

Abstract
Monitored contaminants in drinking water represent a small portion of the total compounds present, many of which may be relevant to human health. To understand the totality of human exposure to compounds in drinking water, broader monitoring methods are imperative. In an effort to more fully characterize the drinking water exposome, point-of-use water filtration devices (Brita® filters) were employed to collect time-integrated drinking water samples in a pilot study of nine North Carolina homes. A suspect screening analysis was performed by matching high resolution mass spectra of unknown features to molecular formulas from EPA’s DSSTox database. Candidate compounds with those formulas were retrieved from the EPA’s CompTox Chemistry Dashboard, a recently developed data hub for approximately 720,000 compounds. To prioritize compounds into those most relevant for human health, toxicity data from the US federal collaborative Tox21 program and the EPA ToxCast program, as well as exposure estimates from EPA’s ExpoCast program, were used in conjunction with sample detection frequency and abundance to calculate a “ToxPi” score for each candidate compound. From ∼15,000 molecular features in the raw data, 91 candidate compounds were ultimately grouped into the highest priority class for follow up study. Fifteen of these compounds were confirmed using analytical standards including the highest priority compound, 1,2-Benzisothiazolin-3-one, which appeared in 7 out of 9 samples. The majority of the other high priority compounds are not targets of routine monitoring, highlighting major gaps in our understanding of drinking water exposures. General product-use categories from EPA’s CPCat database revealed that several of the high priority chemicals are used in industrial processes, indicating the drinking water in central North Carolina may be impacted by local industries.
Keywords: Drinking water, Exposome, High resolution mass spectrometry, Non-target analysis, Suspect screening
GRAPHICAL ABSTRACT

1. Introduction
Safe drinking water supplies are critical for public health and it has been estimated by the World Health Organization (WHO) that a 10% reduction in worldwide disease could be achieved by improvements related to drinking water alone, including sanitation, hygiene, and water resource management (Prüss-Üstün et al., 2008). Furthermore, it is estimated that 70–90% of disease risks are due to differences in environments (Rappaport and Smith, 2010), which includes direct exposures via consumption of drinking water. Chemicals that are present in water supplies can increase risk for disease and adverse health outcomes over long-term exposure periods (WHO, 2013). It has been demonstrated for various chemical classes, including perfluorinated chemicals, that drinking water can be one of the most important pathways for human exposure (Egeghy and Lorber, 2011, Lorber and Egeghy, 2011). Even so, it has been estimated that only 40% of US consumers used any kind of water purification device in 2014 (Anumol et al., 2015). Certain chemicals are regulated under the Safe Drinking Water Act, but these chemicals constitute only a small fraction of the number of chemicals present in drinking water (US EPA, 2016). New compounds can be added to this list if they are discovered and deemed to pose a threat to human health. These additions, however, require developing and validating “targeted” methods, which is a slow and expensive process. Furthermore, this process requires some a priori knowledge of the compounds for which methods should be developed. As of yet, there is no reliable mechanism to identify and prioritize novel compounds. There are needs, then, for: 1) a more complete picture of chemical exposures via drinking water consumption; 2) methods of rapidly identifying emerging chemicals that may be of importance to human health; and 3) means with which to properly assess exposure-disease relationships and risks to human health (Villanueva et al., 2014).
Recent advances in analytical techniques have led to the detection of various contaminants in water which would have otherwise gone undetected using traditional targeted methods (Schymanski et al., 2015, Strynar et al., 2015). These advanced techniques often employ high resolution mass spectrometry (HRMS), or tandem HRMS, to either match unknown sample features to compounds within spectral and/or spectra-less databases (a technique known as suspect screening analysis [SSA]), or elucidate structures of unknowns that may not be contained in a database (a technique known as non-targeted analysis [NTA]). While these two techniques differ, they are often discussed together as they are complimentary to each other. SSA/NTA workflows are rapidly evolving, and are becoming more frequently used to detect differences (or similarities) between two or more groups of samples in case-control style experiments. Example applications include: detecting a chemical spill in a river after a baseline chemical signature has been established (Bader et al., 2016); evaluating the contribution of various tributaries to a river (Ruff et al., 2015); or singling out unknown features that appear in landfill leachate and in downstream drinking water (Müller et al., 2011).
SSA/NTA approaches may also be applied to environmental samples in support of general monitoring – that is, to broadly screen for the occurrence of chemicals in a selected medium. The ability to rapidly identify unknown compounds during routine monitoring is essential to fully explore the exposome, defined as the sum of all exposures (exogenous and endogenous) for an individual over a lifetime (Wild, 2005). In order to sequence the exposome, it is useful and necessary, from an analytical standpoint, to compartmentalize exposures by matrix. Examples of monitoring studies that focus on a specific matrix can be found for dust (Rager et al., 2016), river water (Schymanski et al., 2015), waste water(Schymanski et al., 2014b), etc. but drinking water remains relatively unexplored with regards to SSA/NTA. This is somewhat surprising, as drinking water is a fairly simple matrix to which humans are exposed in similar amounts, in contrast to dust or waste water, which require clean-up steps after extraction, and for which exposure amounts are not well known.
When applied to environmental and biological samples, SSA/NTA methods have the potential to allow rapid chemical characterization without the need for standards or a prioriknowledge of sample constituents. Confidence in the identification of unknowns can be communicated in terms of levels outlined by Schymanski et al. (2014a), where the highest level of confidence (level 1) requires confirmation by an analytical standard, and the next level of confidence (level 2) requires evidence for a probable structure. A goal for researchers using SSA/NTA methods should be to confidently classify as many unknowns as possible into level 2, and not necessarily level 1, as it is not practical, or even possible, to confirm all unknowns with analytical standards. Chemicals of highest concern can then be confirmed with standards, if possible, and categorized into level 1. Confidence in level 2 identifications will most likely come about through the development of several different tools that build increasing confidence of positive detection. As we are in the early years of a burgeoning exposomics field, researchers must find ways to prioritize unknowns into those that they believe are most likely to be relevant to human and environmental health (Sobus et al., 2017). Recently, a method to prioritize the vast number of unknowns in a sample by incorporating toxicity and exposure information was presented by Rager et al. (2016). We have sought to apply this method to drinking water in the Raleigh/Durham/Chapel Hill area of North Carolina, United States, and improve upon it using tools and data available from EPA’s CompTox Chemistry Dashboard (hereafter referred to as “the Dashboard”, https://comptox.epa.gov/dashboard), a newly developed web application that supports SSA/NTA workflows (McEachran et al., 2017b). We have also sought to demonstrate that SSA/NTA methods can rapidly identify contaminants in drinking water that are not routinely monitored and would likely go undetected without these methods.
