

Abstract
A national-scale survey of 247 contaminants of emerging concern (CECs), including organic and inorganic chemical compounds, and microbial contaminants, was conducted in source and treated drinking water samples from 25 treatment plants across the United States. Multiple methods were used to determine these CECs, including six analytical methods to measure 174 pharmaceuticals, personal care products, and pesticides. A three-component quality assurance/quality control (QA/QC) program was designed for the subset of 174 CECs which allowed us to assess and compare performances of the methods used. The three components included: 1) a common field QA/QC protocol and sample design, 2) individual investigator-developed method-specific QA/QC protocols, and 3) a suite of 46 method comparison analytes that were determined in two or more analytical methods. Overall method performance for the 174 organic chemical CECs was assessed by comparing spiked recoveries in reagent, source, and treated water over a two-year period. In addition to the 247 CECs reported in the larger drinking water study, another 48 pharmaceutical compounds measured did not consistently meet predetermined quality standards. Methodologies that did not seem suitable for these analytes are overviewed. The need to exclude analytes based on method performance demonstrates the importance of additional QA/QC protocols.
Keywords: pharmaceuticals, contaminants of emerging concern, drinking water, source water, mass spectrometry
Graphical Abstract
1. Introduction
The frequent detection of pharmaceuticals, personal care products, and other contaminants of emerging concern (CECs) in the environment has become a global problem due to their potential to cause undesirable ecological and human health effects such as endocrine disruption, behavioral alteration, and antibiotic resistance1–5. As interest in these contaminants develops and increases, the need for reliable, sensitive analytical methods also increases. The number of available multi-analyte analytical methods has increased rapidly over the past decade, employing a variety of sample preparation and analysis technologies6–13. Because of the chemical diversity of CECs, no single methodology is capable of being a standard for analysis at this time.
High performance liquid chromatography (HPLC)6–12 and gas chromatography (GC)13 have been used as the primary separation techniques for complex mixtures of CECs, and mass spectrometry is most frequently employed as the detection system6–13. Since many of these CECs are found in the environment in sub-µg/L concentrations, preconcentration prior to analysis has been necessary, most often by solid phase extraction (SPE) for water sample analysis6–9, 13. Direct analysis methods have been recently developed that rely on the specificity and sensitivity of the mass spectrometer system to detect and quantify contaminants at ambient environmental concentrations10–12. Simultaneously analyzing multiple classes of CECs is challenging due to their wide range of chemical and physical properties14, and compromises are often made that could affect quantification, such as reduced recoveries or decreased sensitivity levels. Only a few interlaboratory or intermethod comparison studies of environmental analysis have been performed15–25. Several have reported a range of results for some analytes15, 23, 24, and it has been recommended that increasing the quality assurance measures surrounding sample analysis could reduce variations in reported results15, 26–29. More modeling and risk assessment methods are also relying on published concentrations of contaminants to further our understanding of their environmental behavior30–33. Therefore, providing quality assurance/quality control (QA/QC) context to published data with nonstandard, investigator-specific reporting protocols is increasingly important.
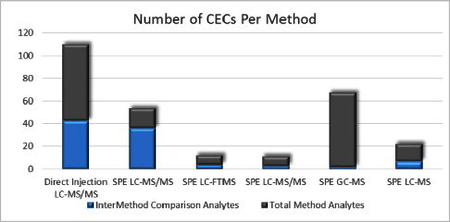
This paper is one of a series that describe the presence, persistence, and concentrations of 247 chemical and microbial CECs in a recent national-scale survey of source and treated drinking water from 25 drinking water treatment plants across the U.S.34A total of 16 methods were required to analyze the full set of 247 CECs, with six of the chemical methods containing 174 pharmaceuticals, personal care products, and pesticides. 46 of the 174 pharmaceuticals and personal care products were analyzed in more than one of these six chemical methods. To address the necessity of using multiple methods to determine such a broad array of contaminants, a QA/QC program was designed that assesses relative performance of the methods used, with the fate of these compounds within drinking water treatment being evaluated in detail elsewhere35, 36. The QA/QC program had three components: a common field sampling protocol and sample design for all chemical analysis methods; a method-specific QA/QC developed by individual investigators to assess within-method performance; and analysis of the suite of 46 organic compounds, referred to as intermethod comparator analytes (ICAs), by two or more analytical methods. This paper provides in-depth evaluation of six individual method performances for the 174 pharmaceuticals and personal care products over a two-year period, then includes an assessment of relative performance of the ICAs present in multiple methods, using the common set of environmental and QC samples. Environmental data for another 48 compounds were not reported as they did not consistently meet predetermined quality standards. Methodologies that did not seem suitable for these analytes are overviewed. Overall, this study demonstrates the necessity of implementing QA/QC programs to accurately compare performance characteristics and integrate results among multiple independent methods used for national-scale, extended-duration contaminant monitoring programs.
2. Methods
2.1. Sample Collection.
Water samples were collected by the staff at each of the 25 drinking water treatment plants (DWTPs) between September 2010 and January 2012 using pre-cleaned sample containers containing the appropriate dechlorination agent and preservatives, as listed in Table 1. Paired source and treated water samples were collected at each DWTP while accounting for residence time of treatment. Each DWTP also was provided with an unopened 4-L bottle of pesticide grade blank water sourced from a single lot to be used as a field blank for each chemical method. A single field blank sample was collected at each DWTP, with the exception of Method 3, which included a separate field blank for source and treated water. After collection, samples were placed in coolers, packed in ice, and shipped via overnight delivery to Denver, CO (all indicated USGS methods) or Cincinnati, OH (all indicated USEPA methods) for each respective analysis34.
Table 1.
A summary of the analytical methods applied to measure chemical contaminants. ACN = acetonitrile, MeOH = methanol
| Method number |
Method name (reference) |
Analytes | Sampling container and dechlorination agent |
Sample holding time |
Sample extraction (Solvents/reagents) [LFB/LFM concentration] |
Sample analysis | Calibration type |
|---|---|---|---|---|---|---|---|
| 1 | USGS Direct Injection (Furlong et al., 2014) |
110 pharmaceuticals and personal care products |
40 mL VOA vial w/ 100 mg ascorbic acid |
96 h | Direct injection of a 100 μL sample aliquot [200 ng/L] |
ESI triple quadrupole LC-MS/MS with MRM |
Isotope dilution with 20 labeled standards |
| 2 | USEPA Pharmaceutical (Batt et al., 2008) |
54 human use prescription pharmaceuticals |
500 mL amber glass w/ 50 mg ascorbic acid |
72 h | SPE with mixed mode OASIS MCX (ACN, MeOH, formic acid, and ammonium hydroxide) [500 ng/L] |
ESI triple quadrupole UPLC-MS/MS with MRM |
Isotope dilution with 32 labeled standards |
| 3 | USEPA Hormone (SI Document 1) |
12 steroids, hormones, bisphenol A, atrazine, triclosan |
600 mL silanized clear glass w/ 6 mg copper sulfate and 7 mg ascorbic acid |
72 h | SPE with C18 Disk (acetone, MeOH, dansyl chloride, hexane, hydroxylamine) [1.7 ng/L] |
ESI LC-FTMS with accurate mass |
Isotope dilution with 12 labeled standards |
| 4 | USGS Antidepressant (Schultz and Furlong, 2008) |
11 antidepressant pharmaceuticals and degradates |
1-L white polyethylene with 100 mg ascorbic acid |
96 h | SPE with OASIS HLB (MeOH, ammonium acetate, acetic acid) [500 ng/L] |
ESI hybrid triple quadrupole/ion trap LC-MS/MS with MRM |
Isotope dilution with 1 isotopically labeled standard |
| 5 | USGS Wastewater (Steven et al., 2002) |
63 wastewater contaminants |
1-L amber glass with 100 mg ascorbic acid |
96 h | SPE with Polystyrene- divinylbenzene (dichloromethane and diethyl ether) [2 μg/L] |
GC–MS with capillary chromatography and electron impact ionization |
Internal calibration with 6 internal standards and 4 surrogate compounds |
| 6 | USGS Pharmaceutical (Cahill et al., 2004) |
14 pharmaceuticals |
1-L amber glass with 100 mg ascorbic acid |
96 h | SPE with OASIS HLB (methanol, trifluoroacetic acid) [250 ng/L] |
ESI single quadrupole LC-MS with SRM |
Internal calibration with 1 internal standard and 1 surrogate compound |
2.2. Sample Analysis.
The targeted analytes were determined by six analytical methods (Table 1). Water samples were collected unfiltered in the field using the indicated sampling containers and were either analyzed (Method 1) or extracted (Methods 2–6) within the indicated holding times. Briefly, for Method 137, each sample was filtered in the laboratory and fortified with a mixture of method-specific, isotopically-labeled internal standards, after which a 100-µL aliquot was analyzed by direct injection. For Methods 28, 3 (full method described in Supporting Information Document 1), 438, 513 and 639, each indicated water sample was filtered in the laboratory, fortified with a method-specific suite of surrogate compounds or procedural internal standard compounds to monitor extraction recovery, and extracted by processing the fortified sample through the indicated SPE cartridge or disk. The cartridge was then eluted with a method-specific sequence of solvents, the extract was concentrated, and any specified aliquot of a method-specific internal standard mixture was added prior to analysis. Samples were analyzed by high-performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS), with multiple reaction monitoring (MRM) of at least two unique ion transitions (Methods 1,2, and 4): liquid chromatography-Fourier transform mass spectrometry (LC-FTMS; Method 3), full-scan capillary gas chromatography-mass spectrometry (GC-MS; Method 5), or LC-MS with selected-ion monitoring of at least two unique fragment ions (Method 6).
