

SUMMARY
Comparative immune response profiling is important for selecting next-generation vaccines. We comprehensively evaluated the antibody responses from a panel of nine respiratory vaccines against Ebola virus (EBOV) derived from human and avian paramyxoviruses expressing EBOV glycoprotein (GP). Most vaccines were protective in guinea pigs but yielded antibody repertoires that differed in proportion targeting key antigenic regions, avidity, neutralizing antibody specificities, and linear epitope preferences. Competition studies with monoclonal antibodies from human survivors revealed that some epitopes in GP targeted for neutralization were vector dependent, while EBOV-neutralizing titers correlated with the response magnitude toward the receptor-binding domain and GP1/GP2 interface epitopes. While an immunogen determines the breadth of antibody response, distinct vaccine vectors can induce qualitatively different responses, affecting protective efficacy. These data suggest that immune correlates of vaccine protection cannot be generalized for all vaccines against the same pathogen, even if they use the exact same immunogen.
Graphical Abstract

In Brief
Meyer et al. developed a panel of second-generation Ebola vaccines using respiratory viruses to deliver a common immunogen. The vaccines were protective against lethal infection in guinea pigs. The different vaccines elicited different antibody profiles, indicating correlates of protection may not be universal among Ebola vaccines.
INTRODUCTION
Ebola virus (EBOV) causes a severe disease in humans, with case fatality rates ranging from 25% to 90% (Kuhn, 2008). The 2013–2016 epidemic in West Africa caused 28,616 suspected, probable, and confirmed cases and 11,310 deaths (CDC, 2016). While there are no approved vaccines or treatments against EBOV, several experimental vaccine strategies are being investigated with varying degrees of success in animal models (Richardson et al., 2010). Leading vaccine candidates, including vesicular stomatitis virus (VSV) and various adenovirus-based vectors, which demonstrated complete protection in non-human primates (NHPs), have advanced into clinical trials (Kennedy etal., 2017).
Previously, we developed a mucosal replicative vaccine based on human parainfluenza type 3 (HPIV3) expressing EBOV glycoprotein (GP) (HPIV3/EboGP). This vaccine was 100% efficacious against lethal EBOV infection when administered as one aerosol dose to the respiratory tract of NHPs (Meyer et al., 2015). Despite the fact that preexisting immunity to HPIV3 was expected to reduce vaccine virus replication, two doses of the vaccine were equally immunogenic in HPIV3-immune and HPIV3-naive NHPs (Bukreyev et al., 2010). Nevertheless, we addressed the concerns of vector immunity by developing alternative approaches. One of them was construction of a chimeric HPIV3/EBOV virus in which the HPIV3 F and HN proteins were deleted and replaced with EBOV GP (HPIV3/ΔFHN/EboGP). A single dose of the construct administered by the intranasal (IN) route protected guinea pigs against lethal EBOV challenge (Bukreyev et al., 2009). The other approach included development of an alternate respiratory vaccine vector based on Newcastle disease virus (NDV), an avian paramyxoviruse (APMV). NDV strains are categorized according to disease severity in birds: (1) lentogenic strains, which cause mild or inapparent infections largely limited to the respiratory tract and that are presently in use as live vaccines for poultry; (2) mesogenic stains, which cause mild systemic infections; and (3) velogenic strains, which cause systemic infections with high mortality rates (Alexander, 2000). Prime-boost vaccinations of NHPs with a constructed mesogenic Beaudette C (BC) strain of NDV expressing GP elicited immunoglobulin A (IgA) titers in respiratory tract secretions and serum EBOV-neutralizing antibody titers equal to those induced by HPIV3/EboGP, suggesting that it may be a suitable vector against EBOV (DiNapoli et al., 2010).
Here, we expanded our respiratory mucosal vaccine repertoire expressing EBOV GP by encompassing other human and avian paramyxoviruses with modifications to the surface glycoproteins. HPIV1 has some sequence homology and shares genome organization with HPIV3. We generated two HPIV1-based constructs expressing EBOV GP from an added transcriptional cassette, one of which mirrors HPIV3/ΔFHN/EboGP by removal of the surface F and HN glycoproteins. We also utilized a lentogenic NDV, strain LaSota (LS), to create two vaccine constructs. The F protein cleavage site (Fc) sequence from one of these constructs was modified to that of APMV type 2 that is avirulent in chickens and has also been isolated from NHPs (Kusagawa et al., 1993). Mesogenic NDV strains replicate in many tissue types and elicit higher levels of immune responses than lentogenic strains. However, due to their lethality in chickens, mesogenic strains are currently listed as a US Department of Agriculture (USDA) Select Agent, which severely limits their practical use as vaccine vectors. Subsequently, we modified the mesogenic BC strain by substituting the polybasic F protein cleavage site sequence to the monobasic F protein cleavage site sequence of the strain LS, replacing the F and HN genes with that of LS or the F and HN ectodomains of APMV3. These changes make this BC strain avirulent in chickens while maintaining its replication ability (Kim et al., 2014).
Comparative studies of EBOV vaccines based on different paramyxoviruses or other types of EBOV vaccines have not been performed. We tested protective efficacy of our mucosal vaccines in a guinea pig challenge model alongside the previously developed HPIV3-based vectors. Comprehensive characterization of the antibody response through serum adsorption studies, biolayer interferometry (BLI) competition assays, and peptide arrays revealed that mucosal vector choice influences the magnitude, quality, and scope of the response, which could ultimately impact the protective outcome. Our results suggest that antibody responses to the same antigen are greatly affected by the vaccine vector employed in antigen delivery.
RESULTS
Generation of Vaccine Constructs
Construction and recovery of HPIV3/EboGP (Bukreyev et al., 2006, 2007) and HPIV3/ΔFHN/EboGP (Bukreyev et al., 2009) was previously described. As with HPIV3-derived constructs, the EBOV GP coding sequence was inserted between the P and M genes of the HPIV1 virus vector, flanked by the transcription start and stop signals of the virus vector backbone (Figures 1 and S1A). The virus was further modified by deletion of the F and HN genes to create HPIV1/ΔFHN/EboGP, which solely relies on the GP protein for attachment and entry (Figures 1 and S1B).
Figure 1. Vectors Utilized for the Vaccine Constructs.

Paramyxovirus-vectored vaccines expressing EBOV GP transcriptional cassette inserted between the P and M genes and the HPIV3 vector control. The genes derived from HPIV3/HPIV1, LS, and BC are shown as white, gray, or black, respectively. One LS backbone virus contained the F protein cleavage sequence of APMV2, KPASRF. BC backbone viruses contained the F protein cleavage sequence of LS (GRQGRL), the F and HN genes from LS, and the F and HN ecotodomains from APMV3 (light blue). See also Figure S1.
Five APMV vaccines were constructed by inserting the GP open reading frame (ORF), flanked by the transcription start and stop signals of NDV, between the P and M genes of (1) LS, (2) LS/APMV2Fc (LS Fc site was substituted with that of APMV2), (3) BC/LSFcHN (the BC Fc site and HN gene were replaced with that of LS), (4) BC/LSFHN (the F and HN genes of BC were replaced with that of LS), and (5) BC/APMV3FHN (the F and HN ecto domains of BC were substituted with that of APMV3) (Figures 1 and S1C). The five recombinant NDVs were recovered as previously described (Bukreyev et al., 2005) and amplified in embryonated chicken eggs. For all constructs, the editing site of GP mRNA was modified to express the membrane-bound full-length GP. Expression of GP was confirmed in rhesus macaque LLC-MK2 and guinea pig CCL158 cells by western blot analysis (Figures S2A and S2B), and replication was confirmed by growth kinetic studies in CCL158 cells (Figure S2C).
Most Constructs Are Protective against EBOV Challenge
Ten groups of guinea pigs (5 animals per vaccine group) were immunized by a single dose of vaccine or an empty HPIV3 vector control via the IN route (Figure 2A). On day 28 post-vaccination, the guinea pigs were intraperitoneally (IP) infected with guinea pig-adapted EBOV. Animals were monitored daily and viremia in serum was measured over the course of infection.
Figure 2. Protection in Guinea Pigs.
