

Abstract
Airway epithelial cells (AECs) orchestrate inflammatory responses to airborne irritants that enter the respiratory system. A viscous mucus layer produced by goblet cells in the airway epithelium also contributes to a physiological defense mechanism through the physical and chemical barriers it provides. Dysregulation or impairment in these functions has been implicated as a cause of the chronic inflammation and tissue remodeling that constitute major pathological features of asthma. In particular, mucus hypersecretion leading to airway obstruction and impaired pulmonary function is associated with morbidity and mortality in asthma patients. Peroxisome proliferator-activated receptor γ (PPARγ) is a ligand-activated transcription factor involved in a variety of cellular processes. Accumulating evidence indicates that PPARγ agonists antagonize exaggerated inflammatory responses, yet PPARγ’s precise role in airway remodeling/mucus hypersecretion has yet to be defined. In this study, we created an AEC-specific PPARγ deletion to investigate PPARγ’s functions in a murine model of allergic airway disease. AEC-specific PPARγ deficiency exaggerated airway hyperresponsiveness, inflammation, cytokine expression, and tissue remodeling. We also found that PPARγ directly bound to a PPAR response element found in MUC5AC and repressed gene expression. Likewise, PPARγ regulated mucin and inflammatory factors in primary human bronchial epithelial cells. In light of the current standard therapies’ limited and inadequate direct effect on airway mucus hypersecretion, our study showing AEC-PPARγ’s role as a transcriptional repressor of MUC5AC highlights this receptor’s potential as a pharmacological target for asthma.
Keywords: Asthma, MUC5AC, Airway remodeling, Transcriptional regulation, Pulmonary disease
Introduction
Asthma, estimated to affect 300 million individuals worldwide (1), is a common inflammatory airway disease and a leading cause of morbidity (2). Currently, corticosteroids and β2-adrenergic receptor agonists constitute standard asthma therapy (3). Although most patients’ symptoms are kept under control and lung function is improved by these drugs, other individuals experience exacerbations or treatment-related adverse effects. Alternative therapies are thus needed.
Epithelial cells protect the airway and act as the first line of defense against airborne pathogens and irritants (4). In addition, a viscous mucus layer produced by goblet cells, secretory cells found in the airway epithelium, and by submucosal glands maintains the health of the airways (4). The pathological aberrations characteristic of asthma, such as chronic inflammation, tissue remodeling, hyperresponsiveness, and mucus hypersecretion, are tied to dysregulation of airway epithelial functions (2, 5–7). In response to allergen exposure, airway epithelial cells (AECs) secrete cytokines (e.g., thymic stromal lymphopoietin [TSLP], interleukin (IL)-25, and IL-33) that recruit various cells of the immune system and thus drive airway inflammation (2). This inflammatory response contributes to airway hyperresponsiveness as well as epithelial tissue remodeling characterized by airway wall thickening, epithelial hyperplasia, and goblet cell metaplasia (2). MUC5AC, one of the two major airway mucin genes (8, 9), becomes significantly upregulated (8, 10), causing mucus hypersecretion that leads to airway obstruction and impaired pulmonary function (4, 6). The Th2 cytokine IL-13 has been implicated as a prominent driver of all these airway epithelial aberrations (9, 11). Thus, AECs are at the core of asthma pathophysiology.
Peroxisome proliferator-activated receptors (PPARs) are a family of ligand-activated transcription factors, comprising PPARα, PPARβ/δ, and PPARγ. Well known for their roles in lipid and glucose metabolism (12), PPARs are also involved in a variety of other cellular processes including inflammatory and immune responses. Accumulating experimental and clinical studies attest to the anti-inflammatory properties of PPAR agonists, highlighting these molecules’ therapeutic potential for treatment of inflammatory lung diseases (12–15). In allergic airway disease (AAD) specifically, PPARγ agonists have been characterized more extensively than those of PPARα or PPARβ/δ; they are known to suppress airway inflammation, remodeling, hyperresponsiveness, and mucus hypersecretion (12, 13, 15–18).