2.3. Quality Control Design.
Both source and treated drinking water were sampled in triplicate. The first sample was analyzed as the primary sample, and reported environmental concentrations for each of the analytes were determined using only the results of the primary sample. The second sample was a duplicate collected to monitor intramethod reproducibility. The third sample was utilized as a matrix spike sample (Laboratory Fortified Matrix, LFM) to assess recovery in ambient water samples and monitor potential matrix enhancement or suppression. This QC design produced two LFM samples (source and treated, water) for every sampling location throughout the study. Recoveries were corrected for any ambient compound contributions. For each method and at each laboratory, an aliquot of reagent blank water was also spiked (Laboratory Fortified Blank, LFB) and analyzed, with each sample set to monitor matrix-free, set-specific, day-to-day method performance. Due to the range of detection limits and expected ambient concentrations for the list of analytes, LFM and LFB samples were not spiked at the same concentration for all methods. The QC samples were generally spiked in mid-calibration range for each method. The QC standards set for this study required matrix and laboratory spike recoveries to fall within 50 to 150%. Laboratory (Laboratory Reagent Blanks, LRB) and field blanks were analyzed with each sample batch to monitor possible contamination from laboratory and field sample collection processes, respectively. For any chemical to be reported as detected in a source or treated water sample, concentrations had to be greater than three times the concentration of the associated field or laboratory blank; environmental results failing this criterion were censored to non-detection.
To facilitate intermethod comparisons, laboratories used a common methodology to calculate a detection limit with the Lowest Concentration Minimum Reporting Level (LCMRL). The LCMRL is defined as the lowest spiking concentration such that the probability of spike recovery in the 50% −150% range is at least 99%40,41, and is determined by analyzing sets of spiked replicates at different concentrations in reagent water with a set of reagent blanks. An online calculator provided regression-based modeling analysis (available at http://water.epa.gov/scitech/drinkingwater/labcert/analyticalmethods_ogwdw.cfm). The appropriate number of spiking levels, spacing between spiking levels, and number of replicates at each spiking level may vary between method and all must be determined as part of the LCMRL process. Iteratively reweighted least squares regression, with weights derived using Tukey’s biweight formula, was used to robustly model the relationship between signal and spike concentration, as well as the relationship between signal variance and spike concentration. These functional relationships were then used to estimate probability of obtaining signal values corresponding to the targeted recovery percentage range at different, potential spike-in concentrations40,41. For the 10 ICAs in which an LCMRL could not be determined, a method detection limit (MDL) was used. The MDL is defined as the minimum concentration of an analyte that can be measured with a 99% confidence that the concentration is greater than zero43. MDLs were calculated by multiplying standard deviation for measured concentrations of the seven replicate samples, at a single spiking level close to the expected MDL, by the Student’s t value for six degrees of freedom.
3. Results and Discussion
For the six methods used to acquire organic CEC concentrations, method performance was assessed by intramethod and intermethod performance. Intramethod performance was evaluated separately for each method and consisted of multiple components. Bias was assessed from analyte recoveries in LFB and LFM samples. Precision at ambient environmental concentrations was evaluated from relative percentage differences, and calculated from concentrations in primary and replicate samples. Long-term precision was determined by calculating relative standard deviations of recoveries from LFB and LFM samples. Contamination in the laboratory or field was assessed from LRB and field blank samples, respectively. Summary recovery statistics of the intramethod performance for LFB samples for the six methods used in this study appear in Tables S1–1 to S1–6.
Overall, intramethod performance can be evaluated from Figure 1, in which distributions of median recoveries for all analytes within a method from LFB samples are compared as boxplots. The number of LFBs used to determine analyte-specific medians varied within each method, and are listed in Tables S1–1—S1–6. Note that the boxplots in Figure 1 contain median recoveries for all compounds determined by each method. Fewer analytes were ultimately used for purpose of interpretation in papers by Glassmeyer et al.34, Furlong et al.35, and Wilson et al.36 There were differences in matrix-free performance between individual methods as seen in Figure 1. Method 1, which included the highest number of analytes and was a direct analysis of the sample, displayed the least bias and narrowest range of median recoveries. Method 2, like Method 1, focused on human-health pharmaceuticals and had a large number of structurally diverse compounds; it also incorporates an SPE isolation step, which may be the source of the more variable recoveries observed. Although Method 3 also uses SPE, it employs isotope dilution, an accurate mass analysis for identification and quantitation, and focuses on a much smaller range of compounds, which may account for the relatively narrow distribution of recoveries observed. Method 4, which included the fewest analytes, was also the most variable, and median recoveries were lower overall when compared to the published method performance38. Methods 5 and 6 both use SPE with chromatography (capillary GC for Method 5 and HPLC for Method 6), followed by a single-stage of low-resolution MS. Both methods had similar distributions of matrix-free median recoveries.
Fig. 1.
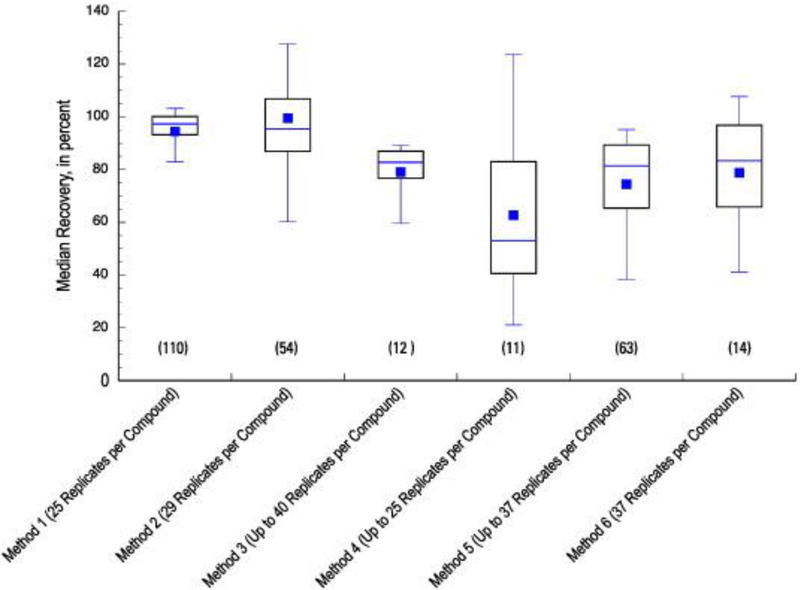
Distribution of the median recoveries of all analytes in each of six methods, determined in laboratory fortified blank samples (LFB). The number of LFBs varied between methods and is indicated as number of replicates analyzed for each. The number of analytes in each method is indicated in parentheses beneath each boxplot. The boxplots display the mean (center dot), 50th percentile (center bar), 25th and 75th percentile (bottom and top of box, respectively), and the 10th and 90th percentile (bottom and top whisker, respectively). Specific method details can be found in Table 1.
The distribution of median recoveries for all methods in up to 25 source water spiked samples (source LFMs) is shown in Figure 2. Source LFMs indicate that similar distributions of median recoveries were observed between the matrix-free LFB samples and source LFMs for Methods 1, 2, and 3. Distributions of median recoveries for Methods 4 and 6 were lower in the source LFM samples compared to LFB recoveries, while the distribution of median recoveries in source LFMs was higher than in LFB samples for Method 5 (the only GC/MS method of six methods employed). The distribution of median recoveries in treated water (Figure 3) was more comparable to the pattern observed in source water than to matrix-free laboratory reagent water for Methods 1, 2, and 3. Methods 4 and 6 displayed distinctly lower recoveries in the two matrix sample types than in reagent water, while Method 5 recoveries were higher in the two matrix types. Method 4 recoveries, low in both matrices, were more consistent in treated water than in source water, as indicated by the narrower boxplot spread in Figure 3 compared to Figure 2. Collectively, results in Figures 1–3 suggest relatively stable performances of Methods 1–3 in matrix or matrix-free samples. Performances of Methods 4 and 6 were lower in matrix samples than matrix-free samples, which suggests either overall matrix interference in the extraction steps used for these two methods or matrix suppression during analysis. In contrast, Method 5 showed overall higher distribution of median recoveries in matrix samples compared to matrix-free LFB samples. This difference may reflect improved recoveries of Method 5 analytes in the presence of sample matrix. Method 5 uses GC/MS and the sample matrix may reduce constituent loss during the extract concentration and solvent exchange steps necessary for sample analysis, or the matrix present in sample extracts may provide a protective effect in the GC injector which reduces analyte loss and results in a matrix enhancement effect.
