

Abstract
Fundamental questions remain about the application of omics in environmental risk assessments, such as the consistency of data across laboratories. The objective of the present study was to determine the congruence of transcript data across 6 independent laboratories. Male fathead minnows were exposed to a measured concentration of 15.8 ng/L 17α-ethinylestradiol (EE2) for 96 h. Livers were divided equally and sent to the participating laboratories for transcriptomic analysis using the same fathead minnow microarray. Each laboratory was free to apply bioinformatics pipelines of its choice. There were 12 491 transcripts that were identified by one or more of the laboratories as responsive to EE2. Of these, 587 transcripts (4.7%) were detected by all laboratories. Mean overlap for differentially expressed genes among laboratories was approximately 50%, which improved to approximately 59.0% using a standardized analysis pipeline. The dynamic range of fold change estimates was variable between laboratories, but ranking transcripts by their relative fold difference resulted in a positive relationship for comparisons between any 2 laboratories (mean R2 > 0.9, p < 0.001). Ten estrogen-responsive genes encompassing a fold change range from dramatic (>20-fold; e.g., vitellogenin) to subtle (∼2-fold; i.e., block of proliferation 1) were identified as differentially expressed, suggesting that laboratories can consistently identify transcripts that are known a priori to be perturbed by a chemical stressor. Thus, attention should turn toward identifying core transcriptional networks using focused arrays for specific chemicals. In addition, agreed-on bioinformatics pipelines and the ranking of genes based on fold change (as opposed to p value) should be considered in environmental risk assessment. These recommendations are expected to improve comparisons across laboratories and advance the use of omics in regulations
Keywords: Endocrine disruptor, Estrogenic compound, Interlaboratory comparison, Risk assessment, Transcriptomics
INTRODUCTION
The use of biological indicators for environmental regulations, risk assessments, or monitoring programs requires careful investigation of the experimental method to generate reliable and reproducible data. Interlaboratory assays are conducted to validate the effectiveness and reliability of a method and to demonstrate how an established protocol performs under different conditions. In the context of ecotoxicology and environmental monitoring, interlaboratory comparisons have been conducted for physiological endpoints such as testosterone, 17β‐estradiol (E2), and 11‐ketotestosterone plasma steroids1,2; ethoxyresorufin‐O‐deethylase 3, 4; plasma vitellogenin 5; as well as acute and short‐term chronic whole‐effluent toxicity test methods 6.
The ability to generate consistent and reliable data widens applicability and improves predictability. More than 60 fish species are used in Canada’s Environmental Effects Monitoring program 7, 8, and each species can show divergent biological responses to endpoint perturbation 9. Because of this inherent species variability, a 25% or 2 standard deviation change from “normal” or baseline data in most whole‐organism endpoints is the recognized environmental effects monitoring threshold which triggers a change in the intensity, frequency, or focus of monitoring to further evaluate potential impacts at a particular site 10. This magnitude of response was found to be consistent using a variety of approaches to define a “critical effects size” 11, although the definition of “unacceptable” change in physiological measurements can be developed via a multistakeholder negotiation process 12.
To date, the inherent variability in physiological endpoints, the inability to narrowly define an ecologically relevant threshold level, and species differences in distribution and sensitivity limit their utility in large‐scale monitoring programs. The acceptable standardization of endpoints commonly used as biomarkers in endocrine‐disrupting compound assessments has been a struggle for nearly 2 decades 13. However, despite variability, physiological endpoints (e.g., plasma vitellogenin) can be successfully linked to ecologically relevant endpoints (e.g., fecundity) using modeling approaches from laboratory experiments 14. Such a linkage requires consistent measurement of physiological endpoints because the predictive model is driven by the magnitude of physiological change.
A concerted effort by researchers to incorporate omics‐based tools in environmental risk assessment is under way, and stakeholders continue to debate the criteria needed to facilitate the acceptance of omics approaches into a regulatory framework. For molecular endpoints, challenges related to reproducibility, variability, and interpretation have been well documented in both laboratory studies 15, 16 and field studies 17. To include molecular tools more broadly in monitoring and risk assessment, it is necessary to standardize and validate methods via interlaboratory studies, to better develop an understanding of the magnitude of change which is meaningful, and to better understand baseline variability 17.
Transcriptomic studies generally identify a list of differentially expressed genes related to specific biological processes that are responsive to a treatment. The successful identification of impacted genes relies heavily on the minimization of technical variation. For example, many nonbiological variables have been identified that can influence interlaboratory comparisons of microarray data, such as sample processing, batch effects, platform type, microarray labeling, hybridization, filtering, data acquisition and normalization, and analysis 18–22. Standard operating procedures and the introduction of reporting standards (minimum information about a microarray experiment) 23 have aided in the cross‐comparison of microarray‐generated data. The comparison of a list of differentially expressed genes and the reproducibility of this list across laboratories appear to be major goals of interlaboratory studies and support the notion that the list of differentially expressed genes is consistent among laboratories. Evidence that the standardization of microarray protocols can result in high levels of technical reproducibility, both within and between laboratories, has been demonstrated by the MicroArray Quality Control study 20.
To our knowledge, only 2 studies in ecotoxicology have conducted an interlaboratory study for microarray data (though it is acknowledged that other fields have conducted similar cross‐laboratory comparisons). Gene expression profiles of HepG cells exposed to benzo[a]pyrene were characterized by 2 individual laboratories using 2 different platforms24. Although the study was limited by a small sample size, it concluded that there existed a significant correlation in the identified differentially expressed transcripts in both laboratories following benzo[a]pyrene exposure as well as congruence in treatment‐specific effects, despite data variability. Building on this, 6 laboratories with experience in microarray analysis conducted a study that exposed amphipods to cyfluthrin‐spiked or control sediments 25. The conclusion of that study was that a subset of genes could be consistently identified as differentially expressed across laboratories, despite variability in factors such as ribonucleic acid (RNA) quality.
The objective of the present study was to evaluate the reproducibility of data collected from microarray analysis across multiple laboratories using the fathead minnow (Pimephales promelas), a widely utilized North American toxicological model in both laboratory ecotoxicology testing 26 and established monitoring programs developed by Canada’s Environmental Effects Monitoring program 8 and the US Environmental Protection Agency 27. Our questions centered on the ability of independent laboratories to generate comparable differentially expressed gene lists as well as to identify a suite of biomarkers of exposure exhibiting different magnitudes of response, despite differences in data processing and bioinformatics analyses. To this end, fathead minnows were exposed to a low, measured dose (∼15 ng/L) of 17α‐ethinylestradiol (EE2) because this pharmaceutical is measured in North American water systems (typically detected in the low nanograms per liter range) 28–30. Livers were subsampled and expression profiles compared among 6 laboratories using platforms from the same production batch. The reasons for the use of this pharmaceutical were as follows: 1) EE2 is a model compound for inducing estrogenic effects in fishes 31–34, specifically in the hepatic transcriptome in a number of small‐bodied fish species 35–37; 2) EE2 has been widely studied using transcriptomics 35–38, and thus, there is a strong body of literature that can be leveraged for future comparisons; 3) EE2 has been demonstrated to affect physiological parameters such as steroid levels 39, 40 and vitellogenin 31, 40, as well as fecundity, secondary sexual characteristics, and gonadal somatic index (GSI) 40. It is also associated with population‐level impacts in fathead minnows in wild populations 41. Thus, the nexus between transcriptomic, physiological, and reproductive impacts using this pharmaceutical are most pertinent to monitoring programs.