2. Materials and methods
2.1. Materials
Information about the materials used in this study can be found in the Supporting Information (SI).
2.2. Sample collection
Samples were collected in a pilot scale study by installing a Brita® Basic Faucet Filter in the homes of nine North Carolina residents. Provided in the SI is a list of chemicals that Brita®Basic Faucet Filters are known to remove from drinking water (SI, Table S1), as well as a table of organic chemicals included in the Safe Drinking Water Act (Table S2). Some residents received drinking water from their local municipalities, while other residents received their drinking water from a private well. Information about the water source and municipality can be found in Table 1. Although the samples are labeled by location, many of the drinking water treatment facilities report purchasing water from other facilities so it is possible the sampling location is not fully indicative of the original drinking water source. The study participants were asked to use the filter for cold water during everyday use until the indicator light on the filter turned red, signaling that the filter was at its maximum capacity. This process took between 1 and 4 months for each sample with an average sampling time of 68 days. The participants were asked to return their filters for analysis upon seeing the red indicator light.
Table 1.
Sample information.
| Sample # | Location | Source Type | Population Served |
|---|---|---|---|
| 1 | Durham | Municipal | 265,472 |
| 2 | Durham | Municipal | 265,472 |
| 3 | Apex | Municipal | 46,831 |
| 4 | Cary | Municipal | 182,088 |
| 5 | Chapel Hill | Municipal | 83,300 |
| 6 | Chapel Hill | Private Well | – |
| 7 | Raleigh | Municipal | 540,000 |
| 8 | Pittsboro | Municipal | 4,401 |
| 9 | Pittsboro | Private Well | – |
2.3. Sample extraction and processing
The filter was removed from the plastic casing using a band saw with a clean blade and placed into a plastic bag for storage until extraction. The filters were individually lyophilized for three days to remove any water which remained in the filter pores. The filters were extracted via Soxhlet using 300 mL of an dichloromethane:methanol (80:20 v/v) mixture for 24 h. Upon completion, the flasks were cooled for 30 min before the solvent was removed under reduced pressure using a rotary evaporator. The extract was re-dissolved in 5 mL of methanol, centrifuged at 12,500 × g for 3 min to remove particles from suspension. One-hundred μL of sample was mixed with 300 μL of 2 mM ammonium acetate buffer in an autosampler vial for analysis.
2.4. Instrumental analysis
Liquid Chromatography (LC) - Time-of-Flight (TOF) HRMS analysis was carried out using an Agilent 1100 HPLC (Agilent Technologies, Palo Alto, CA), interfaced with an Agilent 6210 TOF HRMS. Chromatographic separation was accomplished using an Eclipse Plus C8 column (2.1 × 50 mm, 3.5 μm; Agilent Technologies, Palo Alto, CA). The method consisted of the following conditions: 0.2 mL/min flow rate; column at 30 °C; mobile phase A as ammonium formate buffer (0.4 mM) and DI water:methanol (95:5 v/v), and mobile phase B as ammonium formate (0.4 mM) and methanol:DI water (95:5 v/v); gradient: 0–25 min linear gradient from 75:25 A:B to 15:85 A:B; 25–40 min a linear gradient from 15:85 A:B to 100% B; 40–45 hold at 100% B. The TOF-HRMS was fitted with an electrospray ionization source, which operated in both negative and positive ionization modes (separate injection for each mode), using a fragmentor voltage of 80 V. Data was collected in 4 GHz high resolution mode, collecting ions in m/z range 100–1700 in both centroid and profile data formats. Further details on instrumental parameters can be found in Table S3 (SI).
2.5. Molecular feature detection and chemical formula assignment
Molecular feature extraction and formula assignment was performed according to previously published methods (Rager et al., 2016). Briefly, molecular features (defined as an exact mass, retention time, and isotope cluster of an apparent unknown compound) were identified and extracted using Agilent MassHunter 6.0 Qualitative Software’s molecular feature extractor (MFE). Features were extracted from the method blanks and solvent blanks first and the masses of those features were used in a “mass exclusion list” when extracting features from the samples. MassHunter was then used to match molecular features from the samples to chemical formulas contained in EPA’s Distributed Structure-Searchable Toxicity database V2 (DSSTox_V2). This database contains a list of 16,532 unique formulas (de-salted) which correspond to 33,659 chemicals. Feature matches were scored based on neutral accurate mass, isotope distribution, and isotope ratio. While DSSTox_V2 contains chemical compounds, it was used only to assign molecular formulas since isomers cannot be distinguished using the methods described here (which consider molecular MS spectra only). Newer versions of the DSSTox database, including the version which is accessed by the Dashboard (approximately 760,000 as of November 2017), contain many more chemicals; however, the de-salted forms of the molecular formulas were not available at the time the database matching for this study was conducted. Molecular formulas were only assigned to features which attained a match score of ≥90. Further details on the software settings for the MFE and database search can be found in Table S3 (SI).
2.6. Assignment of probable structure from molecular formulas
The workflow for assigning structures to formulas and prioritizing those structures is shown in Fig. 1. Candidate structures associated with molecular formulas were retrieved from the Dashboard using the Batch Search capability (https://comptox.epa.gov/dashboard/dsstoxdb/batch_search). In this manner, the most likely candidate structures are retrieved and ordered by the number of data sources associated with each structure. Data sources in this context represent the number of times an EPA dataset, database, or list within DSSTox contains a particular chemical. This workflow follows previous reports on the identification of “known unknowns” by Little et al. (2012). Additionally, it has been demonstrated using the EPA Dashboard that candidate compounds with the greatest number of data sources are the correct compound for a given formula in over 80% of cases (McEachran et al., 2017b). Bioactivity and exposure data for some of these structures were available from the Tox21/ToxCast (US EPA, 2015) and ExpoCast (Wambaugh et al., 2013) projects, respectively, and accessible via the Dashboard. Compounds for which toxicity and exposure data were available were labeled as “Group A” compounds, whereas compounds missing one or both of these data types were labeled as “Group B”. Multiple candidate compounds often existed for a given formula, with some being Group A compounds and some being Group B compounds. For Group A compounds, a bioactivity ratio was calculated as the number of assay hits divided by the total number of assays tested. Exposure categories were calculated from ExpoCast daily exposure estimates using the categorization described by Rager et al. (2016):
Fig. 1.