Fig. 2.
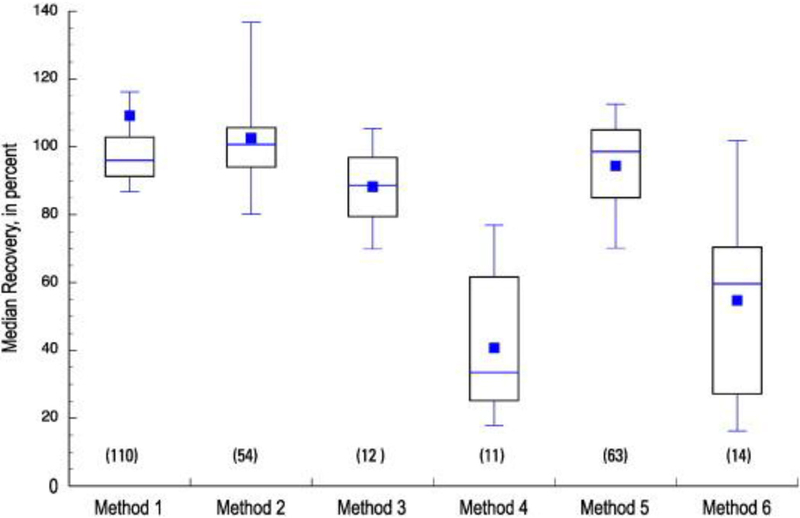
Distribution of median recoveries of all analytes in each of six methods, determined in 25 source water Laboratory Fortified Matrix samples (LFM). The number of analytes per method is indicated in parentheses, and specific method details can be found in Table 1.
Fig. 3.
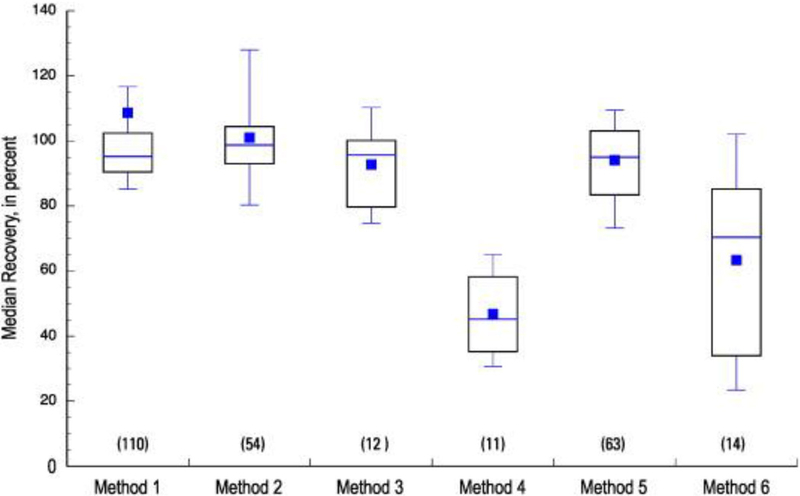
Distribution of median recoveries of all analytes in each of six methods, determined in 25 treated water Laboratory Fortified Matrix samples (LFM). The number of analytes per method is indicated in parentheses, and specific method details can be found in Table 1.
Precision at ambient environmental concentrations was assessed using the replicate source and treated water sample pairs. Duplicates were collected for each environmental sample, with only the primary volume designated to reported environmental values. Precision is expressed as relative percent difference (RPD), calculated as the absolute difference in concentration between two replicate samples, divided by the mean of the two concentrations and expressed as a percentage. All detections quantified in both primary and replicate source and treated water samples are summarized in Table S2 for all methods. Because many detections were at or near the reporting limit, a detection might occur in the primary sample and not the replicate in many cases, and vice-versa. This is reflected in the numbers of quantified detections reported for each method in the primary and replicate samples in Table S2, versus the actual pairs of replicates for which RPDs could be calculated.
Overall, the median RPD observed for all methods in source and treated waters ranged from 5.6 to 15.8%. Typically, the median RPD was lower in treated water samples than source water samples, with the exception of Method 5. Note, however, that the number of pairs was typically lower in treated waters than in source waters, with Method 5 being the exception. Method 5 was used to determine bromoform, a commonly formed disinfection byproduct, which may explain the higher number of replicate pairs and higher median RPD in treated water for this method. Otherwise, there did not appear to be method-specific differences in median RPDs observed among the remaining methods. The 25th and 75th percentiles of RPD provide a measure of the distribution of most RPDs for each method. The largest number of 75th percentile of RPDs occurred for Method 1, at 38.7%. The observed 75th percentile of RPDs for all methods suggests that, in most cases, all methods produced acceptably precise (<30% RPD) results, particularly since calculated RPDs are based on replicate measurement at ambient environmental concentrations, which were in the low ng/L range for most results.
Table S3 shows which compounds occurred in three or more laboratory or field blank samples over the course of our study. The number of field blank samples was constant at 25 for all methods except Method 3, where two field blank samples per site were reported, one for source water and one for treated water. For most methods, overall LRB contamination was low and median LRB detections were typically less than 5 ng/L. The exception to this observation was Method 5 in which median blank detections ranged between 1.25 and 300 ng/L. Method 5 analyzed for a range of anthropogenic waste indicators, many of which are in personal-care products, detergent cleaners, or other products commonly present in laboratory settings which could explain the higher median concentrations detected by this method.
Intermethod performance was first assessed by comparing accuracy and precision of spiked recoveries from the LFBs and source and treated LFMs for the 46 ICAs (Table 2) over the two-year study period. The number of spike samples that did not meet predetermined quality criteria is shown by the number of samples with low (less than 50%) and high (greater than 150%) recoveries. The median recovery in all matrices for the vast majority of the ICAs fell between 70 and 130%, although most compounds had at least one low or high percent recovery. Four compounds (glyburide, prednisone, progesterone, and propoxyphene) typically displayed spiked recoveries greater than 150% for Method 2, as compared to more accurate recoveries with Method 1 (glyburide, prednisone, propoxyphene) or Method 3 for progesterone. These four analytes were not detected with Method 2, however, in any source or treated drinking water samples (Figure 4, Table S4), so the high bias did not affect environmental results. Percent recoveries were not markedly different between the distilled water and drinking water spiked samples in Method 2, indicating the measurements were not heavily influenced by the matrix. Bupropion (Method 4); cotinine, dehydronifedipine, trimethoprim (Method 6); norfluoxetine and desmethylsertraline (Method 2); and promethazine (Method 1) displayed median recoveries below 70%, compared to more accurate recoveries with Methods 2, 4, and 6, as shown in Table 2. Median recoveries for bupropion (Method 4), norfluoxetine and desmethylsertraline (Method 2) were similar in reagent water and matrix water samples, however, median recoveries for cotinine and dehydronifedipine were considerably lower in source and treated water samples than in reagent water, suggesting these two compounds in particular were more susceptible to matrix effects in Method 6 than in Method 1.
Table 2.
A comparative summary of detection levels and intermethod performance, as determined by the percent relative standard deviation (%RSD) in spiked samples for distilled, source, and treated water for each intermethod comparator analyte (ICA) over the two-year study period. LCMRL = Lowest Concentration Minimum Reporting Level, MDL = Method Detection Limit, n = number of samples , n.a. = not available. All concentrations are in ng/L.