The function of AEC-specific PPARγ (AEC-PPARγ) as a negative immunomodulatory agent has been studied extensively, yet data regarding its role in airway remodeling remain scarce. As pathological tissue remodeling is a prominent feature of asthma, better understanding of its pathogenesis could contribute to patients’ improved clinical outcomes. In the current study we further define the role of PPARγ in AEC regulation using an AEC-specific conditional PPARγ knockout in a murine AAD model. We also examine PPARγ’s control of mucin and inflammatory mediators in primary human bronchial epithelial (HBE) cells.
Materials and Methods
Cells
Normal and diseased (asthmatic) HBE (NHBE and DHBE, respectively) cells were obtained from Lonza (Walkersville, MD). NHBE cells were grown and maintained in BEGM (Lonza) supplemented with growth factors and hormones, as described in the manufacturer’s instructions. Incubation was at 37°C in a humidified atmosphere of 5% CO2-95% air using T-75 tissue culture flasks, plates, or dishes. Treatment with OA-NO2 (Cayman Chemical, Ann Arbor, MI) was as described previously (19).
Animals
AEC-specific PPARγ knockout (AEC-PPARγ−/−) mice on a C57BL/6 genetic background were generated by breeding Cc10Cre mice (20) with Ppargfl/fl mice. Wildtype (WT; Cc10wtPpargfl/fl) and AEC-PPARγ−/− (Cc10CrePpargfl/fl) mice were expanded from breeding pairs. Mice were housed in microisolator cages under specific pathogen-free conditions and fed autoclaved food. Male mice aged 6–8 weeks (20–25 g) were used in all experiments. All studies were performed according to protocols reviewed and approved by the VA Pittsburgh Healthcare System Institutional Animal Care and Use Committee.
OVA sensitization, challenge, and specimen collection
Allergen-induced airway disease was produced in mice as described previously (19, 21). Twenty-four hours following the last challenge, airway hyperresponsiveness to methacholine challenge was measured (19). The mice were then euthanized and bronchoalveolar lavage fluid (BALF) and the lungs were harvested for histopathological examination. Staining and pathological scorings were performed as described previously (19). Image segmentation and analysis were performed with MATLAB Image Processing Toolbox (R2017b, MathWorks, Natick, MA).
Measurement of BALF immunoglobulin, cytokine, and chemokine levels
AAD-induced, OVA-specific IgE and IgG1 levels in BALF were measured using ELISA kits (Cayman Chemical) according to the manufacturer’s instructions. Levels of IL-5, IL-13, eotaxin, and TSLP in BALF were measured using ELISA kits (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions.
Laser capture microdissection, RNA isolation, and real-time RT-PCR
AECs were obtained from lung sections prepared on polyethylene naphthalate membrane slides using an LMD7000 laser microdissection system (Leica Microsystems, Buffalo Grove, IL). RNA was isolated using the RNeasy Mini kit (Qiagen, Valencia, CA). cDNA was then generated from 100 ng of total RNA or mRNA using MultiScribe Reverse Transcriptase (Applied Biosystems, Foster City, CA) with random and oligo-dT primers. Real-time RT-PCR was performed using 100 ng of cDNA with 2X SYBR Green Master Mix (Applied Biosystems) and specific primer sets for the genes of interest (Supplemental Table 1) as described previously (22).
Western blotting
Protein concentrations were determined using the BCA Protein Assay Kit (Pierce, Rockford, IL). Western blotting was then performed as described previously (22). Primary antibodies against PPARγ (7196), MUC5AC (20118), NF-κB/p65 (372), Lamin B1 (20682) and β-Actin (1616) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). TSLP antibody (97630) was obtained from Cell Signaling Technology (Beverly, MA). The secondary antibodies, donkey anti-goat IRDye 680RD (926–68074) and goat anti-rabbit IRDye 800CW (925–32211), were from LI-COR Biosciences (Lincoln, NE). The infrared signal was detected using an Odyssey Infrared Imager (LI-COR Biosciences).