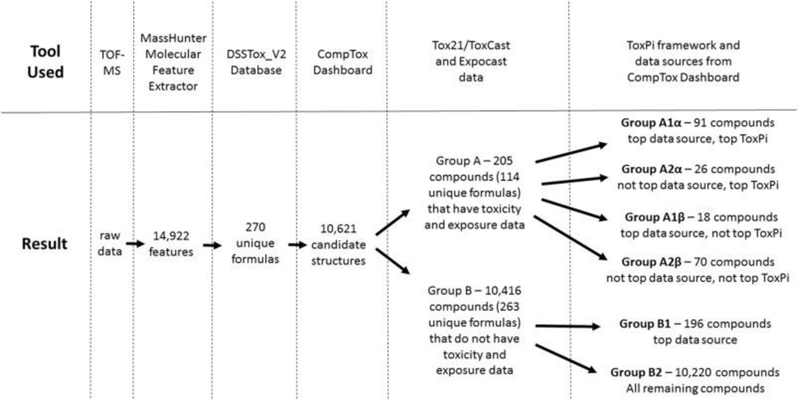
Workflow for processing data and categorizing candidate compounds.
Category 1 <1 × 10−8 mg/kg/day;
Category 2 ≥1 × 10−8 mg/kg/day and <1 × 10−7 mg/kg/day;
Category 3 ≥1 × 10−7 mg/kg/day and <1 × 10−6 mg/kg/day;
Category 4 ≥1 × 10−6 mg/kg/day and <1 × 10−5 mg/kg/day;
Category 5 ≥1 × 10−5 mg/kg/day and <1 × 10−4 mg/kg/day;
Category 6 ≥1 × 10−4 mg/kg/day and <1 × 10−3 mg/kg/day; and
Category 7 ≥1 × 10−3 mg/kg/day and <1 × 10−2 mg/kg/day.
A ToxPi score was calculated for each Group A compound (i) using its bioactivity (B) ratio, exposure category (E), detection frequency (DF), and abundance (average chromatographic peak area, A), according to equation (1). All values for E, DF, and A were log-transformed before applying equation (1) due to the skewed nature of their distributions.
Equal weight was given to each category despite the precedent of weighting some categories differently (Rager et al., 2016).
All compounds were further subcategorized with a “1” if the compound had the highest number of data sources for its formula, or a “2” if it did not. Compounds in Group A were also subcategorized with a “α” if the compound had the highest ToxPi score for its formula, and a “β” if it did not. Thus, all compounds fell into one of six categories: A1α, A2α, A1β, A2β, B1, or B2 (Fig. 1), with A1α compounds being the most likely structures and highest ToxPis for their formulas and thus the highest priority group.
2.7. Literature search
Three databases were searched to assess the prevalence of A1α compounds in the literature: SciFinder®, Google Scholar, and PubMed. As described in Rager et al. (2016), the SciFinder® search (SciFinder, 2017) was performed to determine whether the A1α chemicals have previously been reported as being detected in water. Each chemical’s CASRN was searched by the term “water” within the SciFinder® “Research Topic” menu. The results were then refined to only include journal references and the number of results was recorded. The Google Scholar and PubMed searches were conducted using the same search terms and no filters were applied. All searched were conducted manually. This literature search was not meant to be exhaustive, but rather to provide some indication of each compound’s relative prevalence in the literature and association with water.
2.8. Retention time prediction using OPERA-RT
OPERA-RT is quantitative structure property relationship (QSPR) model that is part of OPERA, a free and open-source suite of models used to predict physicochemical and environmental fate of organic chemicals (download available on Github: https://github.com/kmansouri/OPERA) (Mansouri et al., 2016). OPERA-RT was previously developed as described in McEachran et al. (2017a). The tool uses molecular descriptors as input to predict LC retention times for compounds and is based on the same LC method that was used in this study. Retention times were predicted for A1α compounds and a window of ±10% of the total chromatographic run time (±4.5 min) was used to compare the observed retention time with the predicted retention time of the putative A1α identification. The tool was used to increase confidence in the identification of A1α compounds as recommended by McEachran et al. rather than to exclude compounds that fall outside their retention time window.
2.9. Product-use categories
Product-use categories for A1α compounds were taken from EPA’s CPCat database (Dionisio et al., 2015). These data can be explored through the Dashboard. Principal component analysis (PCA) was performed using a matrix of summed peak areas for A1α compounds in specific samples (observations) and product-use categories (variables). PCA plots were constructed using the caret package (version 6.0–62) in the R programming language (version 3.3.1).
2.10. Quality control and quality assurance
Calibration of the instrument was performed prior to analysis in each mode. Any drift in the mass accuracy of the TOF was continuously corrected by infusion of two reference compounds (purine [m/z = 119.0363] and Hexakis(1H,1H,3H-perfluoropropoxy)phosphazene [identified in the Dashboard as DTXSID90880494, observed as a formate adduct at m/z = 966.0007]) via dual-ESI sprayer. Three unused filters were processed along with the samples as method blanks. The masses of features observed in these methods blanks were used in a blank exclusion list when extracting features from samples. Solvent blanks were also analyzed consisting of a mix of ammonium acetate buffer and methanol.
3. Results and discussion
Approximately 15,000 total features were detected across all samples, with 10,606 found in positive mode and 4,317 in negative mode. The greater number of positive mode features may have been aided by the presence of H+ ions from the slightly acidic mobile phase. Positive mode features tended to be smaller in chromatographic peak area, with the median peak area (190,000) roughly half that of negative mode features (370,000). Four-hundred and thirty features were matched to a formula in the DSSTox_V2 database with a match score of 90 or greater. A greater proportion of negative mode features was matched (4.2%) than positive mode features (2.3%). Across both modes, 2.9% of features were matched yet peak areas for these matched features comprised 16.9% of the total peak area of all features. The number of features matched is similar to that reported by Rager et al. who matched less than 2% of the total number of features in 56 dust samples but did not report the percentage of peak area that was matched. The median peak area of unmatched features was approximately 200,000 while the median peak area of matched features was approximately 1.5 million (Fig. 2). This means that while the number of features being matched is low, matching tends to favor larger peaks. This is not surprising considering that larger features are likely to contain better isotope peaks which play a crucial role in matching to a formula (Kind and Fiehn, 2006). Another possible explanation is that larger peaks tended to be compounds that have been of interest previously and are therefore more likely to be contained within the database from prior study by researchers. Descriptive statistics for features and molecular formula matches can be found in Table 2, and a bubble plot of all features with retention time and m/z can be found in the SI (Fig. S1).