| Analyte | Method | Target detection limit (ng/L) |
LCMRL (ng/L) |
MDL (ng/L) |
Distilled water spike | Source water spike | Treated water spike | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Median | n > 0 | n < 50% | n > 150% | RSD | Median | n > 0 | n < 50% | n > 150% |
RSD | Median | n > 0 | n < 50% | n > 150% | RSD | |||||
| Acetaminophen | 1 | 600,000 | 40 | 3.6 | 97 | 24 | 0 | 0 | 14 | 91 | 25 | 2 | 0 | 24 | 89 | 25 | 2 | 0 | 25 |
| Acetaminophen | 2 | 600,000 | 11 | 1.5 | 103 | 28 | 0 | 0 | 9 | 102 | 24 | 0 | 1 | 14 | 100 | 24 | 0 | 0 | 11 |
| Albuterol | 1 | 460 | 5.3 | 1.2 | 98 | 24 | 0 | 0 | 13 | 91 | 24 | 3 | 1 | 33 | 91 | 25 | 1 | 2 | 32 |
| Albuterol | 2 | 460 | n.a. | 6.2 | 142 | 23 | 0 | 6 | 43 | 140 | 22 | 0 | 9 | 22 | 132 | 23 | 0 | 8 | 20 |
| Alprazolam | 1 | 230 | 27 | 4.3 | 100 | 24 | 0 | 0 | 11 | 93 | 25 | 1 | 0 | 23 | 93 | 25 | 2 | 0 | 25 |
| Alprazolam | 2 | 230 | 7.6 | 2.9 | 92 | 27 | 0 | 0 | 19 | 99 | 24 | 0 | 1 | 23 | 101 | 23 | 0 | 0 | 17 |
| Amitriptyline | 1 | 263 | 69 | 18 | 84 | 24 | 1 | 0 | 22 | 104 | 24 | 2 | 2 | 38 | 108 | 25 | 1 | 3 | 31 |
| Amitriptyline | 2 | 263 | 4.0 | 0.2 | 102 | 28 | 0 | 0 | 14 | 102 | 24 | 0 | 1 | 19 | 102 | 23 | 0 | 0 | 11 |
|
10-hydroxy- amitriptyline |
1 | 797 | 12 | 1.7 | 99 | 24 | 0 | 0 | 14 | 102 | 25 | 2 | 0 | 27 | 99 | 25 | 1 | 2 | 28 |
|
10-hydroxy- amitriptyline |
2 | 797 | 5.0 | 0.2 | 85 | 28 | 2 | 0 | 29 | 94 | 24 | 0 | 1 | 22 | 92 | 24 | 0 | 0 | 16 |
| Amphetamine | 1 | 1428 | 28 | 4.1 | 99 | 24 | 0 | 0 | 12 | 94 | 25 | 2 | 0 | 24 | 91 | 25 | 3 | 1 | 27 |
| Amphetamine | 2 | 1428 | 4.0 | 0.5 | 92 | 28 | 2 | 0 | 26 | 97 | 24 | 0 | 1 | 16 | 96 | 24 | 0 | 1 | 21 |
| Atenolol | 1 | 25,000 | 58 | 2.7 | 95 | 24 | 0 | 0 | 14 | 106 | 25 | 1 | 1 | 24 | 106 | 25 | 2 | 1 | 26 |
| Atenolol | 2 | 25,000 | 23 | 1.9 | 127 | 28 | 0 | 9 | 45 | 112 | 24 | 0 | 2 | 47 | 113 | 24 | 0 | 3 | 23 |
| Atrazine | 1 | n.a. | 22 | 9.7 | 97 | 24 | 0 | 0 | 13 | 94 | 24 | 3 | 0 | 37 | 92 | 25 | 3 | 1 | 29 |
| Atrazine | 3 | n.a. | 0.25 | n.a. | 85 | 40 | 0 | 0 | 9 | 100 | 17 | 6 | 6 | 432 | 99 | 16 | 2 | 6 | 275 |
| Benztropine | 1 | 15 | 68 | 7.9 | 89 | 24 | 2 | 0 | 24 | 73 | 23 | 6 | 2 | 64 | 98 | 25 | 1 | 1 | 28 |
| Benztropine | 2 | 15 | 9.0 | 0.5 | 73 | 25 | 5 | 0 | 48 | 88 | 23 | 4 | 0 | 48 | 87 | 23 | 4 | 1 | 48 |
| Bupropion | 1 | n.a. | 29 | 3.6 | 98 | 24 | 0 | 0 | 10 | 94 | 25 | 2 | 0 | 23 | 96 | 25 | 2 | 1 | 26 |
| Bupropion | 4 | n.a. | n.a. | 0.66 | 71 | 22 | 3 | 0 | 37 | 70 | 23 | 10 | 0 | 60 | 61 | 25 | 11 | 1 | 63 |
| Caffeine | 1 | n.a. | 42 | 18 | 97 | 24 | 0 | 0 | 21 | 97 | 25 | 1 | 0 | 26 | 97 | 24 | 1 | 1 | 28 |
| Caffeine | 5 | n.a. | 100 | n.a. | 93 | 37 | 0 | 0 | 12 | 86 | 25 | 0 | 0 | 21 | 94 | 24 | 0 | 0 | 15 |
| Caffeine | 6 | n.a. | n.a. | 60 | 114 | 37 | 0 | 1 | 17 | 102 | 24 | 2 | 0 | 43 | 101 | 25 | 0 | 0 | 25 |
| Carbamazepine | 1 | 30,000 | 6.1 | 0.84 | 104 | 24 | 0 | 0 | 12 | 99 | 23 | 2 | 0 | 36 | 95 | 23 | 0 | 1 | 22 |
| Carbamazepine | 2 | 30,000 | 7.1 | 1.4 | 93 | 28 | 0 | 0 | 17 | 100 | 24 | 0 | 0 | 17 | 96 | 24 | 0 | 0 | 16 |
| Carbamazepine | 4 | 30,000 | n.a. | 40 | 114 | 23 | 0 | 6 | 49 | 72 | 24 | 6 | 4 | 95 | 72 | 23 | 6 | 5 | 77 |
| Carbamazepine | 6 | 30,000 | 190 | n.a. | 99 | 37 | 0 | 0 | 13 | 55 | 25 | 9 | 0 | 43 | 72 | 25 | 2 | 0 | 32 |
| Clonidine | 1 | 22 | 89 | 30 | 99 | 24 | 0 | 0 | 12 | 102 | 25 | 1 | 0 | 24 | 105 | 25 | 1 | 2 | 27 |
| Clonidine | 2 | 22 | 27 | 11 | 90 | 27 | 0 | 0 | 14 | 98 | 24 | 0 | 0 | 15 | 95 | 24 | 0 | 0 | 15 |
| Codeine | 1 | n.a. | 87 | 44 | 93 | 24 | 0 | 0 | 17 | 93 | 24 | 1 | 1 | 32 | 95 | 25 | 1 | 0 | 23 |
| Codeine | 6 | n.a. | n.a. | 46 | 85 | 37 | 0 | 0 | 11 | 84 | 25 | 0 | 0 | 15 | 85 | 25 | 0 | 0 | 12 |
| Cotinine | 1 | n.a. | 4.4 | 1.3 | 98 | 24 | 0 | 0 | 10 | 97 | 25 | 1 | 0 | 22 | 96 | 25 | 2 | 0 | 24 |
| Cotinine | 6 | n.a. | 180 | n.a. | 89 | 37 | 0 | 0 | 11 | 64 | 25 | 4 | 0 | 29 | 69 | 25 | 6 | 0 | 32 |
| Dehydronifedipine | 1 | n.a. | 12 | 4.9 | 104 | 24 | 1 | 0 | 21 | 100 | 23 | 3 | 0 | 42 | 95 | 22 | 0 | 0 | 22 |
| Dehydronifedipine | 6 | n.a. | n.a. | 80 | 87 | 37 | 3 | 0 | 23 | 103 | 25 | 0 | 0 | 17 | 114 | 25 | 1 | 0 | 20 |
| Diltiazem | 2 | 4350 | 23 | 5.1 | 87 | 28 | 0 | 0 | 14 | 90 | 24 | 0 | 0 | 12 | 91 | 24 | 0 | 0 | 14 |
| Diltiazem | 1 | 4350 | 6.8 | 0.9 | 94 | 24 | 0 | 1 | 17 | 90 | 23 | 3 | 1 | 39 | 88 | 23 | 2 | 1 | 29 |
|
N- desmethyldiltiazem |
1 | 14,500 | 48 | 2.5 | 79 | 24 | 1 | 0 | 19 | 102 | 25 | 1 | 1 | 28 | 101 | 25 | 1 | 2 | 32 |
|
N- desmethyldiltiazem |
2 | 14,500 | 4.4 | 0.5 | 69 | 28 | 1 | 0 | 24 | 80 | 24 | 2 | 0 | 31 | 75 | 24 | 2 | 0 | 25 |
| Diphenhydramine | 1 | n.a. | 6.9 | 2.9 | 98 | 24 | 0 | 0 | 14 | 95 | 23 | 2 | 0 | 36 | 94 | 23 | 1 | 1 | 21 |
| Diphenhydramine | 6 | n.a. | n.a. | 58 | 78 | 37 | 1 | 0 | 15 | 27 | 25 | 22 | 0 | 60 | 34 | 24 | 20 | 0 | 57 |
| Fluoxetine | 1 | 50 | 180 | 5.4 | 95 | 24 | 0 | 1 | 27 | 96 | 25 | 2 | 1 | 35 | 96 | 25 | 2 | 2 | 36 |
| Fluoxetine | 2 | 50 | 2.7 | 0.9 | 91 | 27 | 0 | 0 | 14 | 92 | 24 | 0 | 0 | 18 | 91 | 23 | 0 | 0 | 15 |
|
Fluticasone (propionate) |
1 | 6 | 43 | 0.92 | 98 | 24 | 0 | 0 | 13 | 90 | 25 | 3 | 0 | 27 | 85 | 25 | 3 | 0 | 31 |
|
Fluticasone (propionate) |
2 | 6 | 15 | 6.2 | 101 | 27 | 0 | 0 | 9 | 106 | 24 | 0 | 0 | 9 | 104 | 24 | 0 | 1 | 13 |
| Glipizide | 1 | 158 | 25 | 17 | 95 | 24 | 0 | 0 | 14 | 92 | 25 | 3 | 0 | 25 | 94 | 25 | 3 | 0 | 26 |