Bioinformatics analysis
A genomic DNA sequence of ~2.0 kb located immediately upstream from the transcription start site of the human MUC5AC gene (Gene ID: 4586) was obtained from GenBank. The sequence was analyzed to predict the putative promoter region and to identify putative PPAR response elements (PPREs) by manual inspection using UGENE (Unipro) software or by using MatInspector.
Generation of MUC5AC promoter constructs and transient transfection assay
The ~2.0 kb DNA sequence located immediately upstream from the transcription start site of the human MUC5AC gene, which is the segment harboring the promoter region, was amplified by PCR from human genomic DNA and cloned into the luciferase-containing pGL4 vector (Promega, Madison, WI). In some plasmids, PPRE1, PPRE2, or both elements were deleted. All constructs were sequence-verified and used as indicated. Transient transfection and luciferase activity assays were performed as previously described (22). Briefly, cells were transfected with the indicated plasmids using Lipofectamine 3000 Reagent (Invitrogen) according to the manufacturer’s instructions. Luciferase activity was then measured using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions.
DNA affinity precipitation
Following washing with cold binding and washing (B&W) buffer (B&W buffer: 10 mM Tris-HCl, pH 7.5, 1 mM EDTA, 2 M NaCl, and 1% protease inhibitor mixture), Dynabeads M-280 Streptavidin (Invitrogen) were mixed with 0.5 μM of the indicated biotinylated PPRE-containing oligonucleotides (Supplemental Table 1) and then incubated at room temperature for 30 min. Next, 200 μg of the biotinylated oligonucleotide-coupled beads were added to 30 μg of nuclear proteins and incubated for 30 min at room temperature with rotation. Samples were washed three times with cold B&W buffer. Bead-bound biotinylated oligonucleotide-protein complexes were eluted and subjected to Western blotting as described with indicated antibodies. An aliquot of the nuclear sample that was not incubated with streptavidin beads was used as the input control sample.
EMSA
PPARγ recombinant protein (1 μg) was incubated with 50 nM of double-stranded oligonucleotides end-labeled with infrared dye 700 as indicated (Supplemental Table 1); the incubation medium consisted of binding buffer (100 mM Tris, 500 mM KCl, and 10 mM DTT, pH 7.5), poly(deoxyinosinic-deoxycytidylic) (poly(dI-dC)) (1 μg/μl in 10 mM Tris, and 1 mM EDTA), 25 mM DTT, and 2.5% Tween 20. Samples were then separated on 5% non-denaturing polyacrylamide gels in 1X Tris-Borate EDTA buffer (130 mM Tris, pH 8.3, 45 mM boric acid, and 2.5 mM EDTA). The infrared signal was detected using an Odyssey Infrared Imager (LI-COR).
Nucleic acid-protein docking studies
Docking studies were performed to predict the interactions between PPARγ-DBD and MUC5AC PPRE1 (AGGACAAAGGGCC) or consensus PPRE (AGGTCAAAGGTCA). Discovery Studio software (BIOVIA, San Diego, CA) was used to prepare and apply CHARMM force field to the DNA corresponding to MUC5AC PPRE1 and PPARγ-DBD (PDB ID: 3E00) for modelling. Docking was performed in the nucleic acid-protein docking server as described previously (23) with default docking parameters. Root-mean-square deviation (RMSD) threshold in the clustering procedure was set to 5 Å. After refinement, best-scored decoys from three biggest clusters were analyzed and best pose (highest probability) was used to identify DNA-protein interactions using Discovery Studio software (BIOVIA).
Statistical analysis
Data are presented as the mean ± SD. Differences between groups were analyzed using unpaired t-test or analysis of variance, followed by a Bonferroni multiple comparison correction. Analyses were performed using GraphPad Prism 5.03 (GraphPad Software, La Jolla, CA). P < 0.05 was considered significant.