Fig. 2.
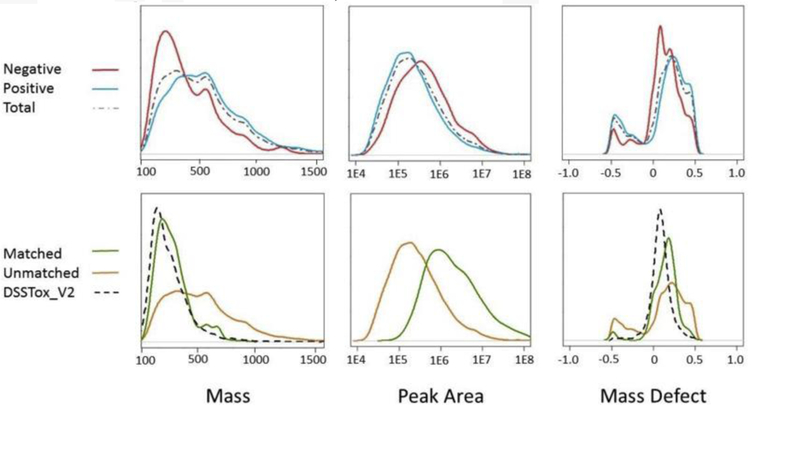
Kernel density plots of mass, peak area, and mass defects for negative, positive, matched (i.e., formula assigned), unmatched features, and the entire DSSTox_V2 database.
Table 2.
Descriptive statistics of features, formulas, and A1α compounds between negative and positive modes.
| Ionization mode | Negative | Positive | Total |
|---|---|---|---|
| Number of features | 4,317 | 10,606 | 14,923 |
| Average (SD) features per sample | 480 (207) | 1,178 (542) | 1,658 (724) |
| Geometric mean peak area | 420,000 | 230,000 | 270,000 |
| Features assigned a formula | 181 | 249 | 430 |
| Unique formulas | 166 | 231 | 270 |
| Percent of features assigned a formula | 4.2% | 2.3% | 2.9% |
| Percent peak area assigned a formula | 12.8% | 19.2% | 16.9% |
| Features with A1α designation | 74 | 74 | 148 |
| Percent peak area of A1α compounds | 8.2% | 7.0% | 7.4% |
Kernel density plots showing the distributions of the masses, volumes, and mass defects of features can be seen in Fig. 2. The mass distribution of features matched to the database was heavily biased towards the distribution of masses in the DSSTox_V2 database. The percentage of features with masses less than 500 Da was 51% for all features, but increased to 90% for features assigned a formula. This is likely due to the fact that 92% of compounds in the DSSTox_V2 database have masses less than 500 Da. The same trend was observed in the distribution of mass defects among features assigned a formula, highlighting the importance of the content of the databases used when performing suspect screening analysis.
Mass and elemental composition of the formulas generated in this study (water filters) were compared to those of the previous study of house dust by Rager et al. (2016) on the basis that the same database and matching algorithm were used. Significant differences (Welch’s two sample t-test, p < 0.001) in mass and number of carbons per formula were observed between the studies, with the house dust containing heavier compounds and 3.4 more carbons per formula, on average, than the water filters. Oxygen and phosphorous were similar in the average number per formula and percentage of formulas in which they were found. Nitrogen, however, was found in 48% of the water filter formulas but only 34% of the house dust formulas. Sulfur was found in 10% of the water filter formulas but 29% of formulas from the house dust. Summary statistics on the elemental composition and mass distributions of the formulas generated in the two studies can be found in the SI (Table S4) as well as a PCA of the element counts, retention times, and masses for each formula (Fig. S2). Despite the differences in carbon, nitrogen, and sulfur content, no clear separation or patterns were observed in the PCA.
The 430 features that were assigned a formula were comprised of 270 unique formulas which generated 10,621 candidate compounds from the Dashboard, giving an average of 39 compounds per formula (range = 1 to 451 compounds per formula). Each candidate compound was then categorized into Group A, containing toxicity and exposure data, or Group B, not containing these data. Of all candidate compounds, 205 contained were categorized into Group A, 91 of which were sub-categorized into Group A1α, which are considered the most likely compounds based on data source rankings (McEachran et al., 2017b) as well as the most important compounds with regards to bioactivity, exposure, abundance, and detection frequency. The SciFinder® search resulted in 59 of the A1α compounds being associated with water in journal articles, meaning 32 have not been associated before with water. Among those with associated journal articles, the average number of articles was 569, highlighting the tendency for researchers to publish on already-known compounds and the need for more work in compound discovery. The PubMed search gave similar results, with 66 compounds associated with water but the Google Scholar search returned 90 compounds associated with water.
The remaining 114 Group A compounds were sub-categorized as follows: 26 into Group A2α, 18 into Group A1β, and 70 into Group A2β. Of the remaining 10,416 Group B compounds, 196 were sub-categorized into Group B1 and 10,220 in Group B2. While the vast majority of candidate compounds fall into Group B2, these compounds are less likely to be the correct compounds for a given set of matched formulas. Group A1α features tend to be larger than most peaks: the median peak area of an A1α feature was approximately 1,900,000 counts whereas the median peak area of non-A1α features that were assigned a formula was 1,300,000 counts, and the median peak area of features that were not assigned a formula was 220,000 counts. Furthermore, 44% of the peak area that was assigned to a formula could be mapped to an A1α compound, which was 7.4% of the total peak area of all features. A list of all A1α compounds along with their bioactivity and exposure values, functional use information, results of the SciFinder® search, and other supplementary data can be found in the SI (Table S5).