| Glipizide | 2 | 158 | n.a. | 22 | 95 | 26 | 0 | 1 | 26 | 104 | 23 | 0 | 0 | 17 | 101 | 22 | 0 | 0 | 14 |
| Glyburide | 1 | 85 | 48 | 0.79 | 100 | 24 | 0 | 0 | 12 | 88 | 25 | 3 | 0 | 25 | 86 | 25 | 3 | 0 | 28 |
| Glyburide | 2 | 85 | n.a. | 102 | 372 | 21 | 1 | 20 | 56 | 165 | 17 | 0 | 10 | 45 | 138 | 17 | 1 | 8 | 57 |
| Hydrocodone | 1 | 605 | 57 | 2.1 | 96 | 24 | 0 | 0 | 14 | 90 | 24 | 2 | 0 | 31 | 89 | 25 | 1 | 0 | 27 |
| Hydrocodone | 2 | 605 | 4.1 | 1.2 | 102 | 28 | 0 | 0 | 13 | 101 | 24 | 0 | 0 | 13 | 100 | 24 | 0 | 1 | 16 |
| Hydrocortisone | 1 | 87 | 190 | 29 | 97 | 23 | 0 | 1 | 19 | 87 | 25 | 1 | 0 | 26 | 93 | 25 | 2 | 2 | 35 |
| Hydrocortisone | 2 | 87 | 17 | 4.6 | 99 | 28 | 0 | 0 | 12 | 98 | 22 | 0 | 0 | 14 | 92 | 23 | 0 | 0 | 12 |
| Metoprolol | 1 | 6250 | 44 | 14 | 97 | 24 | 0 | 5 | 54 | 98 | 22 | 3 | 2 | 129 | 93 | 24 | 3 | 3 | 77 |
| Metoprolol | 2 | 6250 | 4.7 | 4.3 | 160 | 28 | 0 | 16 | 387 | 133 | 24 | 0 | 9 | 25 | 127 | 24 | 0 | 5 | 36 |
| Norethindrone | 1 | 9 | 30 | 2.2 | 98 | 24 | 0 | 0 | 12 | 94 | 25 | 1 | 0 | 22 | 93 | 25 | 2 | 0 | 26 |
| Norethindrone | 2 | 9 | 10 | 2.2 | 124 | 28 | 0 | 5 | 21 | 127 | 24 | 0 | 5 | 17 | 127 | 24 | 0 | 5 | 20 |
| Norfluoxetine | 1 | 100 | 2400 | 39 | 101 | 23 | 0 | 2 | 32 | 103 | 23 | 1 | 1 | 29 | 101 | 22 | 1 | 3 | 43 |
| Norfluoxetine | 2 | 100 | 8.7 | 2.3 | 60 | 28 | 6 | 1 | 45 | 58 | 24 | 7 | 1 | 49 | 50 | 23 | 11 | 1 | 56 |
| Norverapamil | 1 | 2364 | 120 | 1.7 | 87 | 24 | 1 | 0 | 24 | 93 | 23 | 8 | 3 | 68 | 139 | 25 | 1 | 8 | 40 |
| Norverapamil | 2 | 2364 | 8.5 | 1.4 | 76 | 28 | 1 | 1 | 24 | 79 | 24 | 1 | 1 | 47 | 81 | 24 | 2 | 0 | 20 |
| Oxycodone | 1 | 550 | 80 | 4.9 | 94 | 24 | 0 | 0 | 14 | 91 | 24 | 3 | 0 | 32 | 91 | 25 | 3 | 0 | 27 |
| Oxycodone | 2 | 550 | 11 | 0.8 | 107 | 28 | 0 | 0 | 15 | 106 | 24 | 0 | 1 | 14 | 104 | 24 | 0 | 1 | 19 |
| Paroxetine | 1 | 25 | 84 | 4.1 | 82 | 24 | 2 | 0 | 26 | 103 | 25 | 1 | 2 | 33 | 114 | 25 | 1 | 2 | 33 |
| Paroxetine | 2 | 25 | 4.7 | 0.5 | 100 | 26 | 0 | 1 | 19 | 96 | 23 | 0 | 0 | 13 | 99 | 22 | 0 | 0 | 16 |
| Prednisolone | 1 | 500 | 700 | 75 | 103 | 23 | 0 | 0 | 20 | 95 | 24 | 2 | 2 | 37 | 91 | 25 | 2 | 0 | 32 |
| Prednisolone | 2 | 500 | 12 | 3.4 | 96 | 27 | 0 | 1 | 20 | 101 | 22 | 0 | 0 | 14 | 96 | 22 | 0 | 1 | 16 |
| Prednisone | 1 | 1300 | 160 | 84 | 105 | 23 | 0 | 1 | 23 | 95 | 24 | 1 | 3 | 40 | 94 | 25 | 1 | 1 | 29 |
| Prednisone | 2 | 1300 | 9.6 | 9.5 | 134 | 28 | 0 | 10 | 43 | 174 | 23 | 0 | 13 | 27 | 178 | 24 | 0 | 18 | 72 |
| Progesterone | 2 | 0.12 | n.a. | 120 | 107 | 28 | 0 | 2 | 41 | 136 | 24 | 0 | 7 | 25 | 126 | 24 | 1 | 9 | 44 |
| Progesterone | 3 | 0.12 | 0.13 | n.a. | 81 | 40 | 0 | 0 | 12 | 90 | 21 | 1 | 1 | 181 | 97 | 19 | 0 | 3 | 190 |
| Promethazine | 1 | 26 | 94 | 9.9 | 59 | 22 | 7 | 0 | 46 | 70 | 22 | 8 | 1 | 56 | 57 | 23 | 8 | 0 | 45 |
| Promethazine | 2 | 26 | 8.6 | 0.4 | 74 | 27 | 0 | 0 | 17 | 81 | 24 | 0 | 0 | 18 | 81 | 24 | 0 | 0 | 21 |
| Propanolol | 1 | 40 | 11 | 13 | 94 | 24 | 0 | 0 | 10 | 95 | 25 | 1 | 0 | 24 | 99 | 25 | 1 | 1 | 27 |
| Propanolol | 2 | 40 | 1.5 | 1.4 | 87 | 28 | 5 | 3 | 47 | 105 | 24 | 0 | 4 | 37 | 104 | 24 | 0 | 1 | 26 |
| Propoxyphene | 1 | 5520 | n.a. | 3.4 | 92 | 24 | 0 | 0 | 14 | 104 | 25 | 2 | 1 | 31 | 107 | 25 | 1 | 2 | 29 |
| Propoxyphene | 2 | 5520 | 233 | 5.1 | 153 | 28 | 0 | 15 | 75 | 171 | 24 | 0 | 16 | 133 | 175 | 24 | 0 | 15 | 60 |
| Sertraline | 1 | 15 | 94 | 3.3 | 80 | 24 | 2 | 0 | 28 | 100 | 25 | 2 | 3 | 44 | 111 | 25 | 1 | 3 | 40 |
| Sertraline | 2 | 15 | 5.9 | 0.9 | 92 | 27 | 0 | 0 | 23 | 96 | 23 | 0 | 0 | 18 | 92 | 23 | 0 | 1 | 38 |
| Desmethylsertraline | 1 | 150 | n.a. | 38 | 76 | 20 | 2 | 0 | 30 | 107 | 23 | 0 | 3 | 29 | 115 | 24 | 0 | 3 | 30 |
| Desmethylsertraline | 2 | 150 | 6.0 | 3.0 | 60 | 27 | 5 | 0 | 21 | 56 | 22 | 3 | 0 | 34 | 51 | 21 | 9 | 0 | 42 |
| Sulfamethoxazole | 1 | 16000 | 120 | 13 | 94 | 24 | 0 | 0 | 20 | 93 | 24 | 2 | 0 | 35 | 88 | 25 | 3 | 0 | 33 |
| Sulfamethoxazole | 2 | 16000 | 6.5 | 0.5 | 92 | 28 | 0 | 0 | 9 | 98 | 24 | 0 | 0 | 14 | 95 | 24 | 0 | 0 | 15 |
| Testosterone | 2 | 0.40 | 9.6 | 2.0 | 92 | 28 | 0 | 0 | 15 | 94 | 24 | 0 | 0 | 15 | 94 | 24 | 0 | 0 | 14 |
| Testosterone | 3 | 0.40 | 0.1 | n.a. | 82 | 40 | 0 | 0 | 9 | 90 | 21 | 0 | 1 | 29 | 94 | 18 | 1 | 1 | 41 |
| Triclosan | 3 | n.a. | 0.68 | n.a. | 84 | 39 | 1 | 0 | 24 | 87 | 19 | 2 | 3 | 46 | 87 | 19 | 0 | 2 | 30 |
| Triclosan | 5 | n.a. | 1900 | n.a. | 80 | 37 | 0 | 0 | 15 | 91 | 25 | 0 | 0 | 16 | 88 | 24 | 0 | 0 | 11 |
| Trimethoprim | 1 | 1500 | 18 | 3.8 | 102 | 24 | 0 | 0 | 12 | 94 | 24 | 3 | 0 | 31 | 93 | 25 | 3 | 0 | 24 |
| Trimethoprim | 2 | 1500 | 3.5 | 0.8 | 101 | 28 | 0 | 1 | 14 | 105 | 24 | 0 | 1 | 14 | 105 | 24 | 0 | 1 | 16 |
| Trimethoprim | 6 | 1500 | 180 | n.a. | 97 | 37 | 0 | 0 | 11 | 68 | 25 | 2 | 0 | 28 | 82 | 25 | 1 | 0 | 25 |
| Venlafaxine | 1 | n.a. | 6.2 | 0.90 | 99 | 24 | 0 | 0 | 11 | 103 | 25 | 1 | 0 | 24 | 102 | 25 | 0 | 1 | 25 |
| Venlafaxine | 4 | NA | n.a. | 0.30 | 139 | 16 | 6 | 10 | 124 | 85 | 17 | 9 | 6 | 108 | 57 | 15 | 12 | 7 | 127 |
| Verapamil | 1 | 520 | 39 | 3.1 | 89 | 24 | 1 | 0 | 22 | 105 | 25 | 5 | 3 | 61 | 121 | 25 | 1 | 8 | 40 |
| Verapamil | 2 | 520 | 7.8 | 0.8 | 96 | 28 | 0 | 1 | 21 | 98 | 24 | 0 | 3 | 25 | 100 | 24 | 0 | 2 | 20 |
| Warfarin | 1 | 750 | 8.5 | 3.0 | 106 | 24 | 0 | 0 | 10 | 98 | 23 | 2 | 0 | 35 | 96 | 23 | 0 | 0 | 20 |
| Warfarin | 2 | 750 | 15.0 | 3.6 | 100 | 28 | 0 | 3 | 49 | 111 | 24 | 0 | 3 | 36 | 98 | 24 | 0 | 5 | 43 |
Fig. 4.