Results
AEC-PPARγ knockout aggravates AAD-induced airway hyperresponsiveness and inflammation
To investigate the effects of AEC-PPARγ deficiency in AAD induced via OVA sensitization and challenge, as described previously (19, 21), we developed AEC-PPARγ−/− mice. Inflammatory responses, tissue remodeling, hyperresponsiveness, and mucus hypersecretion in these animals’ airways were then examined and compared with those of WT mice.
We first determined the functional effects of AEC-PPARγ knockout by assessing total respiratory resistance (respiratory system resistance to airflow) as a measurement of airway sensitivity, both at baseline and in response to increasing doses of inhaled methacholine. The AEC-PPARγ−/− mice developed substantial responsiveness even at the lowest dose of inhaled methacholine (6.25 mg/ml) (Fig. 1A). Furthermore, the methacholine responsiveness of the AEC-PPARγ−/− mice was considerably higher than that of WT mice at all doses tested (Fig. 1A).
FIGURE 1. AEC-PPARγ knockout aggravates AAD-induced airway hyperresponsiveness to methacholine and inflammatory cell infiltration.
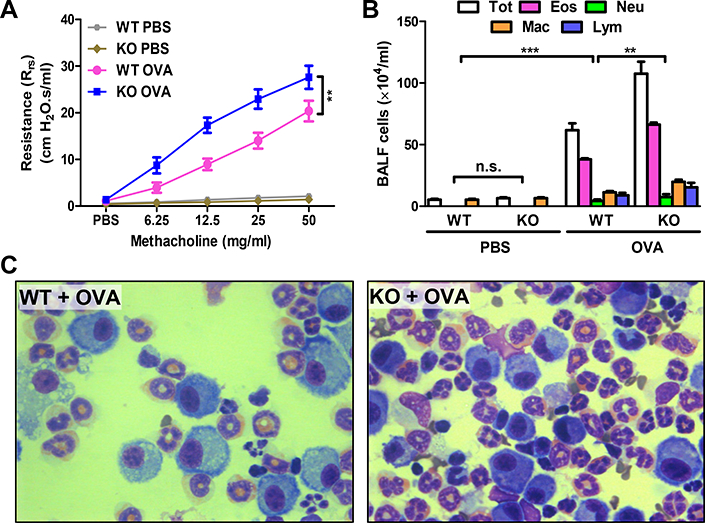
AAD was induced in WT and AEC-PPARγ−/− mice by OVA sensitization on days 0 and 7 and challenge on even-numbered days 14–22. Control mice were similarly treated with PBS. Twenty-four hours later, total respiratory resistance to increasing concentrations of inhaled methacholine was determined. BALF and lung samples were also obtained. (A) Total respiratory resistance. (B) Total (Tot) and differential (Eos = eosinophils; Neu = neutrophils; Mac = macrophages; and Lym = lymphocytes) cell count in BALF. (C) Photomicrographs (100×) of Diff-Quik-stained cells in BALF from the indicated treatment groups. The data are expressed as the mean ± SD with n = 6 mice/group. The results were reproduced at least two times independently; **P < 0.01, ***P < 0.001, n.s. = non-significant.
To determine the effect of AEC-PPARγ deficiency on AAD-associated inflammatory cell infiltration, the number and type of cells in BALF were assessed. Total and differential BALF cell counts were increased by approximately 70% (P < 0.01) compared with those in WT mice (Fig. 1B, 1C). This was attributable to increased numbers of eosinophils, macrophages, and lymphocytes (Fig. 1B). These results suggest that AEC-PPARγ deficiency impairs pulmonary function and exacerbates inflammation in mice with AAD.