3.1. ToxPi scores and confirmation by standards
ToxPi scores for Group A1α compounds ranged from 0.046 to 2.99 out of a maximum possible score of 4. All A1α ToxPis scores are displayed graphically in Fig. 3 with values given for the top 20. In general, the contribution from the four different categories to the total ToxPi score varied greatly from compound to compound.
Fig. 3.
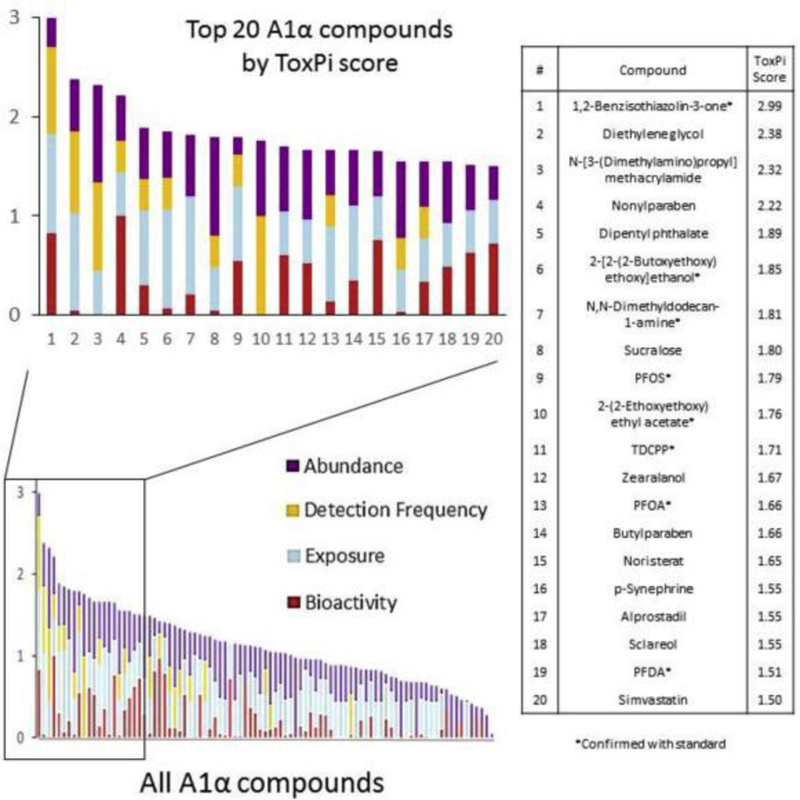
ToxPis of all A1α compounds (bottom left) with the top 20 enlarged (top left) and their corresponding ToxPi scores (right).
To assess correct structure-to-formula assignments and confirm compounds with standards, sample-based formulas were matched with formulas for standards readily available in our laboratory. Sixteen unique compounds had formulas matching those of existing laboratory standards. Thirteen of the standard compounds were categorized as A1α and three as B1. Of the three B1 compounds, no group A compounds existed for those formulas. Fifteen of sixteen compounds were ultimately confirmed with standards via retention time matching and visual inspection of MS spectra. One compound did not match in retention time to its A1α-assigned feature and was therefore considered to be a false positive although its spectrum matched. The formula for this compound was C12H20O7 and the standard with this formula was triethyl citrate. Given the close spectral match but difference in retention time, the sample likely contained an isomer of triethyl citrate. Triethyl citrate was ranked by ToxPi score as 15th among A1α compounds, but removed from Fig. 3 because it was confirmed to be a false positive. All other compounds with this formula were classified as B2 compounds. The twelve A1α compounds confirmed with standards as true positives can be seen in Table S5 (SI), eight of which were among the top 20 highest ToxPis and can be seen in Fig. 3. The three B1 compounds confirmed with standards were Fipronil Sulfone, Perfluorovaleric Acid (PFPeA), and Perfluorohexanoic Acid (PFHxS). The 15 confirmed compounds have a range of log octanol-water partitioning coefficients (log Kow) from 0.8 (1,2-Benzisothiazolin-3-one) to 4.8 (Perfluoroundecanoic acid). The outer bounds of the range of log Kow values for which this method is suitable cannot be fully assessed due to the small number of confirmed compounds but likely extends beyond this range.
The high percentage of correct structure assignments to formulas as confirmed using standards demonstrates the utility of data source ranking described in McEachran et al., where 88% of a test set of 162 compounds ranked first by data source when using the Dashboard (McEachran et al., 2017b). For the confirmed compounds, 8 of the 15 were perfluoroalkylated substances (PFAS), two were chlorinated phosphate flame retardants, and one was a chlorinated pesticide (atrazine). The types of confirmed compounds are a reflection of the types of available standards in our laboratory and not necessarily representative of the types of compounds actually contained in the samples (see section on Product-Use Categories). The percentage of true positives (94%) relative to false positives (6%) is considered very good for SSA and it increases confidence in the method of prioritization but it must be acknowledged that this success rate may not accurately represent the rate of correct prioritization for the rest of the compound-formula mappings due to the fact that standards were not randomly chosen. The standards used in this study were readily available in one of our laboratories and, thus, had previously been purchased due to their environmental relevance.
Eight of the top 20 ToxPi compounds were confirmed with standards, including the compound with the top ToxPi score, 1,2-Benzisothiazolin-3-one. Over 500 product use entries are listed in the EPA’s CPCat database and the Consumer Product InformationDatabase (Consumer Product Information Database, 2017) lists it in many products that are expected to go directly to waste water after use such as hand soap, dish soap, detergent, etc. It was found in 7 of the 9 drinking water samples and was active in 173 of 565 toxicity assays tested. Although the SciFinder® search found 95 journal articles associating this compound with water, it is not regularly monitored for in drinking water and would not have been discovered without an SSA approach.
3.2. Retention time prediction
As described by McEachran et al. (2017a), the OPERA-RT model has a 95% confidence window of ±4.5 min. Of the 91 A1α compounds, 52 were never observed outside this window giving us greater confidence in the correct identification of these compounds (SI, Table S5). These compounds include all 15 true positives that were confirmed with standards. The predicted retention time for triethyl citrate was within the 95% confidence window of the observed feature that was mislabeled as this compound and, thus, OPERA-RT would not have helped to identify this particular false positive. Only through the use of an analytical standard were we able to observe a difference in retention time large enough to confidently label this peak as a false positive, yet small enough to fall within the predicted retention time window from OPERA-RT. To date, the effectiveness and proper implementation of this retention time tool has not been fully evaluated, however, it provides an added layer of confidence for those compounds that fall within their predicted window.