Survival, weight loss, clinical scores, viremia, and humoral response in vaccinated and infected guinea pigs.
(A) Study design.
(B) Survival curves.
(C) Percent weight change.
(D) Illness scores on days 0–28 after EBOV infection.
(E) Viremia after EBOV infection (PFU/mL). Line indicates limit of detection.
(F–H) Total GP-specific IgG (F), IgA (G), and EBOV neutralizing titers (H). Multiple comparisons were performed using one-way ANOVA and Tukey’s post hoc test. Groups that do not share a common letter above them differ at p < 0.05. Shown are mean values ± SE based on 5 animals per group.
(I) Correlation between GP-specific IgG and neutralizing antibody titers (Pearson r = 0.9190, p < 0.0001).
See also Figures S2 and S3.
All animals in the HPIV3 control group developed disease and viremia, and all but one succumbed to infection (Figures 2B–2E). The surviving animal did decline in weight and scored a 2, but later fully recovered. All vaccinated groups were fully protected, with the exception of the BC/APMV3FHN/EboGP group, which demonstrated only 40% survival. The surviving animals within this group demonstrated weight loss and a disease score of 1. Plaque assays revealed detectable viremia in these animals similar to the control group, which peaked at day 6 (average 2.03–105 PFU/mL). All other vaccinated groups showed only low levels of transient viremia with no severe clinical signs of disease; transient low-level disease with a score no more than 1 or 2 was observed in a few animals.
IgG Antibody Responses Do Not Necessarily Correlate with Protection
The total EBOV GP-specific IgG antibody response was highest in animals that received the HPIV3/EboGP vaccine reaching titers of 1:895 27 days post-vaccination. The majority of APMV-vaccinated groups demonstrated comparable IgG titers (Figure 2F), except for the recipients of BC/APMV3FHN/EboGP, which showed almost no IgG response and had the lowest GP expression level in vitro (Figure S2). Recipients of the F/HN-deletion vaccines, HPIV3/ΔFHN/EboGP and especially HPIV1/ΔFHN/EboGP, elicited IgG titers that were lower than the recipients of the parental vectors, yet in the case of the HPIV1 vectors, GP expression levels were comparable (Figures S2A and S2B). In vitro expression of GP varied among the vaccines, but its association with IgG and IgA titers was not significant (Figure S2B). EBOV-neutralizing antibody titers (Figure 2H) mirrored IgG responses, resulting in a strong correlation between the two (Figure 2I). While HPIV1/ΔFHN/EboGP recipients had barely detectable IgG and no neutralizing antibodies, all animals survived EBOV infection and showed no signs of illness and very low levels of viremia. Interestingly, we instead detected GP-specific IgA antibodies in these animals at the levels similar to that in other surviving groups (Figure 2G). Conversely, the group vaccinated with BC/APMV3FHN/EboGP, which had low IgA titers but also had low IgG and no neutralizing titers, demonstrated only 40% survival and high levels of viremia. Cross-neutralization of the additional ebolaviruses Sudan (SUDV) and Bundibugyo (BDBV) was not detected in any vaccinated groups (data not shown).
Antigenic Landscape of Antibodies Targeting GP Forms Varies between Vaccine Vectors
To comparatively characterize the antigenic landscapes of antibodies induced by vaccination, we used BLI to examine the binding of immune sera to four recombinant GPs immobilized on Octet streptavidin biosensors: “full” GP, GPΔmuc, soluble GP (sGP), and cleaved GP (GPcl) (Figure 3A). The full GP (designated GP) represented the complete ectodomain lacking the transmembrane domain and the cytoplasmic tail, which also were absent in all other proteins (Figure 3B). The GPΔmuc protein lacked the complete serine-threonine rich mucin-like domain (MLD), which comprises a large, C-terminal part of GP1 rich in N-glycosylation and O-glycosylation sites (Sanchez et al., 1993; Yang et al., 2000). The sGP represents the abundantly secreted dimeric glycoprotein that is the primary product of the GPgene (Sanchez et al., 1996; Volchkovet al., 1995). sGP shares GP amino acids 31–295 including most of the glycan cap (GC) and the receptor-binding domain (RBD) but bears a unique C terminus that lacks the MLD and GP2 (Volchkova et al., 1998). Upon entry in cells, GP is cleaved by cathepsins, forming GPcl, which has GP1 lacking the GC and MLD, bound to GP2 (Chandran et al., 2005). All serum samples reacted with GP at levels reflective of the IgG ELISA and neutralization results, with the HPIV1/ΔFHN/EboGP and BC/APMV3FHN/EboGP sera showing lower binding (Figure 3C). In general, the sera from HPIV-based vectors (except HPIV1/ΔFHN/EboGP) and not APMV, consistently bound well to all forms of GP, especially GPcl, when compared to empty vector control. The difference between HPIV and APMV groups in binding to GPcl is greater relative to GP, indicating that the majority of GP epitopes bound by APMV sera are lost following enzymatic cleavage of GP.
Figure 3. Distribution of Serum Antibodies Binding to EBOV GP Forms and Antigenic Domains.

(A) Schematic of the BLI assay of serum binding to immobilized GP.
(B) Truncated GP forms.
(C) Maximum response units (RU) of total post-vaccination serum binding to the different truncated GP forms.
(D) Serum dissociation rate from immobilized GP forms.
(E) Schematic of the BLI assay of serum binding to immobilized biotinylated GP forms following serum preadsorption with various GP forms.
(F–I) Binding inhibition to immobilized GP (F), GPΔmuc (G), sGP (H), and GPcl (I) following preadsorption, expressed as a percentage of total binding RU values obtained without serum preadsorption. Shown are mean values ± SE based on 3 representative animals per group in (C), (D), and (F)-(I).
One-way ANOVA with Fisher’s LSD post hoc test was performed. Groups that do not share a common letter above them differ at p < 0.05. See also Figures S3 and S4.
To determine if the different vaccines induced different levels of GP-specific affinity maturation, we measured the dissociation rates of antibody-GP complexes by BLI (Figure 3D), which is independent of the antibody concentration in the sample. Sera from the HPIV groups generally had a higher avidity (lowest off-rate) for GP and GPΔmuc forms compared to the APMV groups. The similar trend observed against GP and GPΔmuc indicates that avidity is not sensitive to the absence of the MLD and is retained by the epitopes on other parts of the GP trimer. This difference in avidity between the HPIV and APMV groups was, however, less pronounced with sGP, while an inverted avidity trend was observed with GPcl, which lacks the GC of trimeric GP. These data indicate that avidity differences between the two paramyxovirus vaccine groups may be attributed to populations of antibodies specific for conformational epitopes, the GC and RBD. Avidity to GP strongly correlated with GP-specific antibody and neutralizing titers (Figures S3A-S3C), indicating the response greater in magnitude was also associated with higher antibody quality.
Proportion of the Antibody Response Targeting GP Antigenic Regions Differs between Vaccinated Groups
To measure the portion of the antibodies that bound key GP surfaces that were either shared or unique among the different GP forms, we performed BLI competition assays; sera from vaccinated animals were preadsorped with the different GP forms, and the residual (non-adsorbed) antibodies were allowed to bind to all indicated GPs immobilized on the sensor (Figure 3E). Serum adsorption with GPΔmuc had considerable effect on the binding to GP but did not completely abolish it (Figure 3F). Serum binding to immobilized GP, which was not removed by GPΔmuc, is most likely specific for the MLD. HPIV1/ΔFHN/EboGP, HPIV3/EboGP, and HPIV3/ΔFHN/EboGP binding to GP was least affected by GPΔmuc; only 55%, 35%, and 60% of antibody binding, respectively, was blocked by GP-Δmuc, indicating that a large part of the antibody response elicited by these vaccines targets the MLD. Other vaccinated groups had at least 70% of antibody binding to GP affected by GPΔmuc. In contrast, preadsorption with GP, which possesses the MLD, blocked binding of sera from all vaccine groups except HPIV1/ΔFHN/EboGP to immobilized GPΔmuc by ~80%. This suggests that only 20% of the response exclusively binds regions on GP obstructed by the MLD, specifically the GC and the RBD (Figure 3G).