METHODS
Experimental animals and EE2 exposure
All experimental procedures were approved by the Animal Care Committee (protocol no. 2013‐3s‐09) and carried out at the Canadian Rivers Institute at the University of New Brunswick, Saint John, NB, Canada. Carbon‐filtered, dechlorinated city water was used for waterborne exposures. Reproductively mature, lab‐reared male fathead minnows aged 1.5 yr were housed in 400‐L fiberglass holding tanks at a density of approximately 60 fish per tank under a 16:8‐h light:dark photoperiod. The temperature of the holding tanks was kept at 24 to 26 °C, and dissolved O2 was 95 to 100%. Fish were fed ad libitum 1 or 2 times a day. We acknowledge that fish were older than those used in laboratory toxicity testing (∼6 mo); however, the response of these animals to EE2 was dramatic and as expected, and the objective was to assess reproducibility of the transcriptome data among laboratories from the same liver.
17α‐Ethinylestradiol (catalog no. E4876; CAS no. 57‐63‐6, ≥98% purity) was purchased from Sigma. A stock solution of 1 mg/mL EE2 in ethanol was diluted to a final nominal concentration of 25 ng/L EE2 and 0.00025% ethanol. Control dose (vehicle control) without EE2 contained 0.00025% ethanol. There were 10 control and 11 treatment 18‐L glass aquaria, each with one male fish, all of equal size. The same temperature, photoperiod, oxygenation, and feeding conditions as stated previously also apply for the exposure and were measured daily. Fish were fed after the completion of each water change. Test waters were renewed at 24, 48, and 72 h by total draining of the tank (100% water change); and the experiment was terminated after 96 h.
Water chemistry
Water samples were collected from 3 randomly chosen tanks for both control and EE2 treatments as fresh preparations of solutions (i.e., immediately after water change) on days 2 and 4. Samples were stored in brown glass bottles with Teflon caps in the dark at 4 °C until chemical analysis, which occurred approximately 1 mo later. Measurements of EE2 were performed in triplicate using an enzyme‐linked immunosorbent assay (ELISA) kit (Abraxis) following the manufacturer’s instructions. Briefly, 1 L of the water was concentrated on ODS‐C18 columns (AccuBond II; Agilent Technologies), eluted with dichloromethane, and evaporated to dryness with nitrogen and a heated water bath. Samples were then reconstituted with methanol followed by water, resulting in 2.5 mL of a 10% methanol solution (400‐fold concentration factor). The working range of this sandwich ELISA is 0.05 to 3 μg/L. Interassay and intra‐assay coefficients of variation (CVs) using positive controls are typically 9.9 and 4.5%, respectively.
Experiment termination
At the conclusion of the exposures, fish were anesthetized using 250 mg/L buffered tricaine methanesulfonate (Sigma), and fish fork length and weight were recorded. Blood samples were collected from the caudal sinus into heparinized hematocrit tubes (Fisher Scientific) for plasma vitellogenin analysis and centrifuged at 2000 g for 10 min, with the resulting plasma being stored at −20 °C for vitellogenin quantification. Fish were then euthanized by spinal severance. Liver and testis weights (±0.01 g) were recorded to calculate morphometric parameters to ensure that there were no differences between treatment groups in GSI (gonad wt/body wt × 100) or liver somatic index (LSI; liver wt/body wt × 100). Each fish liver was then immediately subdivided into 7 equally sized pieces, and each subdivided liver was flash‐frozen in individual Eppendorf® 1.5‐mL centrifuge tubes on dry ice. Multiple pieces of hepatic tissue were harvested from alongside the intestine and scooped into a single pile. Thus, tissue pieces were mixed as effectively as possible prior to distribution into the separate tubes. Tissues for gene expression analysis were stored at −80 °C until RNA extraction. Eight control fish and 8 treated fish were chosen at random to be analyzed by every laboratory for the microarray analysis; one remaining set was kept as a backup. Therefore, every laboratory received a liver subsample from each of 8 control and 8 exposed fish, totaling 16 liver subsamples to be used with 2 slides containing 8 × 60K probes (detailed in the Microarray processing and analysis by each individual laboratory section).
Plasma vitellogenin levels
Plasma vitellogenin was measured at the University of New Brunswick’s Saint John Laboratory using a commercial fathead minnow vitellogenin ELISA kit (Cayman Chemical; no. V01018401). Control samples (n = 8) were diluted 1:1000 and 1:1 000 000, while treatment samples (n = 8) were diluted 1:100 000 and 1:1 000 000 using the provided dilution buffer. Two treatment samples were too concentrated to obtain an absorbance reading and were not used. All samples were analyzed in duplicate, and the CV between duplicates was <20% (average CV 3%). The working range and level of detection of this sandwich ELISA are 0.05 to 50 ng/mL and 0.02 ng/mL, respectively.
Participating laboratories
Sixteen liver samples (8 control and 8 exposed) were shipped on dry ice by courier in numbered (blinded) vials to 6 laboratories with experience in microarray analysis. Participants were asked to check the integrity of the parcel, the presence of dry ice, and the status of the samples immediately on receipt. Each laboratory was given specific instructions on sample processing, detailed in each of the following sections. Microarrays were also provided to each laboratory. The custom 60K probe fathead minnow array Agilent platform used was GPL15775 42. Flexibility was given in the standard operating procedures to realistically model variation present in sample preparation and microarray analysis that is currently present across ecotoxicology laboratories.
RNA isolation and RNA integrity
Each laboratory extracted RNA from liver tissue using a phenol and guanidine isothiocyanate–based method (e.g., TriZOL®; TriReagent, STAT60) according to the manufacturers’ protocols, and pellets were reconstituted based on visual inspection of the pellet. Total RNA was quantified and purity was assessed using the Thermo Scientific NanoDrop by each laboratory. The integrity of the RNA was assessed by each laboratory using the method of their choosing. The RNA integrity values for all 6 laboratories, as well as the method used, are found in Table 1.
Table 1.
Ribonucleic acid (RNA) integrity values and method used by each laboratorya
| Integrity value | |||
|---|---|---|---|
| Method used to assess RNA quality | Mean ± standard deviation |
Range | |
| Lab 1 | Agilent 2100 Bioanalyzer | 9.5 ± 0.1 | Not given |
| Lab 2 | Agilent 2100 Bioanalyzer | 9.7 ± 0.3 | 9.1–10.0 |
| Lab 3 | Agilent 2100 Bioanalyzer | 8.7 ± 0.7 | 7.7–9.7 |
| Lab 4 | 2:1 ratio of 28S:16S bands on a 1.5% agarose gel |
346.2 ng | 16.8–363.1 ng |
| Lab 5 | Bio‐Rad Experion™ Electrophoresis Station | 8.5 ± 1.1 | Not given |
| Lab 6 | Agilent 2100 Bioanalyzer | 9.2 ± 0.3 | 8.8–9.9 |
The Agilent 2100 gives an RNA Integrity Number, whereas the Bio‐Rad Experion Electrophoresis System gives an RNA Quality Index value. Other metrics used to assess sample quality as well as details on the methods for analysis are provided in Supplemental Data, Table S1.
Microarray processing and analysis by each individual laboratory
Each laboratory labeled 100 ng total RNA (laboratory 4 was an exception; 58.5 ng RNA in 1.5 μL was used because of low yields in some samples) and hybridized for 17 h at 65 °C with 2 washes. The Agilent One‐Color Microarray‐Based Gene Expression Analysis (Low input quick Amp Labeling Kit and RNA Spike in Kit) protocol was followed per the manufacturer’s instruction. Each laboratory was asked to scan and report parameters for a 60K microarray. This microarray consists of 65 528 targets, and of these, there were 32 155 unique gene symbols and 20 265 targets on the platform with full gene names. All microarray data have been deposited to the Gene Expression Omnibus at the National Center for Biotechnology Information as a super series (GSE 81544). One laboratory submitted its data independently to the Gene Expression Omnibus (GSE 70807). The letters in the Gene Expression Omnibus submission do not correspond to the laboratory designations in the present study.