3.3. Product-use categories
All A1α compounds were assigned to at least 1 of 15 product-use categories, and some to several categories, as they may have different functional uses. Thirteen of fifteen product use categories contained at least one A1α chemical from the samples. Fig. 4 shows the number of A1α compounds in each sample for a given category. A PCA was performed using the sum of the peak areas of the compounds represented in this matrix. The loadings plot from the PCA is given in the SI (Fig. S3). The first principal component explained 39.2% of the variance and the second explained 27.5%. The two well water samples (Chapel Hill and Pittsboro) were positioned very closely on the PCA score plot, indicating these samples are very similar with regards to product-use categories. Tap water from Apex and Cary plotted closely on the PCA as well, which may be because these towns are very close in proximity and share source water. One outlier on the PCA was the Pittsboro tap water. This sample had the most number of features (3341 compared to an average of 1658 per sample), the most number of formulas assigned to features (108 compared to an average of 48 per samples), and ultimately the most number of A1α chemicals (38).
Fig. 4.
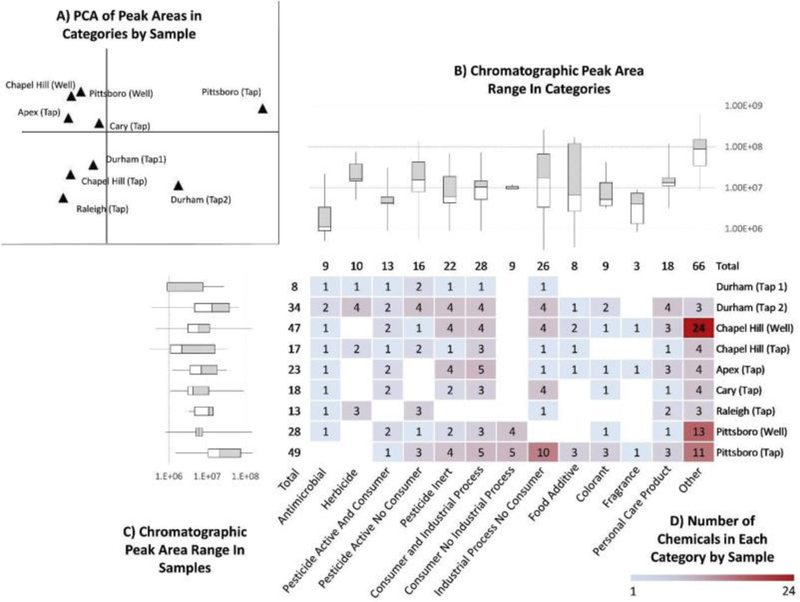
A) First (x-axis) and second (y-axis) principal components in a principal component analysis using summed peak areas for all compounds within a category; B) box and whisker plots representing the range of peak areas for compounds within a category; C) box and whisker plots representing the range of peak areas for compounds within each sample; and D) heat map showing the number of compounds that fall into each category by sample. Blank squares indicate no A1α compound was present for a category in a sample.
Besides the category “other”, the two categories with the most number of A1α chemicals were “Industrial Process No Consumer” followed by “Consumer and Industrial Process”, indicating that drinking water in this area may be impacted by local industries. Other top categories included those containing pesticides (“Pesticide Active and Consumer”, “Pesticide Active No Consumer”, and “Pesticide Inert”). The category “Personal Care Products” was also significant, affecting 8 of the 9 samples.
3.4. Non-targeted analysis (NTA) of unmatched features
An exhaustive NTA is outside the scope of this article, however, some work has been done on identifying features that were not assigned a formula and therefore did not undergo the subsequent steps of our SSA workflow. Emphasis was placed on a mass defect range from −0.2 to 0 as this is indicative of halogenated organic compounds which often contain unique isotope signatures and are often of concern for public health. A focus was also placed on the sample in which the most features was found, the Pittsboro tap water. The most abundant and the fourth most abundant features in the Pittsboro Tap sample that fell into the mass defect range were recognized as being decarboxylated perfluoroalkyl acids. We have previously observed decarboxylation of perfluoroalkyl acids within the ion sourceand fragments would not match to the DSSTox database using this described method. The second largest peak, m/z 564.8848, revealed a high degree of chlorination in its spectra and was found to co-elute with m/z 518.8796, indicating the peak at m/z 564.8848 is a formateadduct. These peaks were found in negative ESI mode, meaning the peak at m/z 518.8796 likely results in the loss of a proton making the neutral mass approximately 519.8869. A chromatogram of these two peaks and the spectrum of the larger peak (m/z 564.8848) is shown in Fig. S4 (SI). Formula generation using MassHunter, which considers relative isotopic abundance and spacing as well as exact mass for the isotope cluster beginning at m/z 518.8796, produced C12H20Cl7O5P with a match score of 99.5 out of a possible 100. No compounds matching this formula were found in public databases such as the Dashboard or PubChem; however, a search using SciFinder® revealed one match for this formula, (2-chloroethyl)-bis[2,2,2-trichloro-1-(1-methylethoxy)ethyl] ester phosphonic acid (CAS 71039–43-5), shown in Fig. 5. This compound is found in a patent and described, along with several other chlorinated phosphonic acids, as plant growth regulators. However, this compound is strikingly similar to other organophosphate compounds, such as TDCPP, also found in this study and commonly used as flame retardants. Further NTA work to identify features which were not assigned a formula will continue using similar approaches as described here.
Fig. 5.