Adsorption with GPcl had no discernable effect on the antibodies that could bind GP (Figure 3F) or GPΔmuc (~10%–15% residual binding) (Figure 3G), indicating a large proportion of antibodies elicited by these vaccines was likely directed toward the GC or MLD, or other epitopes deleted by cleavage. Similarly, sGP was unable to compete with antibody binding to GP (Figure 3F). This may indicate that responses directed to sGP may not cross-react with GP and preferentially require the unique quaternary structure of sGP. Furthermore, antibodies that are specific for sGP and not GP or GPΔmuc may impede binding of cross-reactive antibodies by sterically hindering their access or occupying partially overlapping footprints shared with the GP- or GPΔmuc-specific antibodies.
sGP preadsorption did compete for a small portion of antibodies binding to GPΔmuc (Figure 3G), most likely to the GC and RBD sites that were shielded by the MLD. The proportion of the LS/APMV2Fc/EboGP, BC/LSFcHN/EboGP, and BC/LSFHN/EboGP response directed to the GC and RBD was ~30%, which is higher than the other vaccines. From this, we can also infer that 70% of the response from these APMV vaccines and 85%–90% of the response from the remaining vaccine groups directed to GPΔmuc may target GP2 or require its trimeric structure. However, given the inability of GPcl preadsorption to inhibit binding to GP and GPΔmuc, GP2-specific antibodies likely comprise a very small proportion of the response compared to antibodies targeting the epitopes lost following cleavage (GC and MLD).
While binding to GP (Figure 3F) and GPΔmuc (Figure 3G) was minimally affected by preabsorption with sGP, these two proteins could adsorb antibodies that recognize sGP, to a greater extent (Figure 3H). This apparent discrepancy in cross-reactivity may be explained by preferential binding or higher affinity for GP and GPΔmuc than sGP. The effect of GPΔmuc was greater overall than GP in blocking binding to sGP, suggesting the presence of sGP-cross-reactive epitopes in the GC and RBD sterically shielded by the MLD of GP. We again observed that BC/LSFcHN/EboGP and BC/LSFHN/EboGP elicit a higher proportion of the response targeting the GC and RBD than other vectors. As expected, GPcl did not appreciably affect binding to sGP (Figure 3H), indicating antibodies were likely binding the GC, absent on GPcl. To measure the level of GC antibodies cross-reactive between the sGP and GP, we subtracted the levels of RBD-targeted antibodies from the levels of antibodies targeting RBD and GC (Figure S4A). The levels of RBD-targeting antibodies were defined as the levels of binding to sGP, inhibited by GPcl (Figure 3H). The levels of RBD- and GC-targeting antibodies were defined as the levels of binding to sGP inhibited by GPΔmuc adsorption. We found that GC antibodies elicited by BC/LSFcHN/EboGP and BC/LSFHN/EboGP were significantly higher than the HPIV vaccines (Figure S4B).
Adsorption with GPΔmuc and GP affected the binding of sera to GPcl to a similar extent; removal of the MLD perhaps only exposed epitopes that are lost upon cleavage (Figure 3I). 25%–40% of the sera derived from HPIVI/EboGP, HPIV3/EboGP, and BC/LSFcHN/EboGP were found to target regions shared between all three forms, specifically the RBD and GP2. However, the RBD of sGP had minimal effect on GPcl binding (Figure 3I) and GP2-specific antibodies likely make up a small portion of the total response (Figures 3F and 3G). Therefore, rather than exclusively binding exposed surfaces of the RBD of GP1 or GP2, it is probable that these antibodies bind favorably to epitopes spanning the trimeric structures of GP and GPΔmuc but are unstable toward GP upon cleavage. Conversely, at least 75% of the antibody pool toward GPcl targets epitopes exposed after cathepsin cleavage.
Adsorption of GP-Specific Antibodies Targeting sGP and GPΔmuc Has a Vaccine-Vector-Dependent Effect on the Neutralization Activity of Sera
We next analyzed if preadsorption of immune sera with sGP and GPΔmuc would abrogate its neutralizing capacity. This was tested in a reverse neutralization assay whereby sera were mixed with the purified proteins at varying concentrations to preadsorb antibodies with potential virus neutralization activity and subsequently identify regions on GP that elicit a neutralizing antibody response. The effect of GPΔmuc interference on antibody-mediated EBOV neutralization differed among the vaccine groups (Figures 4A and 4C). GPΔmuc completely removed neutralization activity of sera from animals vaccinated with HPIVI/EboGP (half maximal effective concentration [EC50] 0.064) and APMVs, indicating that the MLD of the GP expressed by these vectors was not required for eliciting a neutralizing antibody response. These data suggest that these vaccines induce a repertoire of neutralizing antibodies that makes contact with GP2 or the regions masked by the MLD, specifically the GC and the RBD, including the epitope recognized by Niemann-Pick C1 (NPC1), the receptor for EBOV. Unexpectedly, adsorption had only a minimal effect on the sera from animals vaccinated with HPIV3/EboGP (EC50 3.545) and HPIV3/ΔFHN/EboGP (EC50 2.8), reducing neutralization activity by only 25% and 32%, respectively, at the highest concentration of GPΔmuc. These data indicate that the HPIV3-derived vaccine vectors generated a major portion of neutralizing antibodies, which target the MLD.
Figure 4. Epitope Specificity of Neutralizing Antibodies Depends on a Vaccine Vector.

(A and B) Neutralizing activity of immune sera preincubated with GPΔmuc (A) or sGP (B) expressed as percentages of activity of the same sera not preincubated with proteins. Sera from vaccinated groups, diluted to achieve 80% of their neutralization activities, was incubated with increasing concentrations of GPΔmuc or sGP. Data are presented as mean values ± SE.
(C) EC50 values of GPΔmuc required to restore infectivity.
The neutralizing activity of sera from all vaccinated groups was not abrogated by sGP (Figure 4B). Over 90% of sGP’s amino acid sequence is derived from the N-terminal region of GP1, including the MLD-shielded RBD; the protein lacks the MLD and GP2. Depletion of neutralizing activity in the immune sera from HPIV1- and APMV-vaccinated animals by GPΔmuc, but not sGP, suggest that it is directed to GP2, the trimeric structure of GP, and the epitopes sterically shielded by the MLD. These data also suggest that antibody populations specific for sGP and cross-reactive for sGP and GP are not involved in neutralization.
Neutralizing Activity of Immune Sera Correlates with Binding to Epitopes Located in RBD and GP1/GP2 Interface
To evaluate the composition of the anti-GP antibodies in serum targeting specific epitopes, we analyzed competition of monoclonal antibodies (mAbs) with the immune sera for binding to GP by BLI (Figure 5A). Six mAbs recognizing the RBD (EBOV520), GC (BDBV289; amino acids 273–310), MLD (EBOV55), the GP1/GP2 interface (KZ52; discontinuous epitope at amino acids 42, 43, 505–514, and 549–556; Lee et al., 2008a), and membrane proximal external region (MPER) (BDBV223 and BDBV317) were used. All but the MLD-specific mAb neutralize EBOV (Flyak et al., 2016; J.E.C. and A.B., unpublished data). Appreciable differences between the vaccinated groups’ ability to block mAb binding to its targeted epitope were seen in the RBD and MPER regions (Figure 5B). Sera from HPIV3/EboGP- and LS/EboGP-vaccinated animals significantly blocked binding to the RBD epitope compared to the HPIV3/ΔFHN/EboGP and BC/LSFcHN/EboGP groups. Differences in binding to the MPER region was only seen against the BDBV223 epitope and not BDBV317. Competition with BDBV223 was generally poor among the vaccinated groups, with only HPIV3/EboGP sera blocking at least 50% of binding. Little or no differences between sera from various vaccine groups were demonstrated for binding to the other epitopes. Testing of correlation between binding competition and neutralizing ability of the sera demonstrated a strong correlation for the RBD epitope (Figure 5C) and weaker but still significant correlation for the epitope spanning the GP1/GP2 interface (Figure 5D).
Figure 5. Epitope Diversity of Immune Sera Characterized by Competition Assays on the BLI System.