After each laboratory had performed microarray hybridization using different data‐processing and analysis methods, sample identifications were released. There were no restrictions on algorithms or methods for microarray analysis because a goal of the project was to assess congruency in the number, identification, fold change, and p value of gene probes identified as differentially and not differentially expressed between control and treatment groups across laboratories. Each laboratory used a variety of methods to analyze the data. Details on the data processing and analysis conducted by individual laboratories are provided in Supplemental Data, Table S1. Both raw and normalized data were then forwarded by each laboratory to laboratory 1 (the coordinating laboratory).
Microarray analysis by laboratory 1
Laboratory 1 was selected as the relative benchmark for interlaboratory comparisons to determine the relative contribution of data processing to the variability in the response. Each individual data set from a laboratory was analyzed separately and identically by laboratory 1. Spot intensity and CV data were calculated by first normalizing the within‐experiment microarrays using quantile normalization. To reduce the effect of noise across arrays and to standardize the limit of detection among laboratories, data sets were filtered to the average intensity of the eighth Agilent spike across all spots and arrays ([+]E1A_r60_a107 [spike 8]). The rationale for using this spike‐in was that, on viewing all quality control reports for all of the laboratories, it consistently marked the final point in linearity of the dynamic range. The limits of detection for intensity, based on 560 array targets, were as follows: laboratory 1 = 4.34, laboratory 2 = 5.25, laboratory 3 = 5.26, laboratory 4 = 4.87 (one array was removed because of poor hybridization), laboratory 5 = 6.5 (one duplicate array was removed from the final analysis), and laboratory 6 = 5.01. Prior to an analysis of variance on normalized data (JMP Genomics; V6), all control spots were removed from each experimental data set, leaving 61 057 targets. A false discovery rate was set at 0.05.
RESULTS
17α‐Ethinylestradiol in the water
The mean concentration (± standard error of the mean) of EE2 in the tanks was 15.67 ± 4.71 ng/L. There was no detectable EE2 in the control water samples.
Morphological and physiological endpoints
There were no significant differences in body weight, body length, GSI, or LSI between the control and EE2‐treated group (Table 2). Plasma vitellogenin levels of EE2‐treated males were 4.0 ± 12.7 μg/mL for control and 21 666 ± 1821 μg/mL for EE2‐treated fish after 96 h, and fathead minnow responded as expected to the exposure.
Table 2.
Morphometrics (± standard error of the mean) in fathead minnows exposed to control (n = 8) and 25 ng/L 17α‐ethinylestradiol (n = 8): There were no significant differences among treatments in any endpoints (α = 0.05)
| Control fish | 17α‐Ethinylestradiol‐exposed fish | |
|---|---|---|
| Length (mm) | 78.63 ± 2.02 | 81.63 ± 1.80 |
| Weight (g) | 6.07 ± 0.35 | 6.16 ± 0.36 |
| Gonadal somatic index | 1.42 ± 0.20 | 0.98 ± 0.16 |
| Liver somatic index | 1.41 ± 0.18 | 1.62 ± 0.16 |
Expression results across laboratories
All laboratories submitted data from RNA extraction/purification methods and RNA quality assessment and stated the method that was used to normalize the microarray intensity data (Table 1; Supplemental Data, Table S1). Laboratory 5 had to omit 2 control samples because of low RNA quality, and instead, 2 replicate samples were hybridized again as technical replicates to confirm that there was >98% reproducibility in signal intensity per target on the microarray between technical replicates (data not shown). Only one technical replicate was chosen at random to represent the single biological replicate. All labeled samples used by each laboratory met the minimum criteria for hybridization per Agilent guidelines (yields >0.825 μg and specific activity >6.0 pmol Cy3 per microgram complementary RNA). Each laboratory submitted data containing the number, identifier, fold change relative to controls, and p value of all gene probes to laboratory 1.
Characterization of probe variability across laboratories
To standardize the analysis approach and to determine how much variability was a result of the hybridization and scanning protocols, technical variability for both mean target probe CV(a measure of interassay variability) and intensity was determined for both control and treatment samples for each laboratory based on the common analysis workflow performed by laboratory 1. Each laboratory showed a comparable level of variability (Figure 1A). Overall, in each group, the liver transcriptome showed a CV (expressed as percentage) that ranged from 6 to 8%. Likewise, the mean log2 spot intensity of all targets on the microarray ranged from 6.0 to 8.0 (Figure 1B).
Figure 1.
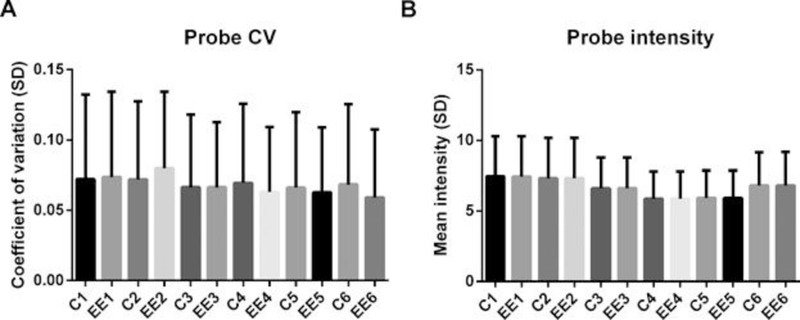
Mean coefficient of variation (CV; ± standard deviation [SD]) of all spots over all microarrays (n = 8 biological replicates) within an experimental group. (A) Coefficient of variation is plotted separately in the control group and 17α‐ethinylestradiol for each laboratory. (B) Mean probe intensity (± SD) in each experimental group. C = control; EE = 17alpha‐ethinylestradiol exposure.
Each laboratory, using the statistical methods of its choice, submitted a list of differentially expressed probes (false discovery rate of 0.05). A total of 641 differentially expressed probes (duplicates removed) were identified as being in common by all 6 laboratories following their own analyses. This number was largely influenced by laboratory 3, whose post hoc corrected p values resulted in a significant reduction in the number of probes identified (only 165 were reported); therefore, non–false discovery rate–corrected data (2375 probes) were used for laboratory 3 in subsequent comparisons. The decision to include data from laboratory 3 in the study, despite their influence on data variability and congruency across laboratories, was because of the goal of the present study, namely, to assess microarray variability across established ecotoxicology laboratories regularly employing this technology. Despite the variability in methods used, 50.6% of nonduplicate probes (using a false discovery rate of 0.05) were shared on average between each laboratory and laboratory 1, ranging from 30 to 77% overlap (Table 3). When laboratory 3 is excluded, the number of nonduplicate probes shared between each laboratory and laboratory 1 increases by 5 to 55.6%. Unsurprisingly, the number of probes any given laboratory shared in common with laboratory 1 is positively correlated with the number of probes identified. That is to say, the more probes identified, the more likely that some of those probes will be identified by another laboratory.
Table 3.
Number of differentially expressed probes (based on spot identification) identified by each laboratory using its own method of normalization (A) and identified using the same analysis pipeline performed by the benchmark laboratory, laboratory 1 (B)a
| Lab 1 | Lab 2 | Lab 3 | Lab 4 | Lab 5 | Lab 6 | |
|---|---|---|---|---|---|---|
| A | 9534 | 11846 | 9889 | 3488 | 6868 | 9743 |
| B | 9534 | 12244 | 6598 | 2498 | 5927 | 7044 |
| C | 6048 | 2375b | 2886 | 3934 | 4632 | |
| D | 77% | 30%b | 37% | 50% | 59% |
Number of probes in common with laboratory 1 (C), given also as a percentage (D). The targets on the fathead minnow microarray represent both duplicate and nonduplicated genes (thus, “probe ID” is the value in Table 3). All data are based on false discovery rate corrected p values.