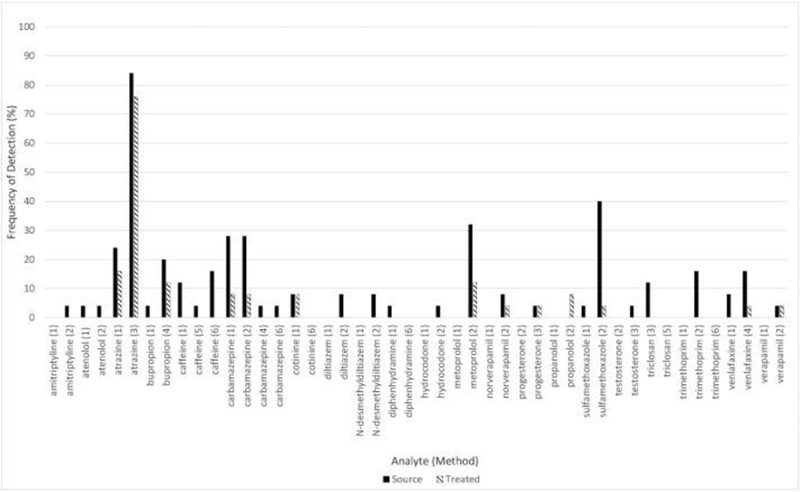
A comparison of the quantitative frequency of detection in source and treated drinking water for 22 intermethod comparator analytes (ICA) quantified in at least one environmental sample (n = 25 for all analytes).
Relative standard deviations (RSDs) greater than 40% were seen for 25 compounds in at least one method, most likely due to the complexity of multi-analyte analysis, the two-year length of the study, and from multiple staff performing sample extraction and analysis for several methods. Metoprolol and propoxyphene (Method 2), atrazine (Method 3), and venlafaxine (Method 4) displayed the most variability with RSDs ranging up to 432%. Variability for atrazine was seen as both low and high recoveries in the source and treated water, however, median recovery in distilled water was 85%, with an RSD of 9%. For the majority of LFM samples, the ambient atrazine concentrations far exceeded the spiking amount (1.7 ng/L, as shown in Table 1) which most likely explains the LFM variation observed. High RSDs of metoprolol, propoxyphene, and venlafaxine, however, were observed in both the LRB and LFM samples, and for venlafaxine, also was reflected in the variable and poor performance for the method in matrix spike samples (Figures 2 and 3). The variability for metoprolol and propoxyphene was seen in Method 2 which uses a solid phase extraction and includes 32 isotopically-labeled procedural internal standards. Unfortunately, at the time of method development, no labeled standards were available for these compounds. The most suitable isotopic compound was assigned, however, it was unable to account for the high bias measured in all three spike matrices. Compounds where a high bias (>150% recovery) was found in a matrix spike, and the analyte was detected in the corresponding sample, were reported as detected but not quantified since a quantitative value could not be confidently determined.
Intermethod performance was also evaluated by comparing environmental sampling results for the 46 ICAs. Of the 46 ICAs, 22 were quantified in at least one environmental sample, and Figure 4 compares the relative frequency of detection for those 22 compounds in source and treated drinking water. Table S4 includes concentrations detected, for each individual sampling location, for all individual ICAs included in the six methods in both primary and duplicate drinking water samples, along with field blanks and matrix spike samples. In general, intermethod comparisons indicated good agreement. For example, carbamazepine was measured in four different methods, and displayed excellent agreement in frequency of qualitative detection (i.e., 28% in source water and 8% in treated water for both Methods 1 and 2, Figure 4) and concentrations measured (i.e., medians of 10.0 and 15.9 ng/L in source water and medians of 13.1 and 17.8 ng/L in treated water for Methods 1 and 2, respectively, Table S4).
Any substantial differences in frequency of detection among the ICAs (Figure 4) was likely a result of the varying detection limits among the methods applied. The maximum concentration of an analyte present in one method commonly fell below the LCMRL of its comparative method (Table S4), which was the case for carbamazepine in Methods 4 and 6, compared to Methods 1 and 2. Remaining inconsistencies in frequency or concentration could be attributed to method performance. Caffeine concentrations varied by up to 70% (from 62 to 130 ng/L) at a few of the source-water sampling locations (DWTP 3, 4, 26, and 27), see Table S4. Reported matrix spike recoveries for these locations, however, also ranged from 4 to 134% and, combined with low concentrations measured near the three various detection limits of the methods (40–100 ng/L), could explain the differences. Venlafaxine concentrations also ranged from not detected to 500 ng/L in a few source-water sampling locations (DWTP 3, 4, and 21; Table S4); again, reported recoveries for venlafaxine ranged from 0 to 145% at these locations. The primary and duplicate sample measurement for venlafaxine by Method 4 in the source water of DWTP 21 had a difference of 192% (515 ng/L and 10 ng/L), which may reflect the overall poorer performance of this method.
As mentioned, the biggest influence in the environmental frequency of detection for the ICAs was detection limit. The LCMRL was chosen as the preferred methodology for determining method detection limits as it takes into account both accuracy and precision at multiple levels, while the more commonly used MDL only takes into account precision of a single low-spiking level. However, because the LCMRL determination requires multiple spiking levels, and the appropriate number and spacing between spiking levels must also be determined, it is a more time-consuming and expensive process. Several methods in this study contained 50 to 110 analytes with substantially different instrument responses, which increased the number of spiking levels to be analyzed, and added to the complexity of the LCMRL determination. For 10 of the ICA analytes, an LCMRL could not be determined and the MDL was used as a detection limit instead (Table 2). Such cases were in methods that contained a lengthy solid-phase extraction sample preparation, and it was not financially feasible to perform all the analysis needed to cover the range of concentrations required for the large number of analytes in these methods. In general, LCMRLs were 2–10 times greater than the MDL (Table 2). Targets for detection limits were previously chosen for a large number of ICAs based on pharmacological potency so as to encourage quantification of concentrations with similar potential for eliciting biological effect.8 The largest number of ICAs existed between Methods 1 and 2. Both methods use triple quadrupole LC-MS/MS with MRM for the instrumental analysis, while Method 1 uses a large volume direct injection and Method 2 uses a more time-consuming solid phase extraction for sample preparation. The LCMRLs usually were lower with Method 2, however, the LCMRLs for most analytes in these two methods were well below the target (Table 2). There were a few cases where lower detection limits may be needed for applications, such as with the antidepressants (fluoxetine, norfluoxetine, paroxetine, sertraline, and desmethylsertraline) and hormones (norethindrone). Two other hormones, testosterone and progesterone, were compared in Methods 2 and 3. Both methods include a solid-phase extraction step, however, Method 3 utilizes FTMS with accurate mass for the instrumental analysis. Method 3 provided LCMRLs for testosterone and progesterone that were more than tenfold lower than Method 2, and considering the high potency of these hormones, the ability to measure concentrations in the sub-ng/L level may be very important.
Environmental data were not reported for any analyte that did not consistently meet predetermined quality standards; a summary of the method performance for those analytes that did not meet these standards is included in Table S5. A compound was excluded from further analysis if there was less than 50% median recovery in LFB or LFM samples, or if the analyte had a recovery outside acceptable limits (i.e., 50 to 150%) in more than 40% of the LFB or LFM samples. Forty-eight compounds did not meet method performance criteria, however, 25 met quality performance criteria in other study methods, so occurrence data are still available35, 36. For Method 1, nine compounds were excluded. Ranitidine consistently performed below 50% median recovery in all matrixes. Tiotropium was poorly recovered from laboratory reagent water, but improved in the source and treated water matrixes, which suggests the sample matrix may have assisted in method performance. The remaining seven compounds in Method 1 (chlorpheniramine, cimetidine, esomeprazole, ketoconazole, nizatidine, orlistat, and quinine) all had acceptable median recoveries in laboratory matrix spike water, but exhibited variably high or low recoveries on a compound-by-compound basis (Table S5). They may reflect sample- or compound-specific effects from sample matrix, such as matrix enhancement or suppression36.