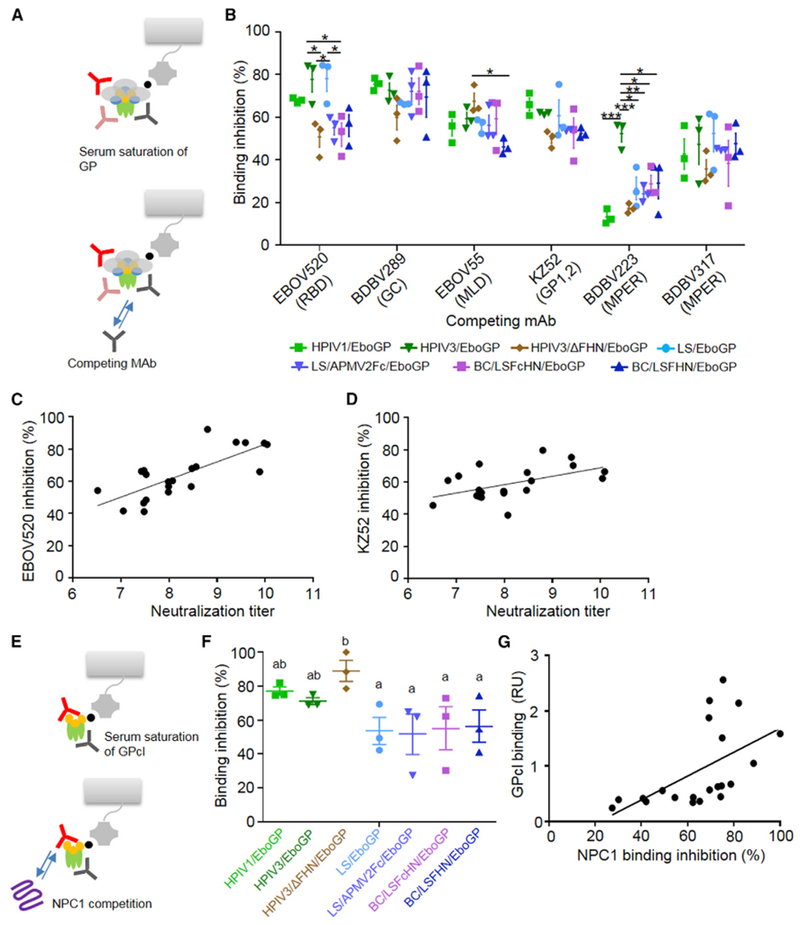
(A) Schematic of BLI-based mAb competition.
(B) Percentage of binding inhibition of mAbs targeting specific regions on GP after saturation with serum. Bars denote average blocking of mAbs by sera from vaccinated groups ± SE. One-way ANOVA with Tukey’s correction (*p < 0.05, **p < 0.01, ***p < 0.001).
(C and D) Positive correlations between neutralizing antibody titers and mAbs EBOV520 (C; Pearson r = 0.7558, p < 0.0001 n = 21) and KZ52 (D; r = 0.5152, p = 0.0084 n = 21).
(E) Schematic of BLI-based NPC1 competition assay.
(F) The average level of NPC1 binding inhibition ± SE, determined as a percentage of blocking activity compared to negative control sera (ANOVA with Fisher’s LSD post hoc test; p < 0.05, groups that do not share a common letter above them).
(G) Positive correlation between serum GPcl binding and NPC1 binding inhibition (Pearson r = 0.5451, p = 0.0053, n = 21).
HPIV Vectors Induce Greater Antibody Response to Blocking Binding to EBOV Receptor NPC1
We used a BLI-based method to analyze the ability of the immune sera to inhibit binding of NPC1 to immobilized GPcl (Figure 5E). Interestingly, immune sera from all three HPIV-based vectors showed greater inhibition than any of the APMV-based vectors (Figure 5F). We observed a correlation between the level of serum binding to GPcl and NPC1 binding inhibition (Figure 5G). The ability of HPIV-based vectors to generate higher levels of GPcl-targeted antibodies (Figure 3C) and greater blocking of NPC1 recognition indicates that they may be more potent in inhibiting the early steps of EBOV life cycle, which includes attachment to the receptor.
Preferential Antibody Reactivity to Linear Epitopes Differs among Vaccine Groups
Serum samples from representative vaccinated groups were examined for their reactivity to linear epitopes by using a set of 167 15-amino-acid-long peptides offset by 4 amino acids, overlapping the entire GP ectodomain, on a microarray platform. The antibody binding patterns, obtained by averaging the peptide mean fluorescent intensities (MFIs) from three animals per group, revealed differences among the vaccine platforms. All vaccinated groups except the BC/APMV3FHN/EboGP, which succumbed to infection, developed peak MFI values of up to 60,000, in two regions, amino acids 417–451 and 465–503, respectively, located in the MLD and just outside the MLD before the furin cleavage site between GP1 and GP2 (Figure 6; Table S1) (the GP regions were mapped according to Lee and Saphire (2009). Three more regions within the MLD were variably recognized (Table S1) and bound at different magnitudes by the vaccinated groups. Most of the regions we identified are consistent with well-characterized immunodominant regions in GP, functioning as footprints for protective or non-protective mouse mAbs (Lee et al., 2008b; Olal et al., 2012; Wilson et al., 2000) and sera from survivors and asymptomatic subjects of the Gabon outbreak (Becquart et al., 2014). However, amino acids 417–451 in the MLD have not yet been defined as a known linear epitope. Other regions recognized by some of the vectors with very weak binding magnitudes with MFI not more than 10,000 were located in the GC and within the MPER and HR2 regions of GP2.
Figure 6. Binding of IgG from Immune Sera to Peptides Encompassing Full-Length GP.

The MFI of binding of immune sera from seven vaccine groups specific to individual GP peptides, plotted against the amino acid start position of each peptide. The vaccine constructs are indicated at top left corner of each plot. Symbols represent the average MFI from 3 vaccinated animals; bars represent ± SE. Black line above GP indicates previously identified epitopes for mAbs and polyclonal serum. See also Figure S5 and Table S1.
To compare the regions preferentially targeted across the different vaccines, we calculated the average MFI of positive peptide binding sorted by regions within GP (Figure S5A). Binding to the C-terminal region of GP1 was significantly greater than to other GP regions for HPIV-based vaccine recipients (p < 0.05 for all comparisons by two-way ANOVA with uncorrected Fisher’s least significant difference [LSD] post hoc test). In contrast, the responses from APMV vaccine groups did not show a preference for the MLD or C terminus of GP1. We examined the breadth of the response by calculating the number of binding sites per GP region for our vaccinated groups (Figure S5B). Despite the preferential binding to the GP1 C-terminal region by HPIV-based recipients compared to APMV recipients, there was no difference in antibody breadth between the groups. Furthermore, more sites were targeted in the MLD by most vaccines, particularly the HPIV3 vectors, yet it was the GP1 C-terminal region that demonstrated a higher magnitude of binding.
DISCUSSION
Deciphering the humoral immune response to vaccination is necessary to understand the requirements for a protective vaccine, and to design and screen next-generation vaccine candidates. Therefore, in addition to efficacy studies, we sought to characterize antibodies elicited upon vaccination with different mucosal vaccine vectors expressing EBOV GP, and subsequently choose the best performers. Typically, vaccine vectors are designed to achieve high-magnitude responses, while vaccine antigens are chosen based on their ability to produce a protective immune response with greater breadth and depth (Stephenson and Barouch, 2013). We challenge this design paradigm by showing here that different mucosal vaccine platforms expressing the same antigen induced qualitatively different antibody responses, which may influence the protective outcome.
Eight out of our nine mucosal vaccines provided 100% protection. High GP-specific IgG and neutralizing antibody titers typically equated with protection. However, HPIV1/ΔFHN/EboGP was the exception, with IgA, rather than IgG or neutralization titers, appearing to be responsible for 100% protection. While this study is in agreement with others that highlight an association between antibodies (Marzi et al., 2013) or specifically IgG (Wong et al., 2012) and survival, we identify a correlation between IgA and protection and suggest IgG may contribute to protection, but they are not an obligatory requirement.
