

Abstract
The production of photochemical atmospheres under controlled conditions in an irradiation chamber permits the manipulation of parameters that influence the resulting air-pollutant chemistry and potential biological effects. To date, no studies have examined how contrasting atmospheres with a similar Air Quality Health Index (AQHI), but with differing ratios of criteria air pollutants, might differentially affect health end points. Here, we produced two atmospheres with similar AQHIs based on the final concentrations of ozone, nitrogen dioxide, and particulate matter (PM2.5). One simulated atmosphere (SA-PM) generated from irradiation of ∼23 ppmC gasoline, 5 ppmC α-pinene, 529 ppb NO, and 3 μg m−3 (NH4)2SO4resulted in an average of 55 μg m−3 PM2.5, 643 ppb NO2, and 430 ppb O3 (AQHI of 99.8). Chemical The other atmosphere (SA-O3) generated from 8 ppmC gasoline, 5 ppmC isoprene, 874 ppb NO, and 2 μg m−3 (NH4)2SO4resulted in an average of 55 μg m−3 PM2.5, 643 ppb NO2, and 430 ppb O3 (AQHI of 99.8). Chemical speciation by gas chromatography showed that photo-oxidation degraded the organic precursors and promoted the de novo formation of secondary reaction products such as formaldehyde and acrolein. Further work in accompanying papers describe toxicological outcomes from the two distinct photochemical atmospheres.
Introduction
It is well-recognized that exposure to specific criteria air pollutants, including O3, PM2.5, and NO2, results in a broad array of adverse health outcomes such as reduced lung function, cardiopulmonary inflammation, increased risk or severity of respiratory and cardiovascular disease, cancer, and, in some instances (e.g., PM), increased mortality.(1–3) Ascribing causality among components of regional complex multipollutant mixtures to health effects in epidemiology studies, however, is challenging because the aforementioned criteria pollutants often track together. Thus, it is not always clear whether the resulting health effects are consequence of additive, antagonistic, or synergistic interactions of the components of air pollution.(4)
The U.S. Environmental Protection Agency (EPA) air quality index (AQI) provides public health guidance on potential hazards of each of the criteria pollutants regulated by the clean air act (O3, PM, SO2, NO2, and CO). Although the AQI is a valuable forecasting tool for public health officials, communities, and the general public, it does not incorporate quantitative information for mixtures of air pollutants. An Air Quality Health Index (AQHI) was developed to provide an integrated assessment of the risk attributed by three air pollutants; O3, PM, and NO2.(5) Although this model appears to be more sensitive than the one-pollutant AQI system,(6) it has not been applied in toxicological investigations to compare the relative potency of air-pollution mixtures.
Health effects of air pollutants in regional and urban atmospheres are associated with primary source emissions and secondary reaction products such as ozone and secondary organic aerosol (SOA). Selected volatile organic compounds (VOCs), including benzene, 1,3-butadiene, formaldehyde (HCHO), and selected higher-molecular-weight (MW) aldehydes, are associated with increased cancer risk.(1,7) Although formaldehyde and acetaldehyde are produced from primary sources, in the atmosphere, their formation is generally dominated by reactive secondary processes of hydrocarbon precursors emitted into the atmosphere.(8) The importance of atmospheric transformation products has been recognized within the U.S. Clean Air Act,(9) which mandates that the EPA “conduct a program of research with respect to sources of hazardous air pollutants and shall include consideration of atmospheric transformation and other factors that can elevate public health risks from such pollutants”.
Earlier studies showed that photochemical reaction with either single VOCs (toluene, acetaldehyde, allyl chloride, or propylene)(10–14) or a complex mixture such as wood smoke(15)produced atmospheres whose gas-phase mutagenicity in Salmonella was due exclusively to the formation of secondary reaction products. Peroxyacetyl nitrates (PAN) were among the important photochemical products identified, and the mutagenic potencies of various forms of PAN-type analogues in Salmonella ranged over a factor of 10.(16,17)
In many airsheds, SOA can comprise a substantial fraction of PM2.5(18,19) resulting in an increasing number of studies investigating the health effects of SOA.(20–28) Laboratory study of these processes typically requires photo-oxidized mixtures to be generated in a controlled manner.(5) Acute and chronic exposures to such atmospheres can thus be used with cellular and selected animal models representing both healthy and at-risk populations.
Automobiles emit a large fraction of urban and regional VOCs and oxides of nitrogen (NOX). In 2011 automobiles and other on-road vehicle sources contributed 33% of the NOX and nearly one-quarter of the VOC emitted in the United States.(29) With the inclusion of off-road motorized vehicles, these values increased by ∼50%.(29) In California (during the 2010 CalNex Study), these precursors were reported to be the major source of secondary PM.(30)
In the present work, chemical mixtures that were designed to approximate urban and regional evaporative pollutant emissions were photo-oxidized, resulting in atmospheres with roughly equivalent AQHI values.(31) The atmospheres were characterized through several analytical techniques and reaction products discussed. In accompanying papers, the atmospheres’ short-term health effects in Salmonella and a variety of animal models are studied to assess whether their toxicological potencies were similar, as predicted by the AQHI.
Experimental Methods
Apparatus
General aspects of the experimental system are similar to those described previously.(32) A photochemical reaction chamber is housed within a 24 ft long trailer and is temperature controlled by two throttled-refrigerant air conditioners. The chamber is a nominal 14.27 m3 rectangular parallelepiped enclosure fabricated from stainless steel with interior walls coated with Teflon. Opposite lengthwise sides consist of clear panels of 5 mil FEP Teflon film. Directly outside the transparent walls are 120 4 ft fluorescent bulbs (60 UVB and 60 black lights) that provide radiation with an energy distribution from 300 to 400 nm, similar to that of solar radiation.(33) The bulbs approximate the relative NO2 and carbonyl photolysis rates as that produced by solar radiation.(34) Radiation intensity was monitored continuously with an integrating radiometer (Eppley Laboratory, Inc. Newport, RI).
Materials
The primary source of VOCs was a gasoline mixture produced by Southwest Research Institute (San Antonio, TX) for use in the EPA’s Tier 3 standard. Thus, the gasoline mixture was formulated with an ethanol component and a relatively high aromatic content. A gas chromatographic (GC) analysis of the fuel indicated an aggregate composition of: 8.1% ethanol, 39.9% paraffin, 7.9% olefin, 40.4% aromatic, 3.4% unknown, and 0.3% fused ring. For the distribution of measured hydrocarbons in the liquid mixture, the average carbon number was 6.5; for the headspace, the average carbon number was 6.0. A cylinder mixture was produced by adding 30 mL of the gasoline mixture to an evacuated high-pressure cylinder and then pressurizing with nitrogen (N2) to approximately 10 000 kPa.