Non–false discovery rate data used.
Congruence in the identification of identical differentially expressed genes
Across the 6 laboratories, 12 491 differentially expressed genes (duplicates removed) were identified as responding to the EE2 treatment, out of a total of 22 010 genes on the array. There was a wide range in how many differentially expressed genes each laboratory detected (2196–6361; Table 4), based on its own analysis using a post hoc corrected cutoff of ±1.5‐fold (except laboratory 3 where uncorrected p values were used). For example, laboratory 2 detected 6152 unique genes (Table 4), of which 80% were shared with laboratory 1 (Table 5). However, there was a wide range in the percentage of differentially expressed genes any given laboratory shared with another (Table 5). Laboratories 1, 2, and 6 were most similar in their differentially expressed gene profiles, sharing an average of at least 60% of the same differentially expressed genes with the other 5 laboratories. Between laboratories, laboratory 1 shared the highest percentage of differentially expressed genes with laboratory 2 (4040 genes in common; 79.8% of laboratory 2’s differentially expressed genes). Supplemental Data, Table S2 also presents the same analysis but when the data are analyzed by standard approach. The influence of using a fold change cutoff of 3.0 on the number of genes both unique and shared across laboratories is presented as Supplemental Data, Table S3.
Table 4.
Number of differentially expressed genes (duplicate probe identifiers removed) shared between each laboratory using its own method of normalization and analysis methods for all genes that were identified as different (post hoc corrected)
| Shared differentially expressed genes | ||||||
|---|---|---|---|---|---|---|
| Laboratorya | 1 | 2 | 3b | 4 | 5 | 6 |
| 1 (5064) | X | |||||
| 2 (6152) | 4040 | X | ||||
| 3b (6361) | 2027 | 2426 | X | |||
| 4 (2196) | 1863 | 1999 | 927 | X | ||
| 5 (4324) | 2551 | 2822 | 1918 | 1305 | X | |
| 6 (5744) | 3443 | 3830 | 2172 | 1852 | 2378 | X |
Total number of differentially expressed genes (given in parentheses).
Non–false discovery rate data used.
indicates 100% as data identical.
Table 5.
Number of differentially expressed genes (duplicate probe identifiers removed) shared between each laboratory demonstrating the percentage of genes each laboratory shared with anothera
| Percentage of shared differentially expressed genes | |||||||
|---|---|---|---|---|---|---|---|
| Laboratoryb | 1 | 2 | 3c | 4 | 5 | 6 | Average |
| 1 (5064) | X | 65.7 | 31.9 | 84.8 | 59.0 | 59.9 | 60.3 |
| 2 (6152) | 79.8 | X | 38.1 | 91.0 | 65.3 | 66.7 | 68.2 |
| 3c (6361) | 40.0 | 39.4 | X | 42.2 | 44.4 | 37.8 | 40.8 |
| 4 (2196) | 36.8 | 32.5 | 14.6 | X | 30.2 | 32.2 | 29.3 |
| 5 (4324) | 50.4 | 45.9 | 30.2 | 59.4 | X | 41.4 | 45.4 |
| 6 (5744) | 68.0 | 62.3 | 34.1 | 84.3 | 55.0 | X | 60.7 |
For example, laboratories 1 and 2 found 4040 genes in common; this number is first divided by the total number of differentially expressed genes identified by laboratory 1 (5064) to give a percentage of 79.8. Alternatively 4040 is also divided by the total number of differentially expressed genes identified by laboratory 2 (6152) to give a percentage of 65.7. The columns and the rows represent two different ways of comparing differentially expressed gene lists between laboratories. Supplemental Data, Table S2 shows the exact same analysis but when the data are analyzed with the same analysis pipeline.
Total number of differentially expressed genes (given in parentheses).
Non–false discovery rate data used.
indicates 100% as data identical.
Each laboratory identified a subset of differentially expressed genes that no other laboratory detected (“unique differentially expressed genes”). This number varied widely, for example, laboratory 4 identified the lowest number of unique differentially expressed genes at 48, whereas laboratory 3 identified the highest number of unique differentially expressed genes at 2741 (Table 6). Across all laboratories, the number of unique differentially expressed genes detected totaled 5364, which represents almost 43% (5364/12 491) of all differentially expressed genes responding to the estrogenic treatment. When examined as percentages, only 2.2% of the differentially expressed genes reported by laboratory 4 were unique genes that no other laboratory identified. Thus, one or more of the other laboratories identified the significant majority of transcripts in its list. Similarly, laboratories 1, 2, 3, 5, and 6 reported the following percentages of unique genes: 6.7, 10.6, 43.1, 15.1, and 16.1%. All 6 laboratories identified the same 587 differentially expressed genes (which was 4.7% of the total 12 491 differentially expressed genes detected in one or more laboratory), and 43% (5364) of the expressed genes were uniquely detected by only one laboratory. The proportion of differentially expressed genes that were shared by all 6 laboratories ranged from 9.2% for laboratory 3 to 26.7% for laboratory 4. Laboratory 3 contributed 21.9% unique differentially expressed genes to the 12 491 differentially expressed genes reported and thus contributed the most to “differentially expressed gene noise.” It is unclear whether differentially expressed gene “noise” is reflected by the number of unique genes detected or whether the increased detection represents increased sensitivity. Resolving the issue of sensitivity versus noise may require a comparative pathway analysis or validation with other methods such as quantitative polymerase chain reaction to more clearly evaluate whether increased detection improves the analysis or confuses the issue, but in either case, it is clear that individual gene responses must be interpreted with caution.
Table 6.
Number of unique differentially expressed genes, out of the 12 491 total differentially expressed genes detected, that any given laboratory shared with one or more other laboratories: Columns represent individual laboratories, whereas rows denote the number of laboratories involveda
| Laboratory | ||||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3b | 4 | 5 | 6 | Total differentially expressed genes |
||
| Number of | 1 | 341 | 653 | 2741 | 48 | 653 | 920 | 5364 (42.9%) |
| laboratories | 2 | 676 | 1101 | 1168 | 97 | 682 | 954 | 2339 (18.7%) |
| sharing | 3 | 1082 | 1345 | 688 | 174 | 652 | 1048 | 1664 (13.3%) |
| differentially | 4 | 1274 | 1334 | 591 | 522 | 784 | 1143 | 1383 (11.1%) |
| expressed | 5 | 1104 | 1132 | 586 | 768 | 966 | 1092 | 1127 (9.0%) |
| genes | 6 | 587 | 587 | 587 | 587 | 587 | 587 | 587 (4.7%) |
| Total | 5064 | 6152 | 6361 | 2196 | 4324 | 5744 | ||
For example, in row 1, the values represent how many laboratories reported the same unique differentially expressed genes; therefore, row 1 denotes unique differentially expressed genes found by each laboratory. In row 2, the values represent how many unique differentially expressed genes were shared by any specific laboratory (column) and at least one other laboratory, for a total of 2 laboratories sharing differentially expressed genes. Row 6 reflects that all 6 laboratories shared 587 unique differentially expressed genes.
Non–false discovery rate data used.
Congruence in the magnitude of response of differentially expressed genes across laboratories
The mean fold change of differentially expressed genes for both increasing (Figure 2A) and decreasing (Figure 2B) transcripts relative to the control group demonstrates significant differences across laboratories in overall results. However, when data are reported as median fold changes (Figure 2C,D) distributions of average differentially expressed gene fold changes across laboratories were not more similar. Using unadjusted p values for laboratory 3 and a post hoc corrected cutoff of ±1.5‐fold for all other laboratories, the fold change distribution of differentially expressed genes for both increasing (Figure 2E) and decreasing (Figure 2F) transcripts demonstrates that the majority of genes fall within the 1.5 to 3.0 fold change range.