It was unexpected to see a clear distinction between the antibody response profiles of our replicative human and avian paramyxovirus vaccines, given they are members of the Paramyxoviridae family and administered via the same route. A possible reason for this phenomenon is the effect of vector-derived epitopes on epitope hierarchy. A recent study demonstrated a strong immunodominance of adenovirus-derived CD8+ T cell epitopes resulting in the lack of transgene-specific responses (Schöne et al., 2017); in our work, differential effects from the antigenically distinct vector-specific epitopes on the B cell epitope hierarchy, can also be expected. In addition, EBOV-specific responses may be modulated by the properties of the paramyxovirus surface F and HN glycoproteins involved in cell entry and spread, the site of vaccine replication in the respiratory tract, and the ability of infected cells to present antigenic GP peptides to the immune system. The F0 protein of HPIV3 possesses a cleavage site processed intracellularly by host proteases, particularly furin, which is present in most cell types. Consequently, growth is enabled in a wide range of tissues and increases spread within and beyond the respiratory tract. HPIV1 and our avirulent NDV vectors, either lentogenic or modified to adopt lentogenic avirulent properties, require secreted proteases such as trypsin, thus restricting replication to the epithelial lining. However, modifications to the F protein of another paramyxovirus, human metapneumovirus, which afforded independence from secreted proteases, did not affect replication (Biacchesi et al., 2006). Replacement of cleavage sites of APMV serotypes with that of other APMV serotypes including virulent NDV strains, could increase cleavability and replication, but not virulence, in chickens (Panda et al., 2004; Subbiah et al., 2011; Wakamatsu et al., 2006), indicating the F cleavage site is not the sole determinant of virulence. The V protein of NDV moderates the interferon response in a species-dependent manner (Park et al., 2003). Moreover, the V protein of the mesogenic BC strain has a greater antagonistic effect than the lentogenic LS strain (Alamares et al., 2010), which may explain why our LS-derived vectors elicited a slightly weaker response against GP than the protective BC vectors. In this study, dispensing with the F and HN proteins from our HPIV3 vaccine reduced the response magnitude but did not greatly alter antibody profiles from its parental derivative. This suggests other host factors such as receptors or the susceptibility to innate immune responses may also constrain the growth and immunogenicity properties of the vaccine vectors.
The magnitude of sera binding to the different truncated forms of GP was influenced by the vaccine vector: HPIV-based vectors, except HPIV1/ΔFHN/EboGP, generally bound well to all forms of GP compared to the APMV groups; the difference was particularly evident against GPcl (Figure 7). The antibody response profiles elicited toward GP were vector dependent, varying in their reactivity and composition targeting the key GP regions. The MLD is known as an immunodominant region (Dowling et al., 2007; Martinez et al., 2011), encompassing epitopes involved in neutralization and infection enhancement (Davidson et al., 2015; Takada et al., 2007; Wilson et al., 2000). It is also the most divergent region among the EBOV strains and sterically shields more conserved epitopes located in GP1. The proportion of the antibody response directed to MLD varied between the vaccine vector groups; the HPIV3 vaccines had a high ratio of MLD-specific antibodies, which also largely dominated the neutralizing antibody population in sera. While a substantial portion of the HPIV1/ΔFHN/EboGP antibody response targeted the MLD, neutralizing activity was unexpectedly undetectable and may be attributed to overall low IgG titers. A lower portion of the response from APMV and HPIV1/EboGP vaccines was directed toward the MLD with no detectable neutralization activity; neutralization may require contact points unique to GP2 or trimeric GP devoid of the MLD. Competition studies with neutralizing mAbs indicate the neutralization antibody repertoire produced by HPIV3 vaccines may also target regions outside the MLD. However, our restoration of infectivity data suggests the presence of non-neutralizing competitor antibodies. Linear MLD epitopes associated with neutralization and recognized by the HPIV3-vaccine immune sera with predominant MLD-specific neutralization activity were also recognized by sera from APMV and HPIV1/EboGP groups. This suggests different antibody populations, with disparate affinity or activity, target the linear MLD epitopes. The weaker avidities of APMV-derived antibodies toward GP compared to the HPIV3 sera may explain their lack of MLD-targeted neutralization. However, the avidity of APMV-derived serum toward GPΔmuc remained weaker in general compared to the HPIV vaccines, despite non-MLD regions largely accounting for the neutralization capacity of the APMV sera. In the case of HPIV3 serum, tight binding to non-MLD regions could diminish their ability to locate GP functional intermediates or domains; weaker binding may be necessary for non-MLD domain targeted neutralization. Alternately, the higher avidity could be attributed to interfering non-neutralizing antibodies that may not be present in APMV sera. This highlights that the human and avian paramyxoviruses also elicit functionally distinct populations of antibodies, which target the non-MLD regions.
Figure 7. Heatmap Summary of Vaccine Vector Performance.
Summary of the strengths of sera-binding characteristics, epitope recognition, and efficacy of each vaccine vector group.
sGP, which comprises 70% of the GP produced during an EBOV infection (Dolnik et al., 2004), can act as a decoy for neutralizing antibodies targeting epitopes that are shared or inaccessible in GP trimer (Ilinykh et al., 2016; Mohan et al., 2012). We found that our vaccine-induced antibodies specific or cross-reactive for sGP epitopes in the RBD or GC were not involved in neutralization. The failure of sGP to measurably adsorb neutralizing antibodies and compete against binding of sera to GP on the BLI system is consistent with the phenomenon of “antigenic subversion,” where cross-reactivity between sGP and GP is dependent on the exposure to sGP during the induction of the anti-GP response (Mohan et al., 2012). Since the GP ORF in our vaccine constructs lacks the editing site required for sGP expression, the majority of the response elicited following vaccination is likely to be directed to epitopes unique to GP. In the reverse setting, we could measure some competition for serum binding to sGP by adsorption with GP and GP-Δmuc. These cross-reactive antibodies may have a higher affinity for the GP trimeric structures or preferentially bind through more extensive contacts with the trimer than the sGP dimer. Induction of a neutralizing response specific for GP and the lack of susceptibility to decoy effects of sGP or MLD may be important criteria for selecting a vaccine vector.
Competition studies with mAbs revealed diverse sera-binding patterns between the vaccine groups to sites with neutralizing activity: the RBD and MPER (Figure 7). The level of neutralizing antibodies in sera correlated with the level of competition for the neutralizing epitopes of the RBD and GP1/GP2 interface, but not within the GC and MPER regions. The BDBV223 epitope in MPER is completely protective against EBOV in mice and partially protective in guinea pigs (Flyak et al., 2016). The low abundance of antibodies toward the MPER and greater competition variances between the vaccine groups observed for the BDBV223 epitope, suggests the contribution of MPER antibodies to neutralization may be poor or dependent on the host species or vaccine vector.
Even though GC- and RBD-specific antibodies shared between sGP and the GP trimer were not involved in neutralization (Figure 4), we observed competition between the immune sera and BDBV289, a GC-specific neutralizing mAb, which can also bind sGP (Flyak et al., 2016). However, BDBV289 targets an epitope, which overlaps with other GC-specific mAbs that do not bind sGP (Flyak et al., 2016). This suggests the presence of non-sGP cross-reactive serum antibodies, which compete with BDBV289 for the overlapping epitope to restrict or displace binding and do not share the same contact points as BDBV289.
Despite vector-dependent differences observed in the magnitude of the response directed to the MLD, no significant differences were observed in the ability of sera from vaccine groups to compete with a non-neutralizing MLD-specific mAb, EBOV55. HPIV3-based vectors had the highest fraction of their response targeted to the MLD, which largely influenced their neutralization capacity. The fraction of the MLD-specific antibody response elicited by each vaccine group may therefore be a reflection of the abundance of MLD-specific antibodies with neutralization activity.
The vaccine-induced antibodies competed with KZ52 (Figure 5B), a mAb that inhibits the cathepsin-cleavage-dependent conformational changes necessary for the formation of fusogenic GP (Shedlock et al., 2010). The immune sera also interfered with NPC1 recognition by the cleaved, fusion-active form of GP required for entry (Figure 5F). The levels of immune sera interfering with KZ52 or receptor binding respectively correlated with the levels of serum-neutralizing antibodies or antibodies recognizing GPcl, suggesting that the immune sera potentially hampered virus post-attachment steps.