For the purposes discussed later, α-pinene or isoprene was added to the chamber along with the gasoline to produce a hydrocarbon mixture giving enhanced PM or O3 concentrations during irradiation. Both biogenic hydrocarbons were obtained from Aldrich Chemical Co. (Milwaukee, WI) at the highest available purity (99%) and used without further purification. The nitrogenous oxidant used in the experiments was nitric oxide (NO), obtained as a 1% mixture in N2 from National Welders, Inc. Finally, to ensure that particle formation occurred through condensation onto preexisting particles, a 10 mg L−1 aqueous solution of ammonium sulfate ((NH4)2SO4) was nebulized into the chamber.
Instrumentation and Sampling
Various chemical properties of the gas-phase constituents were monitored during the course of the experiments. NO and total NOX were measured with a ThermoScientific (Model 42i, Thermo Environmental, Inc., Franklin, MA) oxides-of-nitrogen chemiluminescent analyzer. O3 was monitored with a ThermoScientific (Model 49i) UV photometric analyzer. VOC concentrations in the inlet manifold and the chamber were collected in a 3 L Teflon bag analyzed by GC-flame ionization detection (FID; Model 5890, Hewlett-Packard, Palo Alto, CA) using a cryogenic preconcentrator. Peroxyacetyl nitrate (PAN) compounds were measured by GC–electron capture detector (GC–ECD). In some cases, Teflon bag samples were transferred to 6 L electropolished canisters for later analysis. Additional details of sample collection and analysis are found in the Supporting Information.
Gas-phase carbonyls were determined using derivatization with a 1 mM solution of 2,4-dinitrophenylhydrazine (2,4-DNPH) in acetonitrile and analyzed by high-performance liquid chromatography–ultraviolet absorption spectroscopy (HPLC–UV).(35) Samples were collected by liquid impinger, derivatized, and injected into a Hewlett-Packard 1100 HPLC with an Agilent Zorbax ODS column (4.6 × 250 mm, 5 μm particles) using a ternary mobile phase of acetonitrile, methanol, and water. A diode array detector centered at 360 nm with a bandwidth of 10 nm was used to measure the absorption of the individual analytes. Additional details are found in the Supporting Information.
Temperature and relative humidity (RH) were measured with an Omega digital thermo-hydrometer (Model RH411, Omega Engineering, Inc., Stamford, CT) and maintained at a constant value of 30 or 35% using a proportional-integral-derivative (PID)-controlled peristaltic pump coupled to a high-temperature vaporizer. This allowed the RH in the chamber to remain at a stable value in spite of minor temperature fluctuations.
Physical and chemical properties of the particulate matter formed in the system were measured using a variety of techniques. The only source of organic particulate matter formed in the system was from photochemical reactions leading to the formation of SOA. The size distribution of the SOA was measured over a range from 16.8 to 947.5 nm using a scanning mobility particle sizer (SMPS; model 3081 DMA and model 3775 CPC; TSI, Inc., Shoreville, MN) with an X-ray source neutralizer. Organic mass (OM) concentration of the SOA was determined by collection on preweighed Zefluor PTFE filters (Pall Life Sciences). The OC concentration was determined by collection onto prebaked quartz filters with an activated carbon-strip denuder preceding the filter housing to limit gas-phase adsorption artifacts. A 1.5 cm2 standardized filter punch was measured using an elemental carbon (EC)–OC instrument (Sunset Laboratories, Tigard, OR) utilizing a thermal-optical technique described by Birch and Cary (1996).(36) A schematic of the system is shown in Figure 1.
Figure 1.
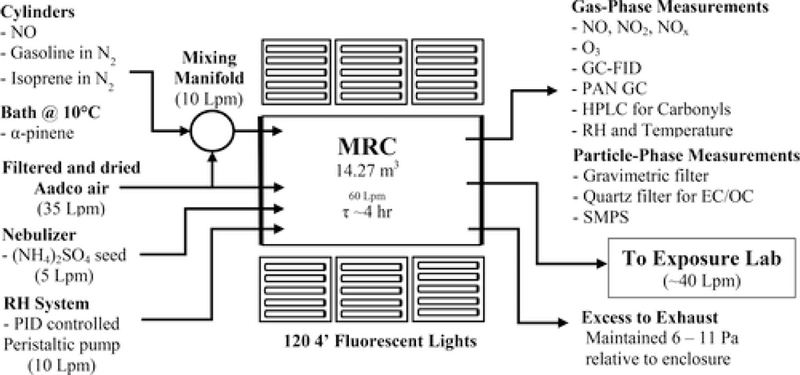
Diagram of MRC and measurement capabilities; not drawn to scale.
Experimental Procedures to Generate Smog
The smog chamber was operated as a continuous stirred tank reactor (CSTR). NO, gasoline mixture, and α-pinene or isoprene were the reactants. Reactants from high-pressure cylinders were continuously added to a mixing by mass-flow controllers. α-Pinene injections were achieved by passing air over the headspace of neat liquid in an impinger immersed in a silicon oil bath maintained at 10 °C. The (NH4)2SO4 solution nebulized into the chamber resulted in a background aerosol concentration of ∼2–4 μg m−3. The reactant mixture was diluted with zero air for a total flow rate of 60 L min−1, resulting in an effluent residence time of ∼4 h. The total flow provided sufficient effluent for both chemical analysis and bioassay exposures.
Reactants were initially introduced until the concentrations reached steady state. The fluorescent lights were turned on, initiating chemical transformations. Reactants continued to convert to products until a steady-state mixture of the reactants and products was attained. The level of conversion depended on the level of irradiation, initial concentrations, and residence time in the chamber. At least three residence times (12 h) were allowed before exposures were initiated.
A pair of sets of experiments were performed. In the first set (SA-PM; experiment nos. MR044, MR050, MR051, MR052, and MR055), the gasoline and α-pinene mixture was irradiated in the presence of NO. As was noted, SA-PM was designed to produce modest amounts of ozone and relatively high PM levels. This type of experiment was conducted multiple times to accommodate various biological assays. In the second set of experiments (SA-O3; MR059 and MR062), the gasoline and isoprene mixture was irradiated in the presence of NO. In contrast, SA-O3 produced relatively high levels of ozone and only modest levels of PM.