Figure 2.
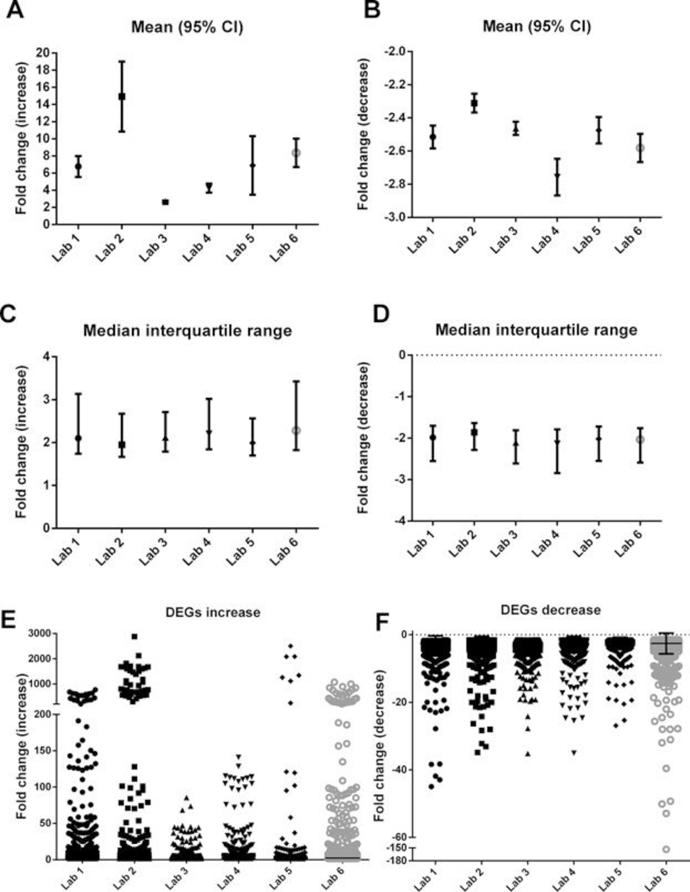
Metrics of variability and spread of fold changes in differentially expressed genes across laboratories. Mean fold change of differentially expressed genes with 95% confidence internals (vertical bars) shown for increasing (A) and decreasing (B) transcripts, relative to the control group. Median fold change of differentially expressed genes with interquartile range (vertical bars) shown for increasing (C) and decreasing (D) transcripts, relative to the control group. Fold change distribution of differentially expressed genes for increasing (E) and decreasing (F) transcripts, relative to the control group. Uncorrected p value for transcripts was used for laboratory 3 instead of corrected p values because the number of differentially expressed genes identified following a post hoc correction was small relative to the other laboratories. CI = confidence interval; DEG = differentially expressed gene.
Congruence in the identification of a suite of known E2‐responsive transcripts
To assess the performance of using microarrays to identify known biomarkers of estrogenic exposure in aquatic species, 10 estrogen‐responsive genes or “E2‐responsive standards” were selected. These E2‐responsive genes were identified using published literature on the expression profiles in the liver of zebrafish following estrogenic treatments 36, 43 as well as input from the Comparative Toxicogenomics Database 44 following searches for transcripts responsive to estrogens. We targeted 10 genes from this list and searched for these within the data sets collected from the 6 laboratories, with the intention of investigating transcripts that showed a broad dynamic range for both fold change and direction of change. These genes included 1) those that may show relatively low response to estrogens, for example, apolipoprotein A‐I‐1 (APOA1), apolipoprotein Eb (APOE), c‐FOS, and block of proliferation 1 (BOP1); 2) a relatively intermediate response to estrogens such as insulin‐like growth factor 1 (IGF1), x‐box binding protein 1 (XBP1), and estrogen receptor 1 (ESR1); and 3) a relatively high response to estrogens such as reticulon 1a (RTN1A), vitellogenin 1 (VTG1), and VTG3. There was strong congruence across laboratories for estimating the fold changes of these known E2‐responsive transcripts, regardless of whether a common analysis for differentially expressed genes was used (Figure 3A) or whether each laboratory used an independent analysis of its choice (Figure 3B). Four laboratories were successful at identifying all 10 E2‐responsive genes as differentially expressed. Laboratory 3 missed 3 of these genes, and laboratory 4 missed one; and this corresponded to the laboratories with the least number of differentially expressed genes identified after a post hoc correction of the data (i.e., may reflect the low number of differentially expressed genes identified initially).
Figure 3.
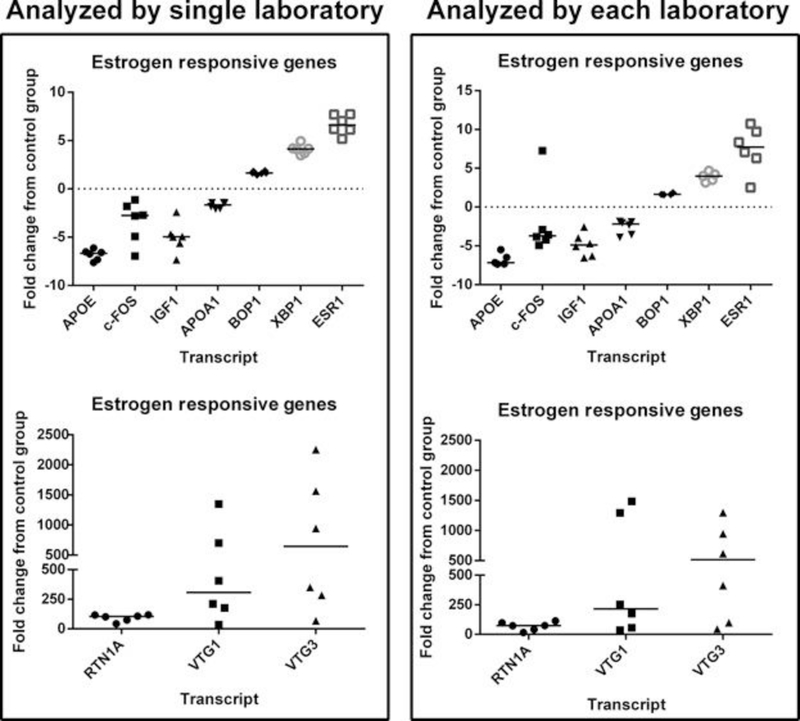
Ten estrogen‐responsive genes arranged in order from decreasing to increasing by fold change. Each point represents a laboratory. Only those genes that were identified as differentially expressed are included in these graphs. Horizontal line represents the median expression. Zero represents no fold change relative to control. APOA1 = apolipoprotein A‐I‐1; APOE = apolipoprotein Eb; BOP1 = block of proliferation 1; ESR1 = estrogen receptor 1; IGF1 = insulin‐like growth factor 1; RTN1A = reticulon 1a; VTG = vitellogenin; XBP1 = x‐box binding protein 1.
DISCUSSION
The present study aimed to assess the reproducibility of microarray data across 6 independent laboratories, following a 96‐h EE2 exposure to fathead minnow. The value of microarrays in environmental monitoring and risk assessment depends on the ability of independent laboratories to reliably distinguish subtle changes in gene expression data (differentially expressed genes relative to control) despite both biological and technical variability. Notwithstanding those probes exhibiting expected high biological variability, the mean probe variability across the transcriptome reported in the present study is consistent with that reported by others (5–10% CV) 45, 46 for a single tissue in fish. The overall mean signal intensity on the arrays after normalization (i.e., range of intensity across all 60K probes) was 6 to 8 depending on the laboratory. This is encouraging, given some flexibility in microarray sample preparation (i.e., different reagents for RNA extraction) afforded to the participating laboratories.