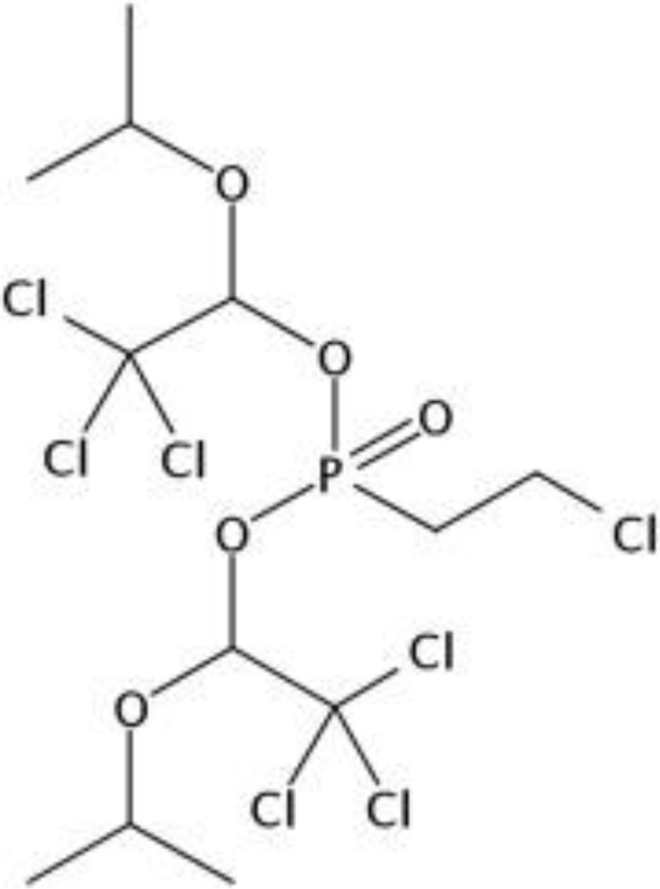
(2-chloroethyl)-bis[2,2,2-trichloro-1-(1-methylethoxy)ethyl] ester phosphonic acid (CAS 71039–43-5), the only discovered structure matching the generated formula of C12H20Cl7O5P for a large unknown peak at m/z 518.8796.
4. Limitations and future directions
The use of an activated charcoal filter to capture contaminants from drinking water likely biased the experimental design towards compounds with sufficiently large Kow values to interact with the filter. It is possible that some compounds which may be of relevance to human health, probably very polar compounds, passed through the filter without capture and, thus, were not retained in the samples. The instrumental analysis could have been expanded in several ways to increase the percent of total features identified. Alternative columns, such as HILIC, can be used to separate compounds that elute in the void volume when using a C8 column. Furthermore, additional ionization sources, such APCI or APPI, could be used to ionize compounds that were not detected under ESI conditions. Future studies should also consider including a gas chromatography (GC) component to explore a larger chemical space. At the time of formula matching, only a limited version of the DSSTox_Database (V2) was available in its de-salted form (de-salted formulas are required to match to mass spectral data). Since then, a much larger, more extensive version of the database has become available in its de-salted form which includes over 720,000 chemicals and can be accessed via the Dashboard’s downloads page (https://comptox.epa.gov/dashboard/downloads). This increase in size would have most likely resulted in a higher percentage of features being assigned formulas. The current method was unable to identify compounds which fragmented in the ionization source, as was observed when the decarboxylated perfluoroalkyl acids were identified. Another limitation to this study, as with most SSA/NTA studies, is the inability to estimate concentration. Future studies should explore ways of estimating instrument responses for compounds without the use of standards. QSPRs appear to be the most viable path to solve this problem. However, a large training set of instrument responses based on chemical standards will be required. Future studies should also focus on better inclusion of tools to mount confidence in level 2 identification on the Schymanski scale, including better implementation of retention time predictors, fragmentation predictors for MS/MS data, etc. In any case, improved access to Open Data sets for integration into our databases will be highly beneficial and the community is encouraged to consider the benefits of such an approach (Schymanski and Williams, 2017).
5. Conclusions
Although there have been abundant research efforts directed at identifying contaminants in drinking water, to the best of our knowledge, this study is the first to use a point-of-use home filter combined with an SSA/NTA approach. Its utility in this pilot scale application is illustrated in our identification of several compounds that would not otherwise be monitored in drinking water. The need for a more comprehensive SSA/NTA approach is highlighted by the large number of features present in the samples, and the limited number of which that were confirmed or tentatively identified.
We have demonstrated that ranking by data source correctly prioritized (Group A1α or B1) 15 out of 16 compounds for which standards were available on hand. Furthermore, ToxPi ranking allowed focus to be placed on compounds of most relevance to human health. Standards are still required for level I identification according to the Schymanski confidence levels (Schymanski et al., 2014a); however, confirmation of all prioritized candidate compounds is impractical therefore researchers should focus on tools that add confidence to level 2 identifications, such as retention time predictors and in silico fragmentors. The retention time prediction model used in this study (OPERA-RT) was unable to identify the one false positive found and thus further development is necessary for larger scale implementation of retention time prediction.
The number of chemicals in the A1α group is very small compared to the number of features extracted, or total chemicals, in the samples. The vast majority of these features are quite small and, thus, may represent chemicals at trace levels. That being said, trace levels of compounds may be of importance to human health. While there was a great degree of variability in the number of features, formulas, and Group A1α compounds in the samples, every sample exhibited some degree of contamination. Given the wide range of retention times and masses observed in this study, as well as the sheer number of features observed, our results indicate that activated carbon point-of-use water filtration systems likely remove compounds spanning a wide range of physicochemical properties.
Supplementary Material
Acknowledgments
The authors thank Kristen Isaacs for providing code to parse formulas and information from the ACToR database; Katherine Phillips for providing product use categories and coding guidance; John Wambaugh for providing ExpoCast data; and Chris Grulke for guidance on the DSSTox database structure.
Footnotes
The authors declare no competing financial interest.
References
- Anumol T, Clarke BO, Merel S, Snyder SA, 2015. Point-of-use devices for attenuation of trace organic compounds in water. J. AWWA 107, 9. [Google Scholar]
- Bader T, Schulz W, Lucke T, 2016. Application of Non-target Analysis with LCHRMS for the Monitoring of Raw and Potable Water: Strategy and Results, Assessing Transformation Products of Chemicals by Non-target and Suspect Screening _ Strategies and Workflows Volume 2. American Chemical Society, pp. 49e70. [Google Scholar]
- Consumer Product Information Database, 2017. CPID.