Results
The major considerations for selecting the conditions for the experiments included (a) having the two experimental types sufficiently different in ozone and PM concentrations to see statistical significance in the reaction mixtures (assuming effects are present), (b) that concentrations in the exposure chambers be sufficiently above background, and (c) that conditions be approximately scalable downward to ambient atmospheric conditions.
As was noted, experiments containing α-pinene (SA-PM) resulted in enhanced production of SOA as compared to isoprene-containing experiments. Double-bonded organics, such as α-pinene, improve the reactivity of the system for the short residence times in the chamber compared to the real atmosphere. The formation of SOA by this biogenic volatile organic compound (BVOC) is well-known, and its yield from ozonolysis products is on the order of 5–10% in the chamber. Although this approach may not produce the range of particulate constituents found typically in the atmosphere, it does fulfill the goal of obtaining a relevant metric for regulatory standards such as PM2.5 mass.
In SA-O3, isoprene was used as a means of enhancing ozone production due to its high OH rate constant and the production of new radicals by the photolysis of light carbonyl compounds, such as formaldehyde. The photo-oxidation of isoprene is known to generate SOA in addition to gas-phase products; however, the yield from such a reaction is generally <2% at concentrations found in the atmosphere.
Simulated Atmosphere with Enhanced Concentration of Particles (SA-PM)
The initial conditions for experiments to produce the atmosphere augmented by particles through the photooxidation of α-pinene are shown in Table 1 (MR044-MR055). Ozone is produced by OH-cycling reactions in the presence of NOX, and SOA is formed by photo-oxidation with OH or ozone as the oxidant, depending on the residual NOX levels. In these experiments, NOX concentrations were in the 0.5–0.6 ppm range except for MR052, which had a value of 0.42 ppm. The total hydrocarbon (α-pinene plus gasoline mix) concentrations for these experiments ranged from 27 to 31 ppmC. The 20 most abundant hydrocarbons in the initial chamber mixture are given in Table S1.
Table 1.
Initial Conditions
| SA-PM | SA-O3 | ||||||
|---|---|---|---|---|---|---|---|
| parameter | MR044 | MR050 | MR051 | MR052 | MR055 | MR059 | MR062 |
| NO (ppb) | 490.6 | 466.8 | 447.9 | 394.6 | 516.8 | 749.5 | 778.6 |
| NOx (ppb) | 528.2 | 512.5 | 491.2 | 420.0 | 547.2 | 861.2 | 887.3 |
| THCb (ppmC) | 29.9 | 28.5 | 31.0 | NAa | 27.2 | 12.4 | 14.0 |
| RH (%) | 29.8 | 35.1 | 34.9 | 34.6 | 34.9 | 35.1 | 35.0 |
| T (°C) | 21.0 | 20.9 | 21.2 | 22.1 | 22.1 | 21.6 | 21.9 |
| HC/NOx | 56.6 | 55.6 | 63.1 | NAa | 49.7 | 14.4 | 15.7 |
| volume concentration (nL m−3) | 3.3 | 3.6 | 3.8 | 3.4 | 2.5 | 1.8 | 2.2 |
| MMD (μm)c | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 |
Not available.
Total hydrocarbon (THC) measured by gas chromatography–flame ionization detector (GC–FID).
δ = 1.0.
The evolution over time of NOX and ozone before and after the chamber irradiation was initiated is shown in Figure 2. Under these high VOC-to-NOX ratios, the hydrocarbons and oxidant decreased rapidly. Before steady state was achieved, a transition period was seen in which the primary products exceed their steady-state concentrations. This effect results from the CSTR, whereby the initial reactant mixture in the chamber rapidly converted to products, while the added steady-state reactants tended to dilute the mixture until an overall steady-state concentration was achieved. The products of these experiments are given in Table 2. The NO was almost entirely consumed, and the remaining low concentration was typical of these experiments once secondary reactions reached steady state. At this point in the irradiation, a photostationary state of NO–NO2–O3existed and continued to generate NO, albeit at low levels. The self-reaction of peroxy radicals (HO2 and RO2) at this extent of reaction was able to compete with NO to form organic peroxides and hydroperoxides. There is also the possibility that RO2 reacts with NO2 to form aerosol as found in the experiments of Chan et al.(37) In this reaction, regime particles form at a more rapid rate due to the presence of these highly oxygenated organics.
Figure 2.
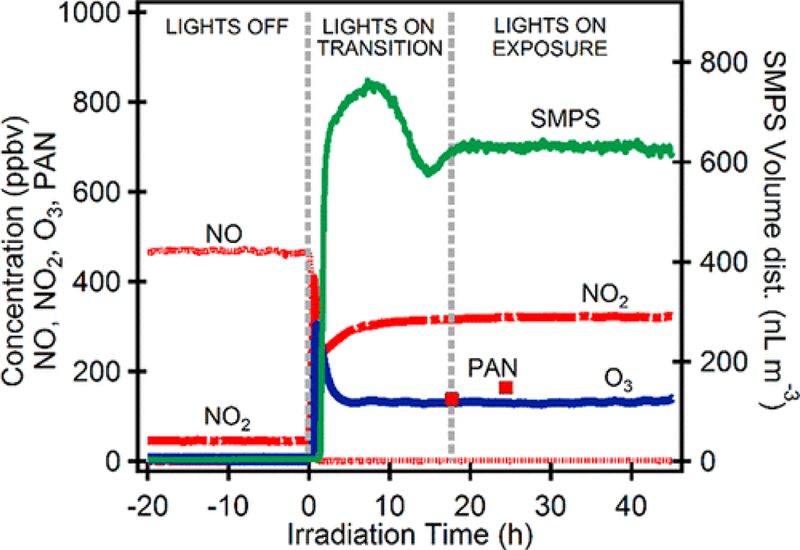
Plot of the SOA volume and major gas-phase components for SA-PM (MR050) for the photo-oxidation of the gasoline mixture with α-pinene. The figure shows the concentrations of the mixture from steady-state lights off, through transition, and, finally, once the system reached a steady-state condition.
Table 2.