This study provides the most comprehensive comparative evaluation of antibody repertoires to a large panel of mucosal EBOV vaccines to date, and reveals important differences in their epitope specificities; profile differences may result from the cooperative interplay between epitope accessibility, specificity, and affinity for epitopes associated with neutralization, the effect of targeting multiple sites, and interference from non-neutralizing antibodies. HPIV3-based vectors appear to generate a more robust and diverse polyclonal antibody response compared to APMV and HPIV1: all conformational forms of GP (including its fusogenic form) were recognized, GP was targeted with higher avidity, different total and neutralizing antibody repertoires were generated and largely directed toward the MLD, linear protective epitopes were targeted with broader specificity, and the response may interfere with virus receptor recognition to a greater extent. APMV-derived antibodies that recognized the different forms of GP, albeit at lower levels, were more evenly distributed over the GP1 domains and produced a neutralizing antibody repertoire against GP devoid of the MLD. We also demonstrated that one of our vaccines could confer full protection despite producing no detectable neutralizing antibodies. Our data suggest that correlates of protective immunity induced by a single EBOV vaccine, and probably any other vaccine, cannot be universally applied to other vaccines against the same pathogen, even if they utilize the exact same immunogen. Moreover, the shift to use next-generation vaccine vectors, genetically modified or derived from rare serotypes that resist the pressures of preexisting immunity and address safety concerns, may yield new response profiles that should be taken into account when establishing protective correlates. Knowing that breadth and potency of a response, in addition to magnitude, are influenced by the vaccine vector is an important consideration for design and selection of an appropriate vector.
EXPERIMENTAL PROCEDURES
Construction and Recovery of Recombinant Vaccines Expressing EBOV GP
The construction of full-length HPIV3EboGP and HPIV3/ΔFHN/EboGP cDNA was previously described (Bukreyev et al., 2009). To construct HPIV1/EboGP, the GP ORF was amplified by PCR using primers that introduced flanking NotI sites, the gene end (GE) and gene start (GS) signals of P and M, respectively, upstream of GP and a SacI site positioned downstream of GP. The GP PCR product was digested with NotI and cloned into the respective site of the HPIV1 full-length cDNA (FLC) genome plasmid, which placed the GP ORF downstream of HPIV1 P ORF. The F and HN genes from HPIV1 were also deleted to create a construct solely expressing GP as its surface protein, HPIV1/ΔFHN/EboGP. First, we amplified a segment from the HPIV1/EboGP FLC downstream of the inserted GP gene. This segment begins at the newly introduced SacI site, encompasses the M ORF, and ends with an introduced KpnI site inserted directly downstream of the M ORF devoid of its gene signal. We also amplify a segment from the FLC plasmid to encompass a KpnI site, the untranslated region, and the GE signal downstream of the HN ORF and 2,598 nt of the L gene ending at SphI found within the L gene. These two fragments were digested with their flanking enzymes and ligated together via the KpnI site in the pUC19 vector. The ligated insert was digested with SacI and SphI and used to replace the SacI/SphI fragment of HPIV1/EboGP, which contains the F and HN genes. The rescue of HPIV1-derived vaccines was performed as described for HPIV3.
The GP DNA flanked with the NDV GS and GE sequence motifs was amplified using specific primers and inserted between the P and M genes in the FLC of NDV strains LS (Huang et al., 2001), the modified BC (Kim et al., 2014), and modified LS (Figure 1). In modified LS cDNA, the LS/APMV2Fc, F protein cleavage site was changed to that of APMV2 (KPASRF). A BC cDNA was modified to BC/LSFcHN by replacing the HN gene and F protein cleavage site with that of strain LS (Kim et al., 2014). Modified BC/LSFHN and BC/APMV3FHN were created by replacing the F and HN genes of BC with that of LS and the F and HN ectodomains of APMV3, respectively. NDV recombinants were recovered as described previously (Krishnamurthy et al., 2000). All of the NDV recombinants generated in this study were plaque-purified and grown once in the allantoic cavities of 9-day-old specific-pathogen-free embryonated chicken eggs.
All gene deletions, substitutions, and GP gene insertions were confirmed by DNA sequencing of the RT-PCR products using the viral genomes purified from virions as templates. HPIV3- and HPIV1-derived viral titers were determined in LLC-MK2 cells (Bukreyev et al., 2006). NDV titers were similarly determined with the exception of the addition of TrypLE select enzyme (Thermo Fisher) to the culture media. GP protein expression was confirmed in both LLC-MK2 and JH4 clone 1 (ATCC CCL158) cells by western blot (Bukreyev et al., 2006), and growth kinetics was examined by infecting the later cell line at an MOI of 0.1 PFU/cell over 3 days.
Guinea Pig Immunization
All animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Texas Medical Branch (UTMB). Groups of five 4- to 8-week-old female Hartley guinea pigs (Charles River Laboratories) were immunized via the IN route on day 0 with 4 × 105 PFU (100 μL per nostril) of each virus. Blood was collected retro-orbitally on days 0 (pre-bleed) and 27.
EBOV Infection
Vaccinated and control guinea pigs were infected IP with 1,000 PFU of guinea pig adapted EBOV, strain Mayinga (Connolly et al., 1999) and monitored daily for 28 days for survival and clinical symptoms. Weights were measured for the first 14days, then every 3 days until day 28. Serum was collected every 3 days for 12 days and at the termination of the protocol.
Analysis of GP-Specific Antibodies by ELISA
EBOV GP-specific antibody titers were measured by an ELISA as previously described (Meyer et al., 2015), with some modifications. GP-specific IgG and IgA ELISA plates coated with 7.25 ng/well or 50 ng/well, respectively, of purified recombinant EBOV GP protein (IBT Bioservices) were blocked with 3% skim milk in PBS for 1 hr at 37°C. Serially diluted sera were added to the plates, and bound complexes were detected with horseradish peroxidase (HRP)-conjugated anti-guinea pig IgG or IgA for 1 hr at 37°C. The plates were washed and developed with HRP peroxidase substrate solution (KPL), and the signal was read at 630 nm. Specific antibody titers were defined as the reciprocal of the endpoint dilution with an optical density (OD) of 0.2.
Analysis of EBOV-Neutralizing Antibodies
Plaque-reduction neutralization assays were performed as previously described (Meyer et al., 2015).
BLI Analysis of Antibody Binding to Various Forms of GP and Competition Assays
A FortéBio Octet Red96 instrument was used to measure sera reactivity to EBOV GP forms. All assays were performed with agitation at 1,000 rpm, at 28°C in black 96-well plates. Dilutions of samples were prepared in 1× Kinetics buffer (FortéBio) with a final volume of 200 μL/well. Biotinylated EBOV GP, GP-Δmuc, sGP, and GPcl were immobilized onto streptavidin sensors at 2 μg/mL for 300 s. Capture levels were between ~1 nm and 1.5 nm, and variability within a row of eight sensors did not exceed 0.1 nm this step. This was followed by a biocytin treatment of tips for 120 s. Biosensor tips were then equilibrated for 300 s in 1× Kinetics buffer prior to binding measurements. Sera were diluted to 1/40, and binding was assessed for 600 s. Sera depletion was carried out using 2.5 μg GP, GPΔmuc, or sGP and 0.5 μg GPcl. Dissociation for 600 s in 1× Kinetics buffer followed. Parallel corrections for baseline drift were made by subtracting measurements recorded with GP loaded sensors in the absence of sera. To determine nonspecific binding responses, binding of sera from HPIV3-vaccinated animals to GP-variant-loaded probes was monitored and set as the background level. Percent inhibition of binding to immobilized GP following serum adsorption was calculated as: % inhibition = [100 – (binding of serum preadsorped with GP form (RU)/binding of serum without preadsorption (RU))] × 100.