Data analysis pipelines contributed to data variability, and there was an increase in differentially expressed gene overlap across laboratories by approximately 9% when a single pipeline was used (an increase to ∼59% from ∼50%). Variability in the analysis also arose, in part, from differences in background intensity of the microarrays. Other interlaboratory microarray studies have substantiated the numerous sources of data variation and have highlighted the role of using standardized protocols in producing highly correlated and reproducible data 18, 20–22. It is important to recognize that a high level of concordance of differentially expressed genes across platforms and laboratories depends mainly on the statistical criteria used to rank and select them; using a fold change or ratio (vs a p value) generated the most robust results 20. Using the ranked log ratios of fold changes, we confirmed that all laboratories were detecting similar overall transcriptome responses that were statistically and highly correlated. In the case of laboratory 3, post hoc corrected pvalues resulted in a significant reduction in the number of differentially expressed genes (and the number of differentially expressed probes) identified and influenced the results. Thus, laboratory 3 appeared to differ most in the bioinformatics approach used to process the data. However, all laboratories used different strategies (Supplemental Data, Table S1) for data processing, editing, averaging of probes, and statistical identification of differentially expressed genes which influences the p value threshold for a decision on whether or not a transcript is differentially expressed.
Analysis of probe variability in each laboratory revealed that there were differences in the overall magnitude of response when mean fold change is plotted. These values can be heavily influenced by transcripts that have large expression changes relative to the control, as in the case of VTG. Thus, the sensitivity of the arrays can vary from laboratory to laboratory in the context of fold change. Plotting the median values instead of the mean revealed that the laboratories do not differ substantially, and the median fold change for the large majority of transcripts is approximately 2‐fold.
The absolute number of differentially expressed genes detected varied considerably across laboratories. Of the 22 010 genes on the array, 12 491 differentially expressed genes responded to the EE2 treatment. Of these, 587 (4.7%) common differentially expressed genes were detected by all 6 laboratories, and 43% (5364) were uniquely detected by only one laboratory. It is difficult to examine the differences and similarity in detail, but we did examine the ability of each laboratory to detect a suite of core transcripts known to be impacted by EE2 exposure (in lieu of number of differentially expressed genes identified) by assessing the ability of independent laboratories to identify common “gold standard” estrogen‐responsive genes. It is noteworthy that, despite flexibility in the standard operating procedures for much of the experimental design, 4 laboratories successfully identified significant changes in all 10 gold standards along a continuum of relative fold changes (decreasing to increasing). This was true irrespective of whether each laboratory independently analyzed the data or whether a single laboratory applied a common analysis. The laboratories could very precisely converge on fold change estimates for some transcripts, for example, APOE and XBP1, whereas other transcripts showed wider variability in fold change estimates across laboratories (e.g., c‐FOS and VTG). Although all 6 laboratories correctly identified VTG1 and VTG3 as up‐regulated following EE2 treatment, the magnitude of induction differed significantly between laboratories. Moving forward, a recommendation is to incorporate a ranking system of transcript response because laboratories will report fold changes differently. Lastly, BOP1 was identified as an estrogen‐responsive gene that was expressed at a low level in every data set (>2 fold) when analyzed by laboratory 1. This suggests that the practice of removing genes based on a fold change cutoff may result in removing genes that are responsive to the treatment, although this is a trade‐off because transcripts that show lower fold changes are perhaps more likely to be false positives. Arbitrary fold change cutoffs for expression data may not be the best approach moving forward and will result in discrepancies of differentially expressed genes reported among laboratories 47.
The ability to also identify core gene targets with fold changes close to background levels suggests that microarrays are capable of measuring a suite of responsive genes with a high degree of precision when sensitive transcriptional networks are identified and assessed a priori. In this example, all 6 laboratories would have almost perfect consensus in their ranking of these 10 E2‐responsive transcripts. For new chemicals, observational transcriptome data would need to be collected first to identify responsive transcripts. Databases such as the Comparative Toxicogenomics Database could then be used to hone the list of responsive transcripts.
Expression profiling generates important information on the chemical mode of action 48, 49. Under Canada’s Environmental Effects Monitoring program, confirmed positive responses can enter into an “investigation of cause” phase, and pathway information can provide valuable insight into potential modes of action to help focus subsequent studies. However, to use transcriptomics in a meaningful and legislatively robust way, it must be able to provide thresholds for specific parameters that can be tested across a range of environments, conditions, and species. At this point, it may be important for risk assessment and environmental monitoring to identify a common subset of transcripts responsive to a chemical or mixture, as opposed to leveraging the entire molecular data set. Because of variability in responses, a recommendation would be to first agree on a suite of biomarker transcripts for a particular chemical, followed by a ranking system based on fold change 50.
To incorporate microarray technology into monitoring programs and risk assessment, there needs to be a definition of what constitutes a significant change in expression (1.5‐fold vs 2). Because different normalization techniques can significantly alter the gene lists created, a decision on a common normalization technique is warranted, and this may influence the estimate of fold change. Until then, we recommend using fold change (vs a p value) as the ranking criterion for gene selection, with a nonstringent p value cutoff based on analyses using human data sets subjected to cross‐laboratory and interplatform comparisons 20. Reproducibility among laboratories is also reported to be highest when analysis was based on biological themes defined by enriched Gene Ontology categories 22, 51, an area of research that warrants further examination in an ecotoxicological context.
CONCLUSIONS
The standardization of transcriptomics and the generation of comparable data via an interlaboratory study are necessary steps prior to their inclusion in regulatory or monitoring programs. What constitutes a significant change in expression levels must therefore be defined. Proper study design will require the identification of a predetermined “magnitude of change” (or “critical effect size”) at the molecular level (i.e., transcripts), which could be the level of change required to have an effect at the individual and/or population level. Using a common suite of genes known to be estrogen‐responsive to various degrees, we demonstrated that there can be high success rate across laboratories. Further evidence of reproducible and expanded differentially expressed gene lists (or subsets) across laboratories at both low and high expression levels is required. We strongly recommend to researchers in ecotoxicology that they present data as fold change ranking and place less emphasis on p values to generate differentially expressed genes, with the caveat that a single individual is not disproportionately driving a biological response. In addition, the generation of biological pathways and/or Gene Ontology terms would be beneficial as anchoring points for molecular data and to reduce the stochastic noise among individual genes, though this was not done in the present study. The role for omics is still at this point best suited for finding mechanisms and screening and not for regulatory or monitoring programs because of the difficulty in their use for pass/fail decision‐making. Currently, no molecular endpoints are used in Canada’s Environmental Effects Monitoring program, and no established methodology exists for using these endpoints in risk assessment. We suggest that we move toward a ranking system for triggering action or regulation based on transcriptomic responses.
Supplementary Material
Acknowledgment
We thank G. Van Aggelen for his assistance with the manuscript. The present study was sponsored by Natural Sciences and Engineering Research Council of Canada Discovery Grant 386275‐2010 (to C.J. Martyniuk) and 238621‐2011 (to K.R. Munkittrick) and a Canada Research Chair (to C.J. Martyniuk and K.R. Munkittrick).
Footnotes
Publisher's Disclaimer: Disclaimer
Publisher's Disclaimer: The views expressed in the present study are those of the authors and do not necessarily represent the views or policies of the US Environmental Protection Agency.
Data Availability
All data are publicly available at the National Center for Biotechnology Information’s Gene Expression Omnibus (GSE 81544 and 70807).