Steady-State Concentrations of Inorganic Products and Reacted Hydrocarbons
| SA-PM | SA-O3 | ||||||
|---|---|---|---|---|---|---|---|
| concentration | MR044 | MR050 | MR051 | MR052 | MR055 | MR059 | MR062 |
| steady state | |||||||
| NO (ppb) | 0.8 | 1.7 | 1.1 | 1.1 | 1.0 | 1.5 | 1.1 |
| NOx (ppb) | 347 | 325 | 301 | 334 | 310 | 660 | 628 |
| O3 (ppb) | 188 | 133 | 141 | 124 | 102 | 447 | 413 |
| Δconcentration (ppmC) | |||||||
| a-pinene | 4.96 | 5.12 | 4.98 | NAa | 4.56 | – | – |
| isoprene | – | – | – | – | – | 5.03 | 4.90 |
| gasoline mix | 4.36 | 5.98 | 4.57 | NAa | 2.28 | 1.45 | 1.11 |
| Σreacted HC | 9.32 | 11.10 | 9.55 | NAa | 6.84 | 6.48 | 6.02 |
| products | 1.44 | 1.61 | 1.98 | NAa | 2.35 | 1.64 | 2.40 |
Not available.
The sum of reacted HC is shown in Table 2 along with the sum of organic products. For these experiments, approximately one-fifth of the reacted hydrocarbon could be identified as products, either as carbonyl products or from the peak growth of new products from the GC. Total organic products in the aerosol phase were determined from the OC steady-state values, as represented in Table 3. Organic carbon for the 5 enhanced PM2.5 experiments ranged from 473 to 517 μg m−3. The values were highly consistent with an independent measure of the organic mass (OM) derived from gravimetric filter samples (Table 3). The calculated OM-to-OC ratio also was highly consistent with values determined previously(38) and, in this case, averaged 2.04. The particle-phase OC concentration was wall-loss corrected and, along with the sum of reacted HC, converted to common units of μgC m−3 to perform a carbon balance estimation of the total gas-phase reaction products, as shown in Table 4. The effective SOA and SOC yields are also presented. Details of these calculations can be found in the Supporting Information.
Table 3.
Organic Aerosol Formed in the Particle Phase during Irradiations
| SA-PM | SA-O3 | ||||
|---|---|---|---|---|---|
| parameter | MR044 | MR050 | MR055 | MR059 | MR062 |
| [OC] (μgC m−3) | NAc | 473.7 | 517.0 | 24.3 | NAc |
| [OM] (μg m−3)a | 1036.2 | 998.6 | 954.7 | 52.2 | 56.7 |
| [OM]/[OC] | NAc | 2.11 | 1.85 | 2.15 | NAc |
| vol concentration (nL m−3) | 709b | 604b | 658b | 46 | 81 |
| MMDa (μm) | 0.59b | 0.59b | 0.60b | 0.28 | 0.31 |
| GSD | 1.52b | 1.52b | 1.52b | 1.64 | 1.64 |
δ = 1.0.
Volume concentration measured by SMPS. Measured size distribution was partially off-scale.
Not available.
Table 4.
Wall-Loss-Corrected Carbon Balance and Yields during Irradiations
| SA-PM | SA-O3 | ||||
|---|---|---|---|---|---|
| parameter | MR044 | MR050 | MR055 | MR059 | MR062 |
| [OC]corrected (μgC m−3)a | NAc | 662 | 722 | 4 | NAc |
| reacted HC (μgC m−3)b | 4542 | 5410 | 3335 | 3159 | 2934 |
| GP products (μgC m−3)d | NAc | 4749 | 2613 | 3125 | NAc |
| Unres. GP (μgC m−3)e | NAc | 3964 | 1467 | 2325 | NAc |
| SOA yield | 0.279 | 0.227 | 0.352 | 0.020 | 0.022 |
| SOC yield | NAc | 0.122 | 0.217 | 0.011 | NAc |
Corrected for wall loss rate of 0.0835 h−1, as determined from previous experiments.
Conversion based on calculated MW and carbon nos. for each experiment’s reacted HC. Details are given in the Supporting Information.
Not available.
Gas-phase reaction products estimated from reacted HC – [OC]corrected.
Unresolved gas-phase reaction products estimated from reacted HC – [OC]corrected – products (see Table 2).
The major carbonyl species detected in the chamber effluent are given in Table S2. Most of the aldehydes and dialdehydes likely originated mainly from the gasoline component of the reactant mixture. Glyoxal and methylglyoxal likely came mainly from the photo-oxidation of aromatic hydrocarbons, although small amounts are also likely to come from α-pinene oxidation.
Simulated Atmosphere with Enhanced Concentration of Ozone (SA-O3)
Initial conditions for experiments to produce the atmosphere with enhanced ozone used isoprene, as shown in Table 1 (MR059 and MR062). Enhanced concentrations of ozone were produced by OH-cycling reactions in the presence of NOX, the extremely high OH rate constant of isoprene, and isoprene’s high yield for low-molecular-weight reactive-carbonyl products aided in this process. The ozonolysis of isoprene was negligible in this reactive system. For these experiments, NOX concentrations averaged 0.87 ppm. Concentrations of isoprene plus the gasoline mix ranged from 12.4 to 14.0 ppmC. The time evolution of this mixture is shown in Figure 3, and the 20 most-abundant hydrocarbons in the initial chamber mixture are given in Table S2.
Figure 3.
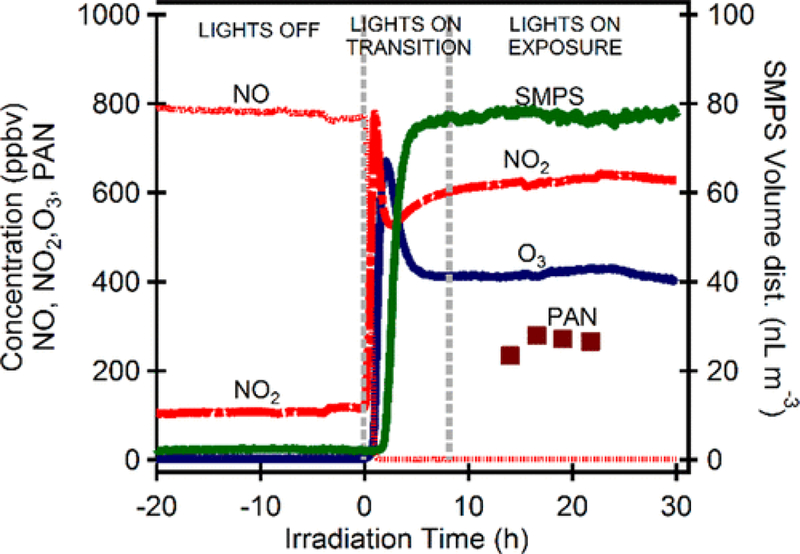
Plot of the SOA volume and major gas-phase components for SA-O3 (MR062) for the photo-oxidation of the gasoline mixture with isoprene. The figure shows the concentrations of the mixture from steady-state lights off, through transition, and finally once the system reached a steady-state condition.