For site-specific antigenicity assessment, biotinylated GP-loaded probes at a capture level of ~0.5 nm were incubated with serum diluted 1/5 in 1× Kinetics buffer for 900 s to generate a saturating signal against the competing mAb. Probes were then washed twice for 60 s before the remaining reactivity of competing mAbs (25 nM BDBV223, EBOV55, and KZ52 and 300 nM BDBV289, EBOV520, and BDBV317) toward GP were assessed for 600 s. Similar steps were performed for competition with NPC1, with the exception that biotinylated GPcl was immobilized at a capture level of ~1.5 nm. Sensors were exposed to 700 nM NPC1-C domain (Sinobiological) following incubation with serum. The level of binding inhibition to GP was calculated as a percentage of the blocking activity of sera from vaccinated animals compared to the negative control sera against the tested mAb or NPC1. Sera from vaccinated groups with barely detectable antibody titers were excluded from octet competition and adsorption assays.
Data analysis and curve fitting were carried out using Octet software, version 7.0. To elucidate dissociation rates, binding equations describing a 1:1 interaction were used.
Diminishing Neutralizing Activity in the Presence of GP Forms
Competition neutralization assays were performed by choosing sera concentrations at which at least 60%–80% neutralization of EBOV was achieved. Vaccinated groups that did not produced neutralizing antibody responses were excluded. The diluted sera were incubated with increasing concentrations of sGP or GPΔmuc. Mixtures were then incubated for 1 hr at 37°C with a recombinant EBOV expressing GFP from an added gene provided by J. Towner and S. Nichol (Centers for Disease Control and Prevention [CDC], Atlanta, GA, USA) (Towner et al., 2005), and a neutralization assay was performed as described above. Restoration of infectivity was determined as a percentage of the plaques formed in the presence of serum incubated with the competing GP forms compared to the serum without the GP forms.
Analysis of Immune Sera for Binding to GP Peptide Array
To identify linear epitopes and the immunodominant sites in GP recognized by the humoral response to vaccination, we performed epitope mapping using peptide microarrays. A microarray slide consisted of 21 blocks to enable analysis of up to 20 samples and one secondary antibody control. Each block was spotted with 167 15-meric peptides offset by 4 amino acids, spanning the 676 amino acids of GP of EBOV Mayinga Zaire 1976 (GenBank: NP 066246), as triplicate subarrays, by J.P.T. Serum diluted 1/200 in wash buffer (J.P.T.) was applied to individual chambers on the slides and incubated for 1 hr at 30°C. Following 4 washes, slides were incubated with 0.1 μg/mL anti-guinea pig Cy5-conjugated antibodies (Jackson ImmunoResearch Laboratories). After additional washes and a final rinse in deionized water, the slide was dried by centrifugation. Slides were scanned with the GenePix 4200AL using the 635 nm laser at 500 PMT and 100 Power settings. The fluorescent intensity for each spot of the array image were analyzed by GenePix Pro 6 (Molecular Devices), and the MFI across the triplicate sub-arrays for each block was calculated and normalized by subtraction from the secondary antibody control. Sera from 3 animals per group were tested, and normalized MFIs for each peptide were corrected for baseline by subtracting the corresponding pre-vaccination MFIs.
Statistics
Statistical comparisons between groups were made using one-way or two-way ANOVA with post hoc Tukey’s or uncorrected Fisher’s LSD tests (Prism version 6, GraphPad Software) as specified in the figure legends. A p value of less than 0.05 was considered significant.
Supplementary Material
Highlights.
Next-generation respiratory Ebola virus vaccines are protective in guinea pigs
Ebola-specific IgA correlates with protection
Different vaccines expressing the same immunogen induce different antibody profiles
Correlates of protection may not be universal among Ebola virus vaccines
ACKNOWLEDGMENTS
This work is supported by the National Institutes of Health (grant number 1R01AI102887-01A1) (A.B.). We would like to thank Jessica Graber, Dr. Curtis Klagis, and the staff of Animal Resources Center at UTMB for contributing to the animal study procedures.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures and one table and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.07.044.
DECLARATION OF INTERESTS
P.L.C., S.S., and A.B. have patents related to this work. The remaining authors declare no competing interests.
REFERENCES
- Alamares JG, Elankumaran S, Samal SK, and Iorio RM (2010). The interferon antagonistic activities of the V proteins from two strains of Newcastle disease virus correlate with their known virulence properties. Virus Res. 147, 153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander DJ (2000). Newcastle disease and other avian paramyxoviruses. Rev. Sci. Tech. 19, 443–462. [DOI] [PubMed] [Google Scholar]
- Becquart P, Mahlako˜ iv T, Nkoghe D, and Leroy EM (2014). Identification of continuous human B-cell epitopes in the VP35, VP40, nucleoprotein and glycoprotein of Ebola virus. PLoS ONE 9, e96360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biacchesi S, Pham QN, Skiadopoulos MH, Murphy BR, Collins PL, and Buchholz UJ (2006). Modification of the trypsin-dependent cleavage activation site of the human metapneumovirus fusion protein to be trypsin independent does not increase replication or spread in rodents or nonhuman primates. J. Virol. 80, 5798–5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukreyev A, Huang Z, Yang L, Elankumaran S, St Claire M, Murphy BR, Samal SK, and Collins PL (2005). Recombinant newcastle disease virus expressing a foreign viral antigen is attenuated and highly immunogenic in primates. J. Virol. 79, 13275–13284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukreyev A, Yang L, Zaki SR, Shieh WJ, Rollin PE, Murphy BR, Collins PL, and Sanchez A (2006). A single intranasal inoculation with a paramyxovirus-vectored vaccine protects guinea pigs against a lethal-dose Ebola virus challenge. J. Virol. 80, 2267–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukreyev A, Rollin PE, Tate MK, Yang L, Zaki SR, Shieh WJ, Murphy BR, Collins PL, and Sanchez A (2007). Successful topical respiratory tract immunization of primates against Ebola virus. J. Virol. 81, 6379–6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukreyev A, Marzi A, Feldmann F, Zhang L, Yang L, Ward JM, Dorward DW, Pickles RJ, Murphy BR, Feldmann H, and Collins PL (2009). Chimeric human parainfluenza virus bearing the Ebola virus glycoprotein as the sole surface protein is immunogenic and highly protective against Ebola virus challenge. Virology 383, 348–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukreyev AA, Dinapoli JM, Yang L, Murphy BR, and Collins PL (2010). Mucosal parainfluenza virus-vectored vaccine against Ebola virus replicates in the respiratory tract of vector-immune monkeys and is immunogenic. Virology 399, 290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC (2016). 2014 Ebola Outbreak in West Africa - Case Counts.