- Dionisio KL, Frame AM, Goldsmith M-R, Wambaugh JF, Liddell A, Cathey T, Smith D, Vail J, Ernstoff AS, Fantke P, Jolliet O, Judson RS, 2015. Exploring consumer exposure pathways and patterns of use for chemicals in the environment. Toxicol. Rep 2, 228e237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeghy PP, Lorber M, 2011. An assessment of the exposure of Americans to perfluorooctan sulfonate: a comparison of estimated intake with values inferred from NHANES data. J. Expo. Sci. Environ. Epidemiol 21, 150e168. [DOI] [PubMed] [Google Scholar]
- Kind T, Fiehn O, 2006. Metabolomic database annotations via query of elemental compositions: mass accuracy is insufficient even at less than 1 ppm. BMC Bioinforma 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little JL, Williams AJ, Pshenichnov A, Tkachenko V, 2012. Identification of “known unknowns” utilizing accurate mass data and chemspider. J. Am. Soc. Mass Spectrom 23, 179e185. [DOI] [PubMed] [Google Scholar]
- Lorber M, Egeghy PP, 2011. Simple intake and pharmacokinetic modeling to characterize exposure of Americans to perfluoroctanoic acid, PFOA. Environ. Sci. Technol 45, 8006e8014. [DOI] [PubMed] [Google Scholar]
- Mansouri K, Grulke CM, Richard AM, Judson RS, Williams AJ, 2016. An automated curation procedure for addressing chemical errors and inconsistencies in public datasets used in QSAR modelling. SAR QSAR Environ. Res 27, 911e937. [DOI] [PubMed] [Google Scholar]
- McEachran AD, Mansouri K, Newton S, Beverly B, Sobus JR, Williams AJ, 2017a. Evaluating Three Gradient HPLC Retention Time Prediction Models: 1) logP, 2) ACD/ChromGenius, and 3) a Quantitative Structure Retention Relationship Model (under review) [Google Scholar]
- McEachran AD, Sobus JR, Williams AJ, 2017b. Identifying known unknowns using the US EPA’s CompTox Chemistry Dashboard. Anal. Bioanal. Chem 409, 1729e1735. [DOI] [PubMed] [Google Scholar]
- Müller A, Schulz W, Ruck WKL, Weber WH, 2011. A new approach to data evaluation in the non-target screening of organic trace substances in water analysis. Chemosphere 85, 1211e1219. [DOI] [PubMed] [Google Scholar]
- Prüss-Üstün A, Bos R, Gore F, Bartram J, 2008. Safer Water, Better Health: Costs, Benefits and Sustainability of Interventions to Protect and Promote Health World Health Organization. [Google Scholar]
- Rager JE, Strynar MJ, Liang S, McMahen RL, Richard AM, Grulke CM, Wambaugh JF, Isaacs KK, Judson R, Williams AJ, Sobus JR, 2016. Linking high resolution mass spectrometry data with exposure and toxicity forecasts to advance high-throughput environmental monitoring. Environ. Int 88, 269e280. [DOI] [PubMed] [Google Scholar]
- Rappaport SM, Smith MT, 2010. Environment and disease risks. Science 330, 460e461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff M, Mueller MS, Loos M, Singer HP, 2015. Quantitative target and systematic non-target analysis of polar organic micro-pollutants along the river Rhine using high-resolution mass-spectrometry e identification of unknown sources and compounds. Water Res 87, 145e154. [DOI] [PubMed] [Google Scholar]
- Schymanski EL, Jeon J, Gulde R, Fenner K, Ruff M, Singer HP, Hollender J, 2014a. Identifying small molecules via high resolution mass spectrometry: communicating confidence. Environ. Sci. Technol 48, 2097e2098. [DOI] [PubMed] [Google Scholar]
- Schymanski EL, Singer HP, Longr_ee P, Loos M, Ruff M, Stravs MA, Ripoll_es Vidal C, Hollender J, 2014b. Strategies to characterize polar organic contamination in wastewater: exploring the capability of high resolution mass spectrometry. Environ. Sci. Technol 48, 1811e1818. [DOI] [PubMed] [Google Scholar]
- Schymanski EL, Singer HP, Slobodnik J, Ipolyi IM, Oswald P, Krauss M, Schulze T, Haglund P, Letzel T, Grosse S, 2015. Non-target screening with high-resolution mass spectrometry: critical review using a collaborative trial on water analysis. Anal. Bioanal. Chem 407, 6237e6255. [DOI] [PubMed] [Google Scholar]
- Schymanski EL, Williams AJ, 2017. Open Science for Identifying “Known Unknown”, Chemicals ACS Publications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SciFinder, 2017. Chemical Abstract Services Columbus, OH. [Google Scholar]
- Sobus JR, Wambaugh JF, Isaacs KK, Williams AJ, McEachran AD, Richard AM, Grulke CM, Ulrich EM, Rager JE, Strynar MJ, Newton SR, 2017. Integrating tools for non-targeted analysis research and chemical safety evaluations at the US EPA. J. Expo. Sci. Environ. Epidemiol (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strynar M, Dagnino S, McMahen R, Liang S, Lindstrom A, Andersen E, McMillan L, Thurman M, Ferrer I, Ball C, 2015. Identification of novel perfluoroalkyl ether carboxylic acids (PFECAs) and sulfonic acids (PFESAs) in natural waters using accurate mass time-of-flight mass spectrometry (TOFMS). Environ. Sci. Technol 49, 11622e11630. [DOI] [PubMed] [Google Scholar]
- US EPA, 2015. Toxicity ForeCaster (ToxCast™) Data. US EPA https://www.epa.gov/chemical-research/toxicity-forecaster-toxcasttm-data.
- US EPA, 2016. Safe Drinking Water Act (SDWA) [Google Scholar]
- Villanueva CM, Kogevinas M, Cordier S, Templeton MR, Vermeulen R, Nuckols JR, Nieuwenhuijsen MJ, Levallois P, 2014. Assessing exposure and health consequences of chemicals in drinking water: current state of knowledge and research needs. Environ. Health Perspect (Online) 122, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wambaugh JF, Setzer RW, Reif DM, Gangwal S, Mitchell-Blackwood J, Arnot JA, Joliet O, Frame A, Rabinowitz J, Knudsen TB, 2013. Highthroughput models for exposure-based chemical prioritization in the ExpoCast project. Environ. Sci. Technol 47, 8479e8488. [DOI] [PubMed] [Google Scholar]
- WHO, 2013. Chemical Safety of Drinking-water World Health Organization. [Google Scholar]
- Wild CP, 2005. Complementing the genome with an “exposome”: the outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol. Biomarkers Prev 14, 1847e1850. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.