Again, the NO was consumed almost entirely, and the remaining low concentration was typical of these experiments, allowing substantial reactive conversion of the carbonyl compound to new radicals by photolysis and OH through the photostationary state of NO–NO2–O3 present. The low NO concentrations allowed the self-reaction of peroxy radicals (HO2 and RO2) or reaction with NO2,(37) which compete favorably with NO to form organic peroxides and hydroperoxides. It is in this reaction regime that ozone formation can be enhanced considerably.
The sum of reacted HC and the sum of organic products are given in Table 2. For these experiments, approximately one-half of the initial hydrocarbon was reacted, and of that, ∼20% of the converted hydrocarbon could be identified as either carbonyl or other products from the peak growth seen in the GC. Organic products in the aerosol phase were determined from the OC steady-state values for MR059 (Table 3). OC measurements were not available in MR062 but would be expected to be similar to those in MR059. For MR059, the particle-phase organic carbon concentration was 24 μgC m−3, a factor of 20 less than the enhanced-particle experiments. Again, the values were highly consistent with the independent measure of the organic mass (OM) derived from gravimetric filter samples. The calculated OM-to-OC ratio was 2.15 for MR059. In this case, modest amounts of methylglyoxal may have come from initial reactions of isoprene with OH. The most-important carbonyls resulting from isoprene photo-oxidation were methacrolein and methyl vinyl ketone.
Discussion
Products from the Photo-oxidation of the Gasoline Components
As was noted in Table S1, the gasoline mixture was composed mainly of paraffins and aromatic hydrocarbons (approximately 40% each) and small concentrations of olefins and ethanol (8%). Although present in large abundance, paraffins react relatively slowly with OH radicals; thus, their conversion to products was small as detected by the GC measurements. Consequently, any observed toxicological effects were not due to specific products from these reactions. Olefins and higher-molecular-weight aromatics (>C6) are highly reactive with the OH radical and also can react rapidly with photochemically formed ozone. However, in our studies, the initial concentrations of olefins, while reactively generating small molecular weight carbonyls, were such that they contributed only modestly to the product mixture. In contrast, aromatic hydrocarbons were present in high abundance and reacted to form both gas and aerosol products. Many studies have examined both first- and later-generation products.(39) First-generation products of toluene include dicarbonyls, OH-ring-retaining compounds (e.g., methyl phenols), furan-type compounds, and unsaturated dicarbonyl compounds (e.g., 4-oxo-2-pentanal and 2-methyl-2-butenedial).(40)C8 and higher-molecular-weight aromatics generate these and additional products, such as diones.(41) The aerosol-phase products have been found largely to form from the condensation of highly oxygenated gas-phase products.(42)
Major Reaction Products
Under the conditions that generated each type of atmosphere, the major first-generation organic products formed were carbonyl compounds. With the high concentration of NOX in the mixture, peroxy radicals formed initially and reacted with NO to generate carbonyl compounds. The range of carbonyl compounds in each system are shown in Table S2. Most of the initial HCHO and CH3CHO were generated from the photooxidation of gasoline components, especially acetaldehyde produced from the photo-oxidation of ethanol. Carbonyl compounds specific to isoprene include methacrolein and methyl vinyl ketone. In addition to alkanes and alkenes, high yields of acetone are known to result from the OH reaction with α-pinene.(43) Dicarbonyl compounds (glyoxal and methylglyoxal) are produced in high yields from the OH reaction with aromatic hydrocarbons.(40,41) Although isoprene and α-pinene generated a modest yield of these dicarbonyls, their high abundance in the initial composition undoubtedly produced a significant fraction observed during these experiments. Linear carbonyls with carbon number greater than 2 (Table S2) were formed from alkanes and alkenes in the gasoline mixture.(39)
For SA-PM, the formation of SOA products included contributions from the photo-oxidation of α-pinene and aromatic hydrocarbons.(42) For SA-O3, the reaction of aromatic hydrocarbons contributed the major portion of the observed SOA with only a modest contribution due to isoprene.
Use of the Selected AQI
The AQI serves as the standard in the United States to which states and localities must adhere to be in compliance with the Clean Air Act. For ozone, the concentration for an 8 h period must be below 70 ppb; for PM2.5, 12 μg m−3 was an annual average, with 35 μg m−3 for a 24 h average; and for NO2, the concentration was 100 ppb over 1 h.(29) These standards function independently without consideration of additive or synergistic effects on human health. Thus, this research has explored the extent to which multiple pollutant mixtures give rise to similar or increase toxicological effects as measured by the selected bioassays.
The AQI is the source of color-coding that informs the general population of the health risks from polluted air on individual days throughout the year. There are seasonal effects that often influence pollutant levels. Except in certain situations, ozone tends to be elevated on hot, meteorologically stagnant days. Particulate matter, particularly PM from primary sources, tends to be increased during wintertime days in which shallow boundary layers and cloudy conditions prevent substantive radiative mixing from occurring. In contrast, PM and ozone can also be elevated in the summertime due to SOA formation.
In contrast to the AQI, the AQHI, which was built from mortality data associated with ambient air pollution, seeks to combine the cumulative effects of the three criteria air pollutants (PM2.5, NO2, and O3).(19,31) The construct for calculating such an index to give a single value comes from inclusion of these pollutants as represented by:
| (1) |
For use in ambient atmospheres, the relationship uses 3 h averages for O3 (ppb), NO2 (ppb), and PM2.5 (μg m−3). For purposes of this work, the equation served as a way to provide a health-based comparison for the constituents. Although the relationship itself gives contributions to the health metric for O3, NO2, and PM2.5, for purposes of the present work, the two gas-phase oxidant constituents were also determined as a single parameter to allow comparisons between the gas-phase criteria and particle-phase criteria. Thus, conventional chamber concentrations for these types of experiments tended to be 1 to 2 orders of magnitude higher than the values of the AQHI determined in ambient atmospheres.
The use of eq 1 for steady-state lights on concentrations measured in this study are found in Table 5. For the SA-PM set of experiments, the concentration of PM2.5 dominated the determination of the AQHI. Moreover, for the two gas-phase constituents, NO2 was weighted higher and had a higher concentration that O3. This resulted in a minor contribution of ozone to the AQHI (Table 5); the total oxidant contribution was compared against the PM2.5 in this evaluation. This approach has limitations, but it does represent a composite metric that begins to answer the question of whether gas-phase oxidants represent a greater hazard than does PM2.5 through the use of bioassays.