For Method 2, six compounds were excluded. Cimetidine, betamethasone, fluocinonide, and theophylline consistently had low recoveries in all matrixes. The compound 2-hydroxy-ibuprofen displayed a median recovery of 85% in spiked distilled water samples, however, overlapping interference was often seen in matrix spike samples that prevented quantitation. Since 2-hydroxy-ibuprofen has only one MRM transition available, an alternate transition could not be used for quantitation. Accurate mass methods may be a better instrument analysis choice in such cases.
Eight analytes were excluded in Method 4, the largest proportion for any method used in this study. Although it had been successfully used and documented by the analyzing laboratory39, recoveries were lower and more variable during the period in which this method was used for this study, particularly in source and treated water matrixes. We have not yet identified a specific reason (e.g., change in solvents, SPE media, or analysis conditions) that would explain this substantial difference.
Seven analytes were excluded from Method 5 (Table S5). All but one performed below acceptable median recovery in laboratory reagent spike water, although five of the six compounds displayed similar or improved recoveries in source and treated water matrixes. Two compounds, 3-tert-Butyl-4-hydroxyanisole (BHA) and isoquinoline, exhibited lower median recoveries in source and treated water matrices. This performance level was also observed in the long-term QC data maintained by the performing laboratory (data not shown). Isoquinoline performed acceptably in reagent water but was not recovered in matrix spike samples, and the reason for this divergence in performance could not be identified.
Six compounds were excluded from consideration in Method 6 (Table S5). Exclusion was compound-specific but in all cases exclusion was because performance in one of the three matrices was unacceptably low. All compounds determined by Method 6 were also present in Method 1, the method of choice for these analytes.
4. Conclusion.
When choosing an analysis method, method performance may not be the only criteria under consideration. The newer available technologies are focusing on greener chemistry, such as fast chromatography techniques, which greatly reduce sample run times and therefore reduce solvent and energy consumption. The same is true for extraction techniques, with solid phase extraction, which again greatly reduces solvent consumption, taking the place of liquid-liquid extraction for many environmental methods, and now direct injection analysis, which allows for little or no solvent use. All of these are important considerations which can both reduce the environmental impact and exposure of analytical chemists to hazardous substances. Although it is outside of the scope of this particular study, various tools are available to evaluate the environmental impact of different methods43.
Collectively, evaluation of the field and laboratory QA/QC results suggest most methods performed accurately and precisely during the course of this study, although differences in method performance or environmental occurrence were observed. For many CECs, including pharmaceuticals, to put any analytical uncertainty into perspective, biological potency and study application should be considered. For a compound such as caffeine, with environmental concentrations in the low ng/L and low potency, a 70% measurement range due to analytical uncertainty may not have environmental significance. However, the same 70% range in low concentration of a higher potency pharmaceutical, such as hormones (e.g., testosterone or progesterone) or some antidepressants, could significantly change interpretation of the resulting data4, 5, 32, 33. Although the majority of analytes met the predetermined method performance criteria, a portion (16% of the original analyte list) did not, and thus environmental results could not be confidently assigned for these compounds within those particular methods. The need to exclude some data due to analytical uncertainty demonstrates the importance of additional QA/QC protocols for national-scale, extended duration contaminant monitoring programs.
Supplementary Material
Acknowledgements
The authors declare no competing financial interest. The information in this document has been funded partially or wholly by the U.S. Environmental Protection Agency. The research described in this article has been funded in part by the U.S. Environmental Protection Agency through Interagency Agreement DW14922330 to the U.S. Geological Survey, and through programmatic support of the USGS’ Toxic Substances Hydrology Program and the USEPA’s Office of Research and Development, Office of Water, Office of Chemical Safety and Pollution Prevention, and Region 8. Information Collection Rule approval for the Phase II Questionnaire was granted under EPA ICR No. 2346.01, OMB Control No. 2080–0078. This manuscript has been reviewed in accordance with USEPA policy and approved for publication. Approval does not signify that the contents reflect the views or policies of the USEPA and mention of trade names or commercial products does not constitute endorsement or recommendation for use by USEPA. This document has also been reviewed in accordance with USGS policy and approved for publication. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government. The authors would like to thank all participating drinking water treatment plants for their involvement in the project and for their assistance in collecting the samples. The authors would like to acknowledge Laura J. Coffey, Michael Eastham, Hayden Miracle, Mary C. Noriega, Laura Rosenblum, and Sarah Watson for their assistance in sample preparation and/or data analysis.
References
- 1.Vasquez MI; Lambrianides A; Schneider M; Kümmerer K; Fatta-Kassinos D, Environmental side effects of pharmaceutical cocktails: What we know and what we should know. Journal of Hazardous Materials 2014, 279, 169–189. [DOI] [PubMed] [Google Scholar]
- 2.Wennmalm Å, Pharmaceuticals: Environmental Effects. In Encyclopedia of Environmental Health, Nriagu JO, Ed. Elsevier: Burlington, 2011; pp 462–471. [Google Scholar]
- 3.Zenker A; Cicero MR; Prestinaci F; Bottoni P; Carere M, Bioaccumulation and biomagnification potential of pharmaceuticals with a focus to the aquatic environment. Journal of Environmental Management 2014, 133, 378–387. [DOI] [PubMed] [Google Scholar]
- 4.Fent K; Weston AA; Caminada D, Ecotoxicology of human pharmaceuticals. Aquatic Toxicology 2006, 76, (2), 122–159. [DOI] [PubMed] [Google Scholar]
- 5.Kidd KA; Blanchfield PJ; Mills KH; Palace VP; Evans RE; Lazorchak JM; Flick RW, Collapse of a fish population after exposure to a synthetic estrogen. Proc Natl Acad Sci USA 2007, 104, (21), 8897–8901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gros M; Rodríguez-Mozaz S; Barceló D, Fast and comprehensive multi-residue analysis of a broad range of human and veterinary pharmaceuticals and some of their metabolites in surface and treated waters by ultra-high-performance liquid chromatography coupled to quadrupole-linear ion trap tandem mass spectrometry. Journal of Chromatography A 2012, 1248, 104–121. [DOI] [PubMed] [Google Scholar]
- 7.Gracia-Lor E; Sancho JV; Hernández F, Multi-class determination of around 50 pharmaceuticals, including 26 antibiotics, in environmental and wastewater samples by ultra-high performance liquid chromatography–tandem mass spectrometry. Journal of Chromatography A 2011, 1218, (16), 2264–2275. [DOI] [PubMed] [Google Scholar]
- 8.Batt AL; Kostich MS; Lazorchak JM, Analysis of ecologically relevant pharmaceuticals in wastewater and surface water using selective solid-phase extraction and UPLC-MS/MS. Anal. Chem 2008, 80, (13), 5021–5030. [DOI] [PubMed] [Google Scholar]
- 9.López-Serna R; Petrović M; Barceló D, Development of a fast instrumental method for the analysis of pharmaceuticals in environmental and wastewaters based on ultra high performance liquid chromatography (UHPLC)–tandem mass spectrometry (MS/MS). Chemosphere 2011, 85, (8), 1390–1399. [DOI] [PubMed] [Google Scholar]
- 10.Berset J-D; Brenneisen R; Mathieu C, Analysis of llicit and illicit drugs in waste, surface and lake water samples using large volume direct injection high performance liquid chromatography – Electrospray tandem mass spectrometry (HPLC–MS/MS). Chemosphere 2010, 81, (7), 859–866. [DOI] [PubMed] [Google Scholar]
- 11.Yu K; Li B; Zhang T, Direct rapid analysis of multiple PPCPs in municipal wastewater using ultrahigh performance liquid chromatography–tandem mass spectrometry without SPE pre-concentration. Analytica Chimica Acta 2012, 738, 59–68. [DOI] [PubMed] [Google Scholar]
- 12.Bayen S; Yi X; Segovia E; Zhou Z; Kelly BC, Analysis of selected antibiotics in surface freshwater and seawater using direct injection in liquid chromatography electrospray ionization tandem mass spectrometry. Journal of Chromatography A 2014, 1338, 38–43. [DOI] [PubMed] [Google Scholar]
- 13.Steven D Zaugg SGS, Michael P. Schroeder, Larry B. Barber, and Mark R. Burkhardt, Methods of Analysis by the U.S. Geological Survey National Water Quality Laboratory—Determination of Wastewater Compounds by Polystyrene-Divinylbenzene Solid-Phase Extraction and Capillary-Column Gas Chromatography/Mass Spectrometry. In U.S. GEOLOGICAL SURVEY: Water-Resources Investigations Report 01–4186, 2002; p 37 p.