- Chandran K, Sullivan NJ, Felbor U, Whelan SP, and Cunningham JM (2005). Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 308, 1643–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly BM, Steele KE, Davis KJ, Geisbert TW, Kell WM, Jaax NK, and Jahrling PB (1999). Pathogenesis of experimental Ebola virus infection in guinea pigs. J. Infect. Dis. 179 (Suppl 1), S203–S217. [DOI] [PubMed] [Google Scholar]
- Davidson E, Bryan C, Fong RH, Barnes T, Pfaff JM, Mabila M, Rucker JB, and Doranz BJ (2015). Mechanism of Binding to Ebola Virus Glycoprotein by the ZMapp, ZMAb, and MB-003 Cocktail Antibodies. J. Virol. 89, 10982–10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNapoli JM, Yang L, Samal SK, Murphy BR, Collins PL, and Bukreyev A (2010). Respiratory tract immunization of non-human primates with a Newcastle disease virus-vectored vaccine candidate against Ebola virus elicits a neutralizing antibody response. Vaccine 29, 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolnik O, Volchkova V, Garten W, Carbonnelle C, Becker S, Kahnt J, Stro¨ her U, Klenk HD, and Volchkov V (2004). Ectodomain shedding of the glycoprotein GP of Ebola virus. EMBO J. 23, 2175–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling W, Thompson E, Badger C, Mellquist JL, Garrison AR, Smith JM, Paragas J, Hogan RJ, and Schmaljohn C (2007). Influences of glycosylation on antigenicity, immunogenicity, and protective efficacy of ebola virus GP DNA vaccines. J. Virol. 81, 1821–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flyak AI, Shen X, Murin CD, Turner HL, David JA, Fusco ML, Lampley R, Kose N, Ilinykh PA, Kuzmina N, et al. (2016). Cross-reactive and potent neutralizing antibody responses in human survivors of natural Ebolavirus infection. Cell 164, 392–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Krishnamurthy S, Panda A, and Samal SK (2001). High-level expression of a foreign gene from the most 3′-proximal locus of a recombinant Newcastle disease virus. J. Gen. Virol. 82, 1729–1736. [DOI] [PubMed] [Google Scholar]
- Ilinykh PA, Shen X, Flyak AI, Kuzmina N, Ksiazek TG, Crowe JE Jr., and Bukreyev A (2016). Chimeric filoviruses for identification and characterization of monoclonal antibodies. J. Virol. 90, 3890–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy SB, Bolay F, Kieh M, Grandits G, Badio M, Ballou R, Eckes R, Feinberg M, Follmann D, Grund B, et al. ; PREVAIL I Study Group (2017). Phase 2 placebo-controlled trial of two vaccines to prevent Ebola in Liberia. N. Engl. J. Med. 377, 1438–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Paldurai A, Xiao S, Collins PL, and Samal SK (2014). Modified Newcastle disease virus vectors expressing the H5 hemagglutinin induce enhanced protection against highly pathogenic H5N1 avian influenza virus in chickens. Vaccine 32, 4428–4435. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Krishnamurthy S, Huang Z, and Samal SK (2000). Recovery of a virulent strain of newcastle disease virus from cloned cDNA: expression of a foreign gene results in growth retardation and attenuation. Virology 278, 168–182. [DOI] [PubMed] [Google Scholar]
- Kuhn JH (2008). Filoviruses, First Edition (Springer; ). [Google Scholar]
- Kusagawa S, Komada H, Mao X, Kawano M, Nishikawa F, Tsurudome M, Matsumura H, Ohta H, Yuasa T, Nishio M, et al. (1993). Antigenic and molecular properties of Murayama virus isolated from cynomolgus monkeys: the virus is closely related to avian paramyxovirus type 2. Virology 194, 828–832. [DOI] [PubMed] [Google Scholar]
- Lee JE, and Saphire EO (2009). Ebolavirus glycoprotein structure and mechanism of entry. Future Virol. 4, 621–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, and Saphire EO (2008a). Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 454, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Kuehne A, Abelson DM, Fusco ML, Hart MK, and Saphire EO (2008b). Complex of a protective antibody with its Ebola virus GP peptide epitope: unusual features of a V lambda × light chain. J. Mol. Biol. 375, 202–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez O, Tantral L, Mulherkar N, Chandran K, and Basler CF (2011). Impact of Ebola mucin-like domain on antiglycoprotein antibody responses induced by Ebola virus-like particles. J. Infect. Dis. 204 (Suppl 3), S825–S832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzi A, Engelmann F, Feldmann F, Haberthur K, Shupert WL, Brining D, Scott DP, Geisbert TW, Kawaoka Y, Katze MG, et al. (2013). Antibodies are necessary for rVSV/ZEBOV-GP-mediated protection against lethal Ebola virus challenge in nonhuman primates. Proc. Natl. Acad. Sci. USA 110, 1893–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, Garron T, Lubaki NM, Mire CE, Fenton KA, Klages C, Olinger GG, Geisbert TW, Collins PL, and Bukreyev A (2015). Aerosolized Ebola vaccine protects primates and elicits lung-resident T cell responses. J. Clin. Invest. 125, 3241–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan GS, Li W, Ye L, Compans RW, and Yang C (2012). Antigenic subversion: a novel mechanism of host immune evasion by Ebola virus. PLoS Pathog. 8, e1003065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olal D, Kuehne AI, Bale S, Halfmann P, Hashiguchi T, Fusco ML, Lee JE, King LB, Kawaoka Y, Dye JM Jr., and Saphire EO (2012). Structure of an antibody in complex with its mucin domain linear epitope that is protective against Ebola virus. J. Virol. 86, 2809–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda A, Huang Z, Elankumaran S, Rockemann DD, and Samal SK (2004). Role of fusion protein cleavage site in the virulence of Newcastle disease virus. Microb. Pathog. 36, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MS, García-Sastre A, Cros JF, Basler CF, and Palese P (2003). Newcastle disease virus V protein is a determinant of host range restriction. J. Virol. 77, 9522–9532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson JS, Dekker JD, Croyle MA, and Kobinger GP (2010). Recent advances in Ebolavirus vaccine development. Hum. Vaccin. 6, 439–449. [DOI] [PubMed] [Google Scholar]
- Sanchez A, Kiley MP, Holloway BP, and Auperin DD (1993). Sequence analysis of the Ebola virus genome: organization, genetic elements, and comparison with the genome of Marburg virus. Virus Res. 29, 215–240. [DOI] [PubMed] [Google Scholar]
- Sanchez A, Trappier SG, Mahy BW, Peters CJ, and Nichol ST (1996). The virion glycoproteins of Ebola viruses are encoded in two reading frames and are expressed through transcriptional editing. Proc. Natl. Acad. Sci. USA 93, 3602–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöne D, Hrycak CP, Windmann S, Lapuente D, Dittmer U, Tenbusch M, and Bayer W (2017). Immunodominance of adenovirus-derived CD8+ T cell epitopes interferes with the induction of transgene-specific immunity in adenovirus-based immunization. J. Virol. 91, e01184–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shedlock DJ, Bailey MA, Popernack PM, Cunningham JM, Burton DR, and Sullivan NJ (2010). Antibody-mediated neutralization of Ebola virus can occur by two distinct mechanisms. Virology 401, 228–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson KE, and Barouch DH (2013). A global approach to HIV-1 vaccine development. Immunol. Rev. 254, 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbiah M, Khattar SK, Collins PL, and Samal SK (2011). Mutations in the fusion protein cleavage site of avian paramyxovirus serotype 2 increase cleavability and syncytium formation but do not increase viral virulence in chickens. J. Virol. 85, 5394–5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada A, Ebihara H, Feldmann H, Geisbert TW, and Kawaoka Y (2007). Epitopes required for antibody-dependent enhancement of Ebola virus infection. J. Infect. Dis. 196 (Suppl 2), S347–S356. [DOI] [PubMed] [Google Scholar]
- Towner JS, Paragas J, Dover JE, Gupta M, Goldsmith CS, Huggins JW, and Nichol ST (2005). Generation of eGFP expressing recombinant Zaire ebolavirus for analysis of early pathogenesis events and high-throughput antiviral drug screening. Virology 332, 20–27. [DOI] [PubMed] [Google Scholar]
- Volchkov VE, Becker S, Volchkova VA, Ternovoj VA, Kotov AN, Netesov SV, and Klenk HD (1995). GP mRNA of Ebola virus is edited by the Ebola virus polymerase and by T7 and vaccinia virus polymerases. Virology 214, 421–430. [DOI] [PubMed] [Google Scholar]
- Volchkova VA, Feldmann H, Klenk HD, and Volchkov VE (1998). The nonstructural small glycoprotein sGP of Ebola virus is secreted as an antiparallel-orientated homodimer. Virology 250, 408–414. [DOI] [PubMed] [Google Scholar]
- Wakamatsu N, King DJ, Seal BS, Peeters BP, and Brown CC (2006). The effect on pathogenesis of Newcastle disease virus LaSota strain from a mutation of the fusion cleavage site to a virulent sequence. Avian Dis. 50, 483–488. [DOI] [PubMed] [Google Scholar]
- Wilson JA, Hevey M, Bakken R, Guest S, Bray M, Schmaljohn AL, and Hart MK (2000). Epitopes involved in antibody-mediated protection from Ebola virus. Science 287, 1664–1666. [DOI] [PubMed] [Google Scholar]
- Wong G, Richardson JS, Pillet S, Patel A, Qiu X, Alimonti J, Hogan J, Zhang Y, Takada A, Feldmann H, and Kobinger GP (2012). Immune parameters correlate with protection against ebola virus infection in rodents and nonhuman primates. Sci. Transl. Med 4, 158ra146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang ZY, Duckers HJ, Sullivan NJ, Sanchez A, Nabel EG, and Nabel GJ (2000). Identification of the Ebola virus glycoprotein as the main viral determinant of vascular cell cytotoxicity and injury. Nat. Med. 6, 886–889. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.