Table 5.
Comparison of the Calculated AQHI (Dimensionless) Using Eq 1 for Chamber Mixtures Having High Oxidant and Low PM vs Low Oxidant and High PM Concentrations
| SA | experiment | O3contribution | NO2 contribution (max) |
O3 + NO2contribution |
PM contribution |
totalb |
|---|---|---|---|---|---|---|
| PM | MR044 | 10.2 | 33.8 | 44.1 | 63.1 | 107.2 |
| PM | MR050 | 7.12 | 31.3 | 38.4 | 60.2 | 98.6 |
| PM | MR051 | 7.56 | 28.7 | 36.3 | 58.8 | 95.0 |
| PM | MR052 | 6.62 | 32.3 | 39.0 | 58.8 | 97.7 |
| PM | MR055 | 5.41 | 29.7 | 35.1 | 56.9 | 92.0 |
| O3 | MR059 | 26.1 | 74.5 | 100.6 | 2.48 | 103.0 |
| O3 | MR062 | 23.9 | 69.8 | 93.7 | 2.48 | 96.2 |
NO2 concentrations were an upper limit based on the NOX measurement.
AQHI values calculated based on 1 min average concentrations over the duration of the individual experiments.
Limitations of These Experiments
For toxicity assessment, previous testing using whole-air mixtures generated in a smog chamber generally involved the use of the chamber in a batch mode.(9–16) Although this approach allowed a detailed time course during which the formation of the reaction products could be studied, the usable volume was that of the chamber itself. Thus, only in vitro assays could realistically be used with the batch-reactor mode. In contrast, the flow-reactor mode used here provided an indefinite volume of chamber air, permitting the exposure of the in vivo assays (along with an in vitro assay) described in the accompanying papers. Although the flow reactor can supply indefinite air for exposure, there are two limitations. First, for the extent of the reaction used in these specific experiments, the available flow was limited to 50 L min−1, which was enough for use with only a single whole-body rodent-exposure chamber. At greater extents of reaction, the total flow would be less than the 50 L min−1 used in these experiments. Second, the use of the chamber as a CSTR produced a broad distribution of reactants and products, given that the reactants were flowing continuously into the chamber. This situation made it difficult to examine differences in toxicological effects of primary versus secondary (or higher-order) products.
Due to the flow-reactor configuration of the MRC in this study, gas-phase wall losses were insignificant because all surfaces were well-conditioned. Hence, corrections for gas-phase wall losses were not made in determining SOA or SOC yields. Because the atmospheres (SA-PM or SA-O3) were generated with different precursor hydrocarbon mixtures (α-pinene or isoprene, respectively), differences in the PM composition were likely and may have influenced toxicological effects and complicated the interpretation of PM-derived health effects. However, for the purpose of this study, changes to PM composition were not considered.
The constraints of these experiments, along with the reaction mixtures used, resulted in measured PM2.5 and oxidant levels that far exceeded those found in ambient air, with perhaps the exception of Eastern China or India.(1) However, these high concentrations were required given the sensitivities of the assays during short-term exposures. With the development of more sensitive biological assays, air pollution concentrations generated experimentally that are more-representative of urban atmospheres can be evaluated for health effects.
Supplementary Material
Acknowledgments
The U.S. Environmental Protection Agency through its Office of Research and Development funded and collaborated in the research described here under contract nos. EP-D-10–070 to Alion Science and Technology and EP-C-15–008 to Jacobs Technology, Inc. The manuscript has been subjected to external peer review and has been cleared for publication. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.
References
- 1.IARC Working Group on the Evaluation of Carcinogenic Risk to Humans. Outdoor air pollution. Lyon (FR): International Agency for Research on Cancer; 2016. (IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, No. 109.) Available from: https://www.ncbi.nlm.nih.gov/books/NBK368024/.
- 2.Karagulian F; Belis CA; Dora CFC; Prüss-Ustün AM; Bonjour S; Adair-Rohani H; Amann M Contributions to cities’ ambient particulate matter (PM): A systematic review of local source contributions at global level. Atmos. Environ 2015, 120, 475– 483, 10.1016/j.atmosenv.2015.08.087 [DOI] [Google Scholar]
- 3.Pope CA III; Burnett RT; Thurston GD; Thun MJ; Calle EE; Krewski D; Godleski JJ Cardiovascular mortality and long-term exposure to particulate air pollution: Epidemiological evidence of general pathophysiological pathways of disease. Circulation 2004, 6, 71– 77 [DOI] [PubMed] [Google Scholar]
- 4.Pöschl U Atmospheric aerosols: Composition, transformation, global climate, and health effects. Angew. Chem., Int. Ed 2005, 44, 7520– 7546, 10.1002/anie.200501122 [DOI] [PubMed] [Google Scholar]
- 5.Stieb DM; Burnett RT; Smith-Doiron M; Brion O; Shin HH; Economou V A new multipollutant health index based on short-term associations observed in daily time-series analyses. J. Air Waste Manage. Assoc 2008, 58, 435– 450 [DOI] [PubMed] [Google Scholar]
- 6.Hu J; Ying Q; Wang Y; Zhang H Characterizing multi-pollutant air pollution in China: Comparison of three air quality indices. Environ. Int 2015, 84, 17– 25, 10.1016/j.envint.2015.06.014 [DOI] [PubMed] [Google Scholar]
- 7.Zhou Y; Li C; Huijbregts MAJ; Mumtaz MM Carcinogenic air toxics exposure and their cancer-related health impacts in the United States. PLoS One 2015, 10, 1– 15, 10.1371/journal.pone.0140013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parrish DD; Ryerson TB; Mellqvist J; Johansson J; Fried A; Richter D; Walega JG;Washenfelder RA; de Gouw JA; Peischl J; Aikin KC; McKeen SA; Frost GJ; Fehsenfeld FC;Herndon SC Primary and secondary sources of formaldehyde in urban atmospheres: Houston Texas region. Atmos. Chem. Phys 2012, 12, 3273– 3288, 10.5194/acp-12-3273-2012 [DOI] [Google Scholar]
- 9. 42 U.S. Code Ch. 85, subchapter. I-Clean Air Act Amendments of 1990 – (Public Law 101–549).