- 14.Petrovic M, Methodological challenges of multi-residue analysis of pharmaceuticals in environmental samples. Trends in Environmental Analytical Chemistry 2014, 1, e25–e33. [Google Scholar]
- 15.de Boer J; Cofino WP, First world-wide interlaboratory study on polybrominated diphenylethers (PBDEs). Chemosphere 2002, 46, (5), 625–633. [DOI] [PubMed] [Google Scholar]
- 16.Farré M; Petrovic M; Gros M; Kosjek T; Martinez E; Heath E; Osvald P; Loos R; Le Menach K; Budzinski H; De Alencastro F; Müller J; Knepper T; Fink G; Ternes TA; Zuccato E; Kormali P; Gans O; Rodil R; Quintana JB; Pastori F; Gentili A; Barceló D, First interlaboratory exercise on non-steroidal anti-inflammatory drugs analysis in environmental samples. Talanta 2008, 76, (3), 580–590. [DOI] [PubMed] [Google Scholar]
- 17.Grover DP; Zhang ZL; Readman JW; Zhou JL, A comparison of three analytical techniques for the measurement of steroidal estrogens in environmental water samples. Talanta 2009, 78, (3), 1204–1210. [DOI] [PubMed] [Google Scholar]
- 18.Heath E; Kosjek T; Farre M; Quintana JB; de Alencastro LF; Castiglioni S; Gans O; Langford K; Loos R; Radjenović J; Rocca LM; Budzinski H; Tsipi D; Petrovic M; Barcelo D, Second interlaboratory exercise on non-steroidal anti-inflammatory drug analysis in environmental aqueous samples. Talanta 2010, 81, (4–5), 1189–1196. [DOI] [PubMed] [Google Scholar]
- 19.Lindström G; Kärrman A; van Bavel B, Accuracy and precision in the determination of perfluorinated chemicals in human blood verified by interlaboratory comparisons. Journal of Chromatography A 2009, 1216, (3), 394–400. [DOI] [PubMed] [Google Scholar]
- 20.Onghena M; Moliner-Martinez Y; Picó Y; Campíns-Falcó P; Barceló D, Analysis of 18 perfluorinated compounds in river waters: Comparison of high performance liquid chromatography–tandem mass spectrometry, ultra-high-performance liquid chromatography–tandem mass spectrometry and capillary liquid chromatography–mass spectrometry. Journal of Chromatography A 2012, 1244, 88–97. [DOI] [PubMed] [Google Scholar]
- 21.Van Leeuwen SPJ; De Boer J; Van Leeuwen SPJ; Van Bavel B, First worldwide UNEP interlaboratory study on persistent organic pollutants (POPs), with data on polychlorinated biphenyls and organochlorine pesticides. TrAC Trends in Analytical Chemistry 2013, 46, 110–117. [Google Scholar]
- 22.Weiss JM; van der Veen I; de Boer J; van Leeuwen SPJ; Cofino W; Crum S, Analytical improvements shown over four interlaboratory studies of perfluoroalkyl substances in environmental and food samples. TrAC Trends in Analytical Chemistry 2013, 43, 204–216. [Google Scholar]
- 23.Vanderford BJ; Drewes JE; Eaton A; Guo YC; Haghani A; Hoppe-Jones C; Schluesener MP; Snyder SA; Ternes T; Wood CJ, Results of an Interlaboratory Comparison of analytical methods for contaminants of emerging concern in water. Anal. Chem 2013, 86, (1), 774–782. [DOI] [PubMed] [Google Scholar]
- 24.Konieczka P; Wolska L; Namieśnik J, Quality problems in determination of organic compounds in environmental samples, such as PAHs and PCBs. TrAC Trends in Analytical Chemistry 2010, 29, (7), 706–717. [Google Scholar]
- 25.Konieczka P; Linsinger TJ; Namieśnik J, Determination of POPs in environmental matrices – proficiency tests for Polish laboratories. Accreditation and Quality Assurance 2006, 11, (11), 584–589. [Google Scholar]
- 26.Linsinger TJ; Voorspoels S; Liebich A, Results of an interlaboratory comparison on the determination of polybrominated flame retardants in poly(ethyleneterephthalate). Analytical and Bioanalytical Chemistry 2008, 390, (1), 399–409. [DOI] [PubMed] [Google Scholar]
- 27.Hanke G; Mariani G; Comero S; Loos R; Bidoglio G; Polesello S; Valsecchi S; Rusconi M; Wollgast J; Castro-Jiménez J; Patrolecco L; Ademollo N, Chemical-monitoring on-site exercises to harmonize analytical methods for priority substances in the European Union. TrAC Trends in Analytical Chemistry 2012, 36, 25–35. [Google Scholar]
- 28.Konieczka P, The role of and the place of method validation in the quality assurance and quality control (QA/QC) system. Critical Reviews in Analytical Chemistry 2007, 37, (3), 173–190. [Google Scholar]
- 29.Konieczka P; Namieśnik J, Estimating uncertainty in analytical procedures based on chromatographic techniques. Journal of Chromatography A 2010, 1217, (6), 882–891. [DOI] [PubMed] [Google Scholar]
- 30.Kostich MS; Batt AL; Glassmeyer ST; Lazorchak JM, Predicting variability of aquatic concentrations of human pharmaceuticals. Science of The Total Environment 2010, 408, (20), 4504–4510. [DOI] [PubMed] [Google Scholar]
- 31.Kostich M; Flick R; Martinson J, Comparing predicted estrogen concentrations with measurements in US waters. Environmental Pollution 2013, 178, 271–277. [DOI] [PubMed] [Google Scholar]
- 32.Kostich MS; Batt AL; Lazorchak JM, Concentrations of prioritized pharmaceuticals in effluents from 50 large wastewater treatment plants in the US and implications for risk estimation. Environmental Pollution 2014, 184, 354–359. [DOI] [PubMed] [Google Scholar]
- 33.Kostich MS; Lazorchak JM, Risks to aquatic organisms posed by human pharmaceutical use. Science of The Total Environment 2008, 389, (2–3), 329–339. [DOI] [PubMed] [Google Scholar]
- 34.Glassmeyer ST; Furlong ET; Kolpin DW; Batt AL; Benson B; Boone S; Conerly O; Donohue M; King D; Kostich M; Mash H; Pfaller S; Schenck K; Simmons JE; Varughese E; Vesper S; Villegas E; Wilson V, Nationwide reconnaissance study of contaminants of emerging concern in source and treated drinking water. Submitted to Science of the Total Environment 2015. [DOI] [PMC free article] [PubMed]
- 35.Furlong ET; Batt AL; Mash H; Noriega MC; Rosenblum L; Schenck K; Glassmeyer S; Kolpin DW, Contaminants of emerging concern in source and treated drinking waters from 25 drinking water treatment plants: pharmaceuticals. Submitted to Science of the Total Environment 2015. [DOI] [PMC free article] [PubMed]
- 36.Wilson V; Mash H; Evans N; Schenck K; Rosenblum L; Glassmeyer ST; Furlong ET; Kolpin DW, Comparison of in vitro Assessment of estrogenic activity and measured hormone concentrations in source and treated drinking water. Submitted to Science of the Total Environment 2015. [DOI] [PubMed]
- 37.Furlong ET; Noriega MC; Kanagy CJ; Kanagy LK; Coffey LJ; Burkhardt MR, Determination of human-use pharmaceuticals in filtered water by direct aqueous injection–high-performance liquid chromatography/tandem mass spectrometry: U.S. Geological Survey Techniques and Methods In Methods Research and Development Program, N. W. Q. L., Ed. USGS: 2014; p 49 p. [Google Scholar]
- 38.Schultz MM; Furlong ET, Trace analysis of antidepressant pharmaceuticals and their select degradates in aquatic matrixes by LC/ESI/MS/MS. Anal. Chem 2008, 80, (5), 1756–1762. [DOI] [PubMed] [Google Scholar]
- 39.Cahill JD; Furlong ET; Burkhardt MR; Kolpin D; Anderson LG, Determination of pharmaceutical compounds in surface- and ground-water samples by solid-phase extraction and high-performance liquid chromatography–electrospray ionization mass spectrometry. Journal of Chromatography A 2004, 1041, (1–2), 171–180. [DOI] [PubMed] [Google Scholar]
- 40.Martin JJ; Winslow SD; Munch DJ, A new approach to drinking-water-quality data: Lowest-Concentration Minimum Reporting Level. Environmental Science & Technology 2007, 41, (3), 677–681. [DOI] [PubMed] [Google Scholar]
- 41.Technical Basis for the Lowest Concentration Minimum Reporting Level (LCMRL) Calculator In http://water.epa.gov/scitech/drinkingwater/labcert/analyticalmethods_ogwdw.cfm, 2010; p 24 pgs.
- 42.Definition and Procedure for the Determination of the Method Detection Limit. In U.S. Environmental Protection Agency: 40 CFR 104.1, Part 136, App B., 2012; pp 343–346.
- 43.Gałuszka A; Migaszewski ZM; Konieczka P; Namieśnik J, Analytical Eco-Scale for assessing the greenness of analytical procedures. TrAC Trends in Analytical Chemistry 2012, 37, 61–72. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.