References
- 1.McMaster ME, Jardine JJ, Ankley GT, Benson WH, Greeley MS, Gross TS, Guillette LJ, MacLatchy DL, Orlando EF, Van Der Kraak GJ. 2001. An interlaboratory study on the use of steroid hormones in examining endocrine disruption. Environ Toxicol Chem 20:2081–2087. [DOI] [PubMed] [Google Scholar]
- 2.Feswick A, Ankley GT, Denslow N, Ellestad LE, Fuzzen M, Jensen KM, Kroll K, Lister A, MacLatchy DL, McMaster ME, Orlando EF, Servos MR, Tetreault GR, Van Den Heuvel MR, Munkittrick KR. 2014An inter‐laboratory study on the variability in measured concentrations of 17beta‐estradiol, testosterone, and 11‐ketotestosterone in white sucker: Implications and recommendations. Environ Toxicol Chem 33:847–857. [DOI] [PubMed] [Google Scholar]
- 3.Munkittrick K, van den Heuvel MR, Metner D, Lockhart W, Stegeman J. 1993. Interlaboratory comparison and optimization of hepatic microsomal ethoxyresorufin O‐deethylase activity in white sucker (Catostomus commersoni) exposed to bleached kraft pulp mill effluent. Environ Toxicol Chem 12:1273–1282. [Google Scholar]
- 4.Van Den Heuvel MR, Dixon DG, Munkittrick KR, Stegeman JJ. 1995. Second‐round interlaboratory comparison of hepatic ethoxyresorufin‐O‐deethylase activity in white sucker (Catostomus commersoni) exposed to bleached‐kraft pulp mill effluent. Environ Toxicol Chem 14:1513–1520. [Google Scholar]
- 5.US Environmental Protection Agency. 2003. Comparative evaluation of vitellogenin methods 68‐W‐ 01–023. Washington, DC. [Google Scholar]
- 6.US Environmental Protection Agency. 2001. Final report: Interlaboratory variability study of EPA short‐term chronic and acute whole effluent toxicity test methods 821‐B‐01‐004. Washington, DC. [Google Scholar]
- 7.Environment Canada. 2010. Pulp and paper environmental effects monitoring (EEM) technical guidance document Gatineau, QC, Canada. [Google Scholar]
- 8.Barrett TJ, Munkittrick KR. 2010. Seasonal reproductive patterns and recommended sampling times for sentinel fish species used in environmental effects monitoring programs in Canada. Environ Rev 18:115–135. [Google Scholar]
- 9.Munkittrick K, Kraak G, McMaster M, Portt C. 1992. Reproductive dysfunction and MFO activity in three species of fish exposed to bleached kraft mill effluent at Jackfish Bay, Lake Superior. Water Qual Res J Can 27:439–446. [Google Scholar]
- 10.Munkittrick KR, McGeachy SA, McMaster ME, Courtenay SC. 2002. Overview of freshwater fish studies from the pulp and paper environmental effects monitoring program. Water Qual Res J Can 37:49–77. [Google Scholar]
- 11.Munkittrick KR, Arens CJ, Lowell RB, Kaminski GP. 2009. A review of potential methods of determining critical effect size for designing environmental monitoring programs. Environ Toxicol Chem 28:1361–1371. [DOI] [PubMed] [Google Scholar]
- 12.Larsson Å, Förlin L, Grahn O, Landner L, Lindesjöö E, Sandström O. 2000. Guidelines for interpretation and biological evaluation of biochemical, physiological and pathological alterations in fish exposed to industrial effluents SSVL, Stockholm, Sweden. [Google Scholar]
- 13.Hutchinson TH, Ankley GT, Segner H, Tyler CR. 2006. Screening and testing for endocrine disruption in fish‐biomarkers as “signposts,” not “traffic lights,” in risk assessment. Environ Health Perspect 114:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller DH, Jensen KM, Villeneuve DL, Kahl MD, Makynen EA, Durhan EJ, Ankley GT. 2007. Linkage of biochemical responses to population‐level effects: A case study with vitellogenin in the fathead minnow (Pimephales promelas). Environ Toxicol Chem 26:521–527. [DOI] [PubMed] [Google Scholar]
- 15.Van Aggelen G, Ankley GT, Baldwin WS, Bearden DW, Benson WH, Chipman JK, Collette TW, Craft JA, Denslow ND, Embry MR. 2010. Integrating omic technologies into aquatic ecological risk assessment and environmental monitoring: Hurdles, achievements, and future outlook. Environ Health Perspect 118:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hook S 2010. Promise and progress in environmental genomics: A status report on the applications of gene expression–based microarray studies in ecologically relevant fish species. J Fish Biol 77:1999–2022. [DOI] [PubMed] [Google Scholar]
- 17.Bahamonde PA, Feswick A, Isaacs MA, Munkittrick KR, Martyniuk CJ. 2016. Defining the role of omics in assessing ecosystem health: Perspectives from the Canadian environmental monitoring program. Environ Toxicol Chem 35:20–35. [DOI] [PubMed] [Google Scholar]
- 18.Yauk CL, Berndt ML. 2007. Review of the literature examining the correlation among DNA microarray technologies. Environ Mol Mutagen 48:380–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muller C, Schillert A, Rothemeier C, Tregouet DA, Proust C, Binder H, Pfeiffer N, Beutel M, Lackner KJ, Schnabel RB, Tiret L, Wild PS, Blankenberg S, Zeller T, Ziegler A. 2016. Removing batch effects from longitudinal gene expression—Quantile normalization plus combat as best approach for microarray transcriptome data. PLoS One 11:e0156594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi L, Reid LH, Jones WD, Shippy R, Warrington JA, Baker SC, Collins PJ, De Longueville F, Kawasaki ES, Lee KY. 2006. The MicroArray Quality Control (MAQC) project shows inter‐ and intraplatform reproducibility of gene expression measurements. Nat Biotechnol 24:1151–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scott D, Devonshire A, Adeleye Y, Schutte M, Rodrigues M, Wilkes T, Sacco M, Gribaldo L, Fabbri M, Coecke S. 2011. Inter‐ and intra‐laboratory study to determine the reproducibility of toxicogenomics datasets. Toxicology 290:50–58. [DOI] [PubMed] [Google Scholar]
- 22.Weis B 2005. Standardizing global gene expression analysis between laboratories and across platforms. Nature Methods 2:351–356. [DOI] [PubMed] [Google Scholar]
- 23.Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C, Aach J, Ansorge W, Ball CA, Causton HC, Gaasterland T, Glenisson P, Holstege FC, Kim IF, Markowitz V, Matese JC, Parkinson H, Robinson A, Sarkans U, Schulze‐Kremer S, Stewart J, Taylor R, Vilo J, Vingron M. 2001. Minimum information about a microarray experiment (MIAME)—Toward standards for microarray data. Nat Genet 29:365–371. [DOI] [PubMed] [Google Scholar]
- 24.Hockley SL, Mathijs K, Staal YC, Brewer D, Giddings I, van Delft JH, Phillips DH. 2009Interlaboratory and interplatform comparison of microarray gene expression analysis of HepG2 cells exposed to benzo[a]pyrene. Omics 13:115–125. [DOI] [PubMed] [Google Scholar]
- 25.Vidal‐Dorsch DE, Bay SM, Moore S, Layton B, Mehinto AC, Vulpe CD, Brown‐Augustine M, Loguinov A, Poynton H, Garcia‐Reyero N, Perkins EJ, Escalon L, Denslow ND, Cristina CR, Doan T, Shukradas S, Bruno J, Brown L, Van Agglen G, Jackman P, Bauer M. 2015. Ecotoxicogenomics: Microarray interlaboratory comparability. Chemosphere 144:193–200. [DOI] [PubMed] [Google Scholar]