- 10.Kleindienst TE; Shepson PB; Edney EO; Cupitt LT; Claxton LD The mutagenic activity of the products of propylene photooxidation. Environ. Sci. Technol 1985, 19, 620– 627, 10.1021/es00137a007 [DOI] [PubMed] [Google Scholar]
- 11.Kleindienst TE; Smith DF; Hudgens EE; Cupitt LT; Bufalini JJ; Claxton LD The generation of mutagenic transformation products during the irradiation of simulated urban atmospheres. Environ. Sci. Technol 1992, 26, 320– 329, 10.1021/es00026a012 [DOI] [Google Scholar]
- 12.Dumdei BE; Kenny DV; Shepson PV; Kleindienst TE; Nero CM; Cupitt LT; Claxton LD MS/MS analysis of the products of toluene photooxidation and measurement of their mutagenic activity. Environ. Sci. Technol 1988, 22, 1493– 1498, 10.1021/es00177a017 [DOI] [PubMed] [Google Scholar]
- 13.Shepson PB; Kleindienst TE; Edney EO; Nero CM; Cupitt LT; Claxton LD Acetaldehyde: the mutagenic activity of its photooxidation products. Environ. Sci. Technol 1986, 20, 1008– 1013, 10.1021/es00152a007 [DOI] [PubMed] [Google Scholar]
- 14.Shepson PB; Kleindienst TE; Nero CM; Hodges DN; Cupitt LT; Claxton LD Allyl chloride: the mutagenic activity of its photooxidation products. Environ. Sci. Technol 1987, 21, 568– 573, 10.1021/es00160a007 [DOI] [PubMed] [Google Scholar]
- 15.Kleindienst TE; Shepson PB; Edney EO; Claxton LD; Cupitt LT Wood smoke: measurement of the mutagenic activities of its gas- and particulate-phase photooxidation products. Environ. Sci. Technol 1986, 20, 493– 501, 10.1021/es00147a009 [DOI] [PubMed] [Google Scholar]
- 16.Kleindienst TE; Shepson PB; Smith DF; Hudgens EE; Nero CM; Cupitt LT; Bufalini JJ;Claxton LD; Nestman FR Comparison of mutagenic activities of several peroxyacyl nitrates. Environ. Mol. Mutagen 1990, 16, 70– 80, 10.1002/em.2850160204 [DOI] [PubMed] [Google Scholar]
- 17.DeMarini DM; Shelton ML; Kohan MJ; Hudgens EE; Kleindienst TE; Ball LM; Walsh D;de Boer JG; Lewis-Bevan L; Rabinowitz JR; Claxton LD; Lewtas J Mutagenicity in lung of Big Blue mice and induction of tandem-base substitutions in Salmonella by the air pollutant peroxyacetyl nitrate (PAN): predicted formation of intrastrand cross-links. Mutat. Res., Fundam. Mol. Mech. Mutagen 2000, 457,41– 55, 10.1016/S0027-5107(00)00121-4 [DOI] [PubMed] [Google Scholar]
- 18.Zhang Q; Jimenez JL; Canagaratna MR; Allan JD; Coe H; Ulbrich I; Alfarra MR; Takami A;Middlebrook AM; Sun YL; Dzepina K; Dunlea E; Docherty K; DeCarlo PF; Salcedo D;Onasch T; Jayne JT; Miyoshi T; Shimono A; Hatakeyama S; Takegawa N; Kondo Y; Schneider J;Drewnick F; Borrmann S; Weimer S; Demerjian K; Williams P; Bower K; Bahreini R; Cottrell L;Griffin RJ; Rautiainen J; Sun JY; Zhang YM; Worsnop DR Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes. Geophys. Res. Lett 2007, 34 (1–6), L13801, 10.1029/2007GL029979 [DOI] [Google Scholar]
- 19.Jimenez JL; Canagaratna MR; Donahue NM; Prevot ASH; Zhang Q; Kroll JH; DeCarlo PF;Allan JD; Coe H; Ng NL; Aiken AC; Docherty KS; Ulbrich IM; Grieshop AP; Robinson AL;Duplissy J; Smith JD; Wilson KR; Lanz VA; Hueglin C; Sun YL; Tian J; Laaksonen A;Raatikainen T; Rautiainen J; Vaattovaara P; Ehn M; Kulmala M; Tomlinson JM; Collins DR;Cubison MJ; Dunlea EJ; Huffman JA; Onasch TB; Alfarra MR; Williams PI; Bower K;Kondo Y; Schneider J; Drewnick F; Borrmann S; Weimer S; Demerjian K; Salcedo D; Cottrell L;Griffin R; Takami A; Miyoshi T; Hatakeyama S; Shimono A; Sun JY; Zhang YM; Dzepina K;Kimmel JR; Sueper D; Jayne JT; Herndon SC; Trimborn AM; Williams LR; Wood EC;Middlebrook AM; Kolb CE; Baltensperger U; Worsnop DR Evolution of organic aerosols in the atmosphere. Science 2009, 326, 1525– 1529, 10.1126/science.1180353 [DOI] [PubMed] [Google Scholar]
- 20.McDonald JD; Doyle-Eisele M; Kracko D; Lund A; Surratt JD; Hersey SP; Seinfeld JH;Rohr AC; Knipping EM Cardiopulmonary response to inhalation of secondary organic aerosol derived from gas-phase oxidation of toluene. Inhalation Toxicol 2012, 24, 689– 697, 10.3109/08958378.2012.712164 [DOI] [PubMed] [Google Scholar]
- 21.Lund AK; Doyle-Eisele M; Lin Y-H; Arashiro M; Surratt JD; Holmes T; Schilling KA;Seinfeld JH; Rohr AC; Knipping EM; McDonald JD The effects of α-pinene versus toluene-derived secondary organic aerosol exposure on the expression of markers associated with vascular disease. Inhalation Toxicol 2013, 25, 309– 324, 10.3109/08958378.2013.782080 [DOI] [PubMed] [Google Scholar]
- 22.McWhinney RD; Zhou S; Abbatt JPD Naphthalene SOA: redox activity and naphthoquinone gas-particle partitioning. Atmos. Chem. Phys 2013, 13, 9731– 9744, 10.5194/acp-13-9731-2013 [DOI] [Google Scholar]
- 23.Arashiro M; Lin Y-H; Sexton KG; Zhang Z; Jaspers I; Fry RC; Vizuete WG; Gold A;Surratt JD In vitro exposure to isoprene-derived secondary organic aerosol by direct deposition and its effects on COX-2 and IL-8 gene expression. Atmos. Chem. Phys 2016, 16, 14079– 14090, 10.5194/acp-16-14079-2016 [DOI] [Google Scholar]
- 24.Kramer AJ; Rattanavaraha W; Zhang Z; Gold A; Surratt JD; Lin Y Assessing the exidative potential of isoprene-dervied epoxides and secondary organic aerosol. Atmos. Environ 2016, 130, 211– 218, 10.1016/j.atmosenv.2015.10.018 [DOI] [Google Scholar]
- 25.Tuet WY; Chen Y; Xu L; Fok S; Gao D; Weber RJ; Ng NL Chemical oxidative potential of secondary organic aerosol (SOA) generated from the photooxidation of biogenic and anthropogenic volatile organic compounds. Atmos. Chem. Phys 2017, 17, 839– 853, 10.5194/acp-17-839-2017 [DOI] [Google Scholar]
- 26.Tuet WY; Chen Y; Fok S; Champion JA; Ng NL Inflammatory responses to secondary organic aerosols (SOA) generated from biogenic and anthropogenic precursors. Atmos. Chem. Phys 2017, 17,11423– 11440, 10.5194/acp-17-11423-2017 [DOI] [Google Scholar]
- 27.Tuet WY; Chen Y; Fok S; Gao D; Weber RJ; Champion JA; Ng NL Chemical and cellular oxidant production induced by naphthalene secondary organic aerosol (SOA): effect of redox-active metals and photochemical aging. Sci. Rep 2017, 7, 15157, 10.1038/s41598-017-15071-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang N; Kostenidou E; Donahue N; Pandis SN Multi-generation Chemical Aging of α-Pinene Ozonolysis Products by Reaction with OH. Atmos. Chem. Phys. Discuss 2017, 10.5194/acp-2017-746 [DOI] [Google Scholar]
- 29.U.S. National Archives and Records Administration. National Primary and Secondary Ambient Air Quality Standards, Code of Federal Regulations Title 40 Part 50, 2009.
