [Google Scholar]
- Field HJ. Development of clinical resistance to acyclovir in herpes simplex virus-infected mice receiving oral therapy. Antimicrob Agents Chemother. 1982;21:744–752. doi: 10.1128/aac.21.5.744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field HJ, Darby G, Wildy P. Isolation and characterization of acyclovir-resistant mutants of herpes simplex virus. J Gen Virol. 1980;49:115–124. doi: 10.1099/0022-1317-49-1-115. [DOI] [PubMed] [Google Scholar]
- Field HJ, Tewari D, Sutton D, Thackray AM. Comparison of efficacies of famciclovir and valaciclovir against herpes simplex virus type 1 in a murine immunosuppression model. Antimicrob Agents Chemother. 1995;39:1114–1119. doi: 10.1128/aac.39.5.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudreau A, Hill E, Balfour HH, Erice A, Boivin G. Phenotypic and genotypic characterization of acyclovir-resistant herpes simplex viruses from immunocompromised patients. J Infect Dis. 1998;178:297–303. doi: 10.1086/515626. [DOI] [PubMed] [Google Scholar]
- Hall JD, Coen DM, Fisher BL, Weisslitz M, Randall S, Almy RE, Gelep PT, Schaffer PA. Generation of genetic diversity in herpes simplex virus: an antimutator phenotype maps to the DNA polymerase locus. Virology. 1984;132:26–37. doi: 10.1016/0042-6822(84)90088-6. [DOI] [PubMed] [Google Scholar]
- Hill EL, Hunter GA, Ellis MN. In vitro and in vivo characterization of herpes simplex virus clinical isolates recovered from patients infected with human immunodeficiency virus. Antimicrob Agents Chemother. 1991;35:2322–2328. doi: 10.1128/aac.35.11.2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang CC, Chen HH. An altered spectrum of herpes simplex virus mutations mediated by an antimutator DNA polymerase. Gene. 1995;152:191–193. doi: 10.1016/0378-1119(94)00712-2. [DOI] [PubMed] [Google Scholar]
- Jacobson JG, Ruffner KL, Kosz-Vnenchak M, Hwang CBC, Knipe DM, Coen DM. Herpes simplex virus thymidine kinase and specific stages of latency in murine trigeminal ganglia. J Virol. 1993;67:6903–6908. doi: 10.1128/jvi.67.11.6903-6908.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarisky RT, Nguyen TT, Duffy KE, Wittrock RJ, Leary JJ. Difference in incidence of spontaneous mutations distinct between herpes simplex virus types 1 and 2. Antimicrob Agents Chemother. 2000;44:1524–1529. doi: 10.1128/AAC.44.6.1524-1529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarisky RT, Quail MR, dark PE, Nguyen TT, Halsey WS, Wittrock RJ, Bartus JO, Van Horn MM, Sathe GS, Van Horn S, Kelly MD, Bacon TH, Leary JJ. Characterization of herpes simplex viruses selected in culture for resistance to penciclovir or acyclovir. J Virol. 2001;75:1761–1769. doi: 10.1128/JVI.75.4.1761-1769.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasadeusz JJ, Tufaro F, Safrin S, Schubert K, Hubinette MM, Cheung PK, Sacks S. Homopolymer mutational hot spots mediate herpes simplex virus resistance to acyclovir. J Virol. 1997;71:3872–3878. doi: 10.1128/jvi.71.5.3872-3878.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soullawton J, Seaber E, On N, Wootton R, Rolan P, Posner J. Absolute bioavailability and metabolic disposition of valaciclovir, the l-valyl ester of acyclovir, following oral administration to humans. Antimicrob Agents Chemother. 1995;39:2759–2764. doi: 10.1128/aac.39.12.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton D, Boyd MR. Comparative activity of penciclovir and acyclovir in mice infected intraperitoneally with herpes simplex virus type 1 SC16. Antimicrob Agents Chemother. 1993;37:642–645. doi: 10.1128/aac.37.4.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenser RB, Edris WA. Trigeminal ganglion infection by thymidine kinase-negative mutants of herpes simplex virus after in vivo complementation. J Virol. 1987;61:2171–2174. doi: 10.1128/jvi.61.7.2171-2174.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thackray AM, Field HJ. Differential effects of famciclovir and valaciclovir on the pathogenesis of herpes simplex virus in a murine infection model including reactivation from latency. J Infect Dis. 1996;173:291–299. doi: 10.1093/infdis/173.2.291. [DOI] [PubMed] [Google Scholar]
- Vere HR, Sutton D, Boyd MR, Harnden MR, Jarvest RL. Selection of an oral prodrug (BRL 42810; famciclovir) for the antiherpesvirus agent BRL 39123 [9-(4-hydroxy-3-hydroxymethylbut-1-yl)guanine; penciclovir]. Antimicrob Agents Chemother. 1989;33:1765–1773. doi: 10.1128/aac.33.10.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]