- 26.Ankley GT, Villeneuve DL. 2006. The fathead minnow in aquatic toxicology: Past, present and future. Aquat Toxicol 78:91–102. [DOI] [PubMed] [Google Scholar]
- 27.US Environmental Protection Agency. 2002. Methods for measuring the acute toxicity of effluents and receiving waters to freshwater and marine organisms, 5th ed. EPA‐821‐R‐02‐012. Washington, DC. [Google Scholar]
- 28.Barel‐Cohen K, Shore LS, Shemesh M, Wenzel A, Mueller J, Kronfeld‐Schor N. 2006. Monitoring of natural and synthetic hormones in a polluted river. J Environ Manage 78:16–23. [DOI] [PubMed] [Google Scholar]
- 29.Fernandez MP, Ikonomou MG, Buchanan I. 2007. An assessment of estrogenic organic contaminants in Canadian wastewaters. Sci Total Environ 373:250–269. [DOI] [PubMed] [Google Scholar]
- 30.Atkinson SK, Marlatt VL, Kimpe LE, Lean DR, Trudeau VL, Blais JM. 2012. The occurrence of steroidal estrogens in south‐eastern Ontario wastewater treatment plants. Sci Total Environ 430:119–125. [DOI] [PubMed] [Google Scholar]
- 31.Pawlowski S, Van Aerle R, Tyler C, Braunbeck T. 2004. Effects of 17α‐ethinylestradiol in a fathead minnow (Pimephales promelas) gonadal recrudescence assay. Ecotox Environ Safe 57:330–345. [DOI] [PubMed] [Google Scholar]
- 32.Parrott JL, Blunt BR. 2005. Life‐cycle exposure of fathead minnows (Pimephales promelas) to an ethinylestradiol concentration below 1 ng/L reduces egg fertilization success and demasculinizes males. Environ Toxicol 20:131–141. [DOI] [PubMed] [Google Scholar]
- 33.Martyniuk CJ, Kroll KJ, Doperalski NJ, Barber DS, Denslow ND. 2010. Environmentally relevant exposure to 17alpha‐ethinylestradiol affects the telencephalic proteome of male fathead minnows. Aquat Toxicol 98:344–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salierno JD, Kane AS. 2009. 17α‐Ethinylestradiol alters reproductive behaviors, circulating hormones, and sexual morphology in male fathead minnows (Pimephales promelas). Environ Toxicol Chem 28:953–961. [DOI] [PubMed] [Google Scholar]
- 35.Hoffmann J, Torontali S, Thomason R, Lee D, Brill J, Price B, Carr G, Versteeg D. 2006. Hepatic gene expression profiling using Genechips in zebrafish exposed to 17α‐ethynylestradiol. Aquat Toxicol 79:233–246. [DOI] [PubMed] [Google Scholar]
- 36.Martyniuk CJ, Gerrie ER, Popesku JT, Ekker M, Trudeau VL. 2007. Microarray analysis in the zebrafish (Danio rerio) liver and telencephalon after exposure to low concentration of 17alpha‐ethinylestradiol. Aquat Toxicol 84:38–49. [DOI] [PubMed] [Google Scholar]
- 37.Doyle MA, Bosker T, Martyniuk CJ, Maclatchy DL, Munkittrick KR. 2013. The effects of 17‐alpha‐ethinylestradiol (EE2) on molecular signaling cascades in mummichog (Fundulus heteroclitus). Aquat Toxicol 134–135:34–46. [DOI] [PubMed] [Google Scholar]
- 38.Skillman AD, Nagler JJ, Hook SE, Small JA, Schultz IR. 2006. Dynamics of 17alpha‐ethynylestradiol exposure in rainbow trout (Oncorhynchus mykiss): Absorption, tissue distribution, and hepatic gene expression pattern. Environ Toxicol Chem 25:2997–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.MacLatchy DL, Courtenay SC, Rice CD, Van der Kraak GJ. 2003. Development of a short‐term reproductive endocrine bioassay using steroid hormone and vitellogenin end points in the estuarine mummichog (Fundulus heteroclitus). Environ Toxicol Chem 22:996–1008. [PubMed] [Google Scholar]
- 40.Runnalls TJ, Beresford N, Kugathas S, Margiotta‐Casaluci L, Scholze M, Scott AP, Sumpter JP.2015. From single chemicals to mixtures—Reproductive effects of levonorgestrel and ethinylestradiol on the fathead minnow. Aquat Toxicol 169:152–167. [DOI] [PubMed] [Google Scholar]
- 41.Kidd KA, Blanchfield PJ, Mills KH, Palace VP, Evans RE, Lazorchak JM, Flick RW. 2007. Collapse of a fish population after exposure to a synthetic estrogen. P Natl Acad Sci USA 104:8897–8901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garcia‐Reyero N, Kennedy AJ, Escalon BL, Habib T, Laird JG, Rawat A, Wiseman S, Hecker M, Denslow N, Steevens JA, Perkins EJ. 2014. Differential effects and potential adverse outcomes of ionic silver and silver nanoparticles in vivo and in vitro. Environ Sci Technol 48:4546–4555. [DOI] [PubMed] [Google Scholar]
- 43.Levi L, Pekarski I, Gutman E, Fortina P, Hyslop T, Biran J, Levavi‐Sivan B, Lubzens E. 2009. Revealing genes associated with vitellogenesis in the liver of the zebrafish (Danio rerio) by transcriptome profiling. BMC Genomics 10:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davis AP, Murphy CG, Johnson R, Lay JM, Lennon‐Hopkins K, Saraceni‐Richards C, Sciaky D, King BL, Rosenstein MC, Wiegers TC, Mattingly CJ. 2013. The Comparative Toxicogenomics Database: Update 2013. Nucleic Acids Res 41:D1104–D1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dreier DA, Loughery JR, Denslow ND, Martyniuk CJ. 2016. The influence of breeding strategy, reproductive stage, and tissue type on transcript variability in fish. Comp Biochem Physiol D Genomics Proteomics 19:151–158. [DOI] [PubMed] [Google Scholar]
- 46.Wang RL, Bencic DC, Garcia‐Reyero N, Perkins EJ, Villeneuve DL, Ankley GT, Biales AD. 2014. Natural variation in fish transcriptomes: Comparative analysis of the fathead minnow (Pimephales promelas) and zebrafish (Danio rerio). PLoS One 9:e114178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dalman MR, Deeter A, Nimishakavi G, Duan ZH. 2012. Fold change and p‐value cutoffs significantly alter microarray interpretations. BMC Bioinformatics 13(Suppl. 2):S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomas RS, Rank DR, Penn SG, Zastrow GM, Hayes KR, Pande K, Glover E, Silander T, Craven MW, Reddy JK, Jovanovich SB, Bradfield CA. 2001. Identification of toxicologically predictive gene sets using cDNA microarrays. Mol Pharmacol 60:1189–1194. [DOI] [PubMed] [Google Scholar]
- 49.Mancia A, Abelli L, Kucklick JR, Rowles TK, Wells RS, Balmer BC, Hohn AA, Baatz JE, Ryan JC.2015. Microarray applications to understand the impact of exposure to environmental contaminants in wild dolphins (Tursiops truncatus). Mar Genomics 19:47–57. [DOI] [PubMed] [Google Scholar]
- 50.Dembele D, Kastner P. 2014. Fold change rank ordering statistics: A new method for detecting differentially expressed genes. BMC Bioinformatics 15:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang L, Zhang J, Yang G, Wu D, Jiang L, Wen Z, Li M. 2013. Investigating the concordance of Gene Ontology terms reveals the intra‐ and inter‐platform reproducibility of enrichment analysis. BMC Bioinformatics 14:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.