- 30.Bahreini R; Middlebrook AM; de Gouw JA; Warneke C; Trainer M; Brock CA; Stark H;Brown SS; Dube WP; Gilman JB; Hall K; Holloway JS; Kuster WC; Perring AE;Prevot ASH; Schwarz JP; Spackman JR; Szidat S; Wagner NL; Weber RJ; Zotter P;Parrish DD Gasoline emissions dominate over diesel in formation of secondary organic aerosol mass. Geophys. Res. Lett 2012, 39 (1–6), L06805, 10.1029/2011GL050718 [DOI] [Google Scholar]
- 31.Chen H; Copes R Review of Air Quality Index and Air Quality Health Index; Ontario Agency for Health Protection and Promotion (Public Health Ontario): Toronto, Canada, 2013. [Google Scholar]
- 32.Kleindienst TE; Edney EO; Lewandowski M; Offenberg JH; Jaoui M Secondary organic carbon and aerosol yields from the irradiations of isoprene and α-pinene in the presence of NOX and SO2. Environ. Sci. Technol 2006, 40, 3807– 3812, 10.1021/es052446r [DOI] [PubMed] [Google Scholar]
- 33.Edney EO; Kleindienst TE; Jaoui M; Lewandowski M; Offenberg JH; Wang W; Claeys M Formation of 2-methyl tetrols and 2-methylglyceric acid in secondary organic aerosol from laboratory irradiated isoprene/NOX/SO2/air mixtures and their detection in ambient PM2.5 samples collected in the eastern United States. Atmos. Environ 2005, 39, 5281– 5289, 10.1016/j.atmosenv.2005.05.031 [DOI] [Google Scholar]
- 34.Black F; Tejada S; Kleindienst T Preparation of automobile organic emission surrogates for photochemical model validation. Atmos. Environ 1998, 32, 2443– 2451, 10.1016/S1352-2310(98)00045-4 [DOI] [Google Scholar]
- 35.Smith DF; Kleindienst TE; Hudgens EE Improved high-performance liquid chromatographic method for artifact-free measurements of aldehydes in the presence of ozone using 2,4-dinitrophenylhydrazine. J. Chromatog. A 1989, 483, 431– 436, 10.1016/S0021-9673(01)93146-2 [DOI] [Google Scholar]
- 36.Birch ME; Cary RA Elemental carbon-based method for monitoring occupational exposures to particulate diesel exhaust. Aerosol Sci. Technol 1996, 25, 221– 241, 10.1080/02786829608965393 [DOI] [PubMed] [Google Scholar]
- 37.Chan AWH; Chan MN; Surratt JD; Chhabra PS; Loza CL; Crounse JD; Yee LD;Flagan RC; Wennberg PO; Seinfeld JH Role of aldehyde chemistry and NOx concentrations in secondary organic aerosol formation. Atmos. Chem. Phys 2010, 10, 7169– 7188, 10.5194/acp-10-7169-2010 [DOI] [Google Scholar]
- 38.Kleindienst TE; Jaoui M; Lewandowski M; Offenberg JH; Lewis CW; Bhave PV; Edney EO Estimates of the contributions of biogenic and anthropogenic hydrocarbons to secondary organic aerosol at a southeastern US location. Atmos. Environ 2007, 41, 8288– 8300, 10.1016/j.atmosenv.2007.06.045 [DOI] [Google Scholar]
- 39.Atkinson R Atmospheric chemistry of VOCs and NOX. Atmos. Environ 2000, 34, 2063– 2101, 10.1016/S1352-2310(99)00460-4 [DOI] [Google Scholar]
- 40.Smith DF; McIver CD; Kleindienst TE Primary product distribution from the reaction of hydroxyl radicals with toluene at ppb NOX mixing ratios. J. Atmos. Chem 1998, 30, 209– 238, 10.1023/A:1005980301720 [DOI] [Google Scholar]
- 41.Smith DF; Kleindienst TE; McIver CD Primary product distributions from the reaction of OH with m-, p-xylene, 124- and 1,3,5-trimethylbenzene. J. Atmos. Chem 1999, 34, 339– 364, 10.1023/A:1006277328628 [DOI] [Google Scholar]
- 42.Kleindienst TE; Smith DF; Li W; Edney EO; Driscoll DJ; Speer RE; Weathers WS Secondary organic aerosol formation from the oxidation of aromatic hydrocarbons in the presence of dry submicron ammonium sulfate. Atmos. Environ 1999, 33, 3669– 3681, 10.1016/S1352-2310(99)00121-1 [DOI] [Google Scholar]
- 43.Reissell C; Harry C; Aschmann SM; Atkinson R; Arey J Formation of acetone from the OH radical- and O3-initiated reactions from a series of monoterpenes. J. Geophys. Res 1999, 104, 13869– 13879, 10.1029/1999JD900198 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.