AEC-PPARγ knockout aggravates AAD-induced airway inflammation and airway remodeling
Pathological features of asthma include airway inflammatory cell infiltration, remodeling, and goblet cell metaplasia. Microscopic examination of H&E stained lung sections showed that the airways and blood vessels in AEC-PPARγ−/− mice were lined with infiltrating leukocytes (Fig. 2A, yellow arrowheads), whereas leukocytes were rarely observed along the airways in WT mice. Examination at higher magnification showed that infiltrating leukocytes were mostly eosinophils (Fig. 2B, yellow arrowheads). Histological examination of lung sections from AEC-PPARγ−/− mice also displayed focal bronchiolar epithelial cell hyperplasia and subepithelial fibrosis (Fig. 2A, red arrowheads). In addition, we observed that goblet cell metaplasia often accompanied epithelial cell hyperplasia (Fig. 2B). To further examine this goblet cell metaplasia, we stained lung sections with PAS. Although PAS-positive cells were detected in areas of goblet cell metaplasia and in the airways of both groups following AAD induction (Fig. 2C), airways containing granulated goblet cells and mucus plugs were more abundant in AEC-PPARγ−/− mice than in WT mice (Fig. 2D). These results indicate that AAD-associated inflammation and airway remodeling, represented by epithelial cell hyperplasia and goblet cell metaplasia, are significantly pronounced in the absence of AEC-PPARγ.
FIGURE 2. AEC-PPARγ knockout aggravates AAD-induced inflammatory cell infiltration, airway remodeling, and mucus production.
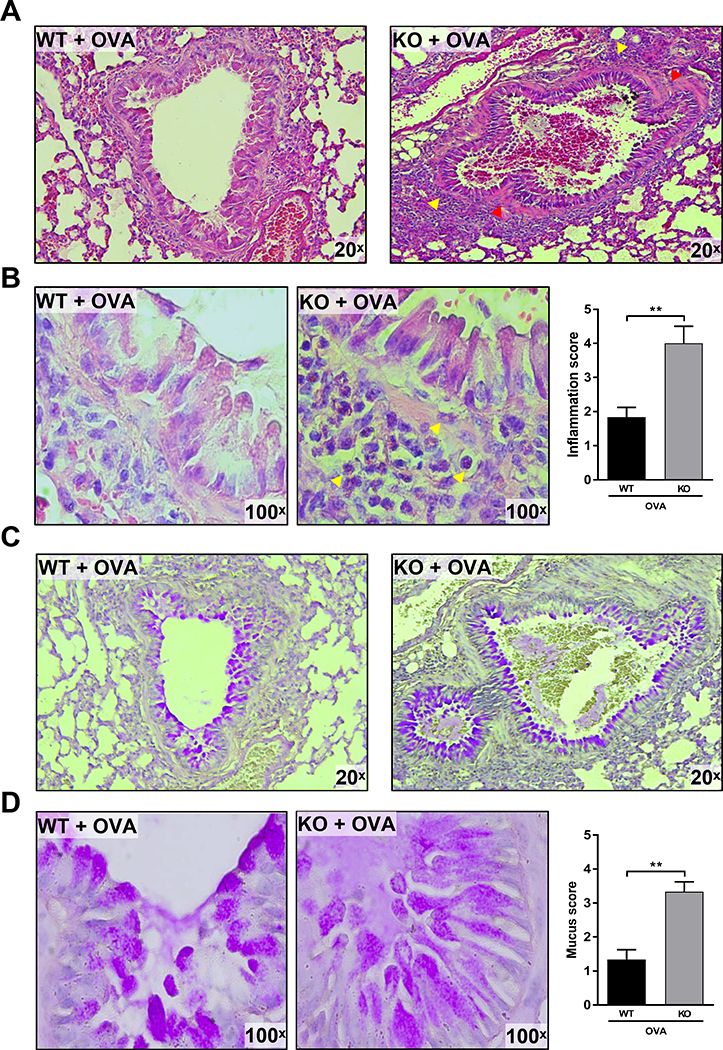
AAD was induced in WT and AEC-PPARγ−/− mice by OVA sensitization on days 0 and 7 and challenge on even-numbered days 14–22. Control mice were similarly treated with PBS. Twenty-four hours later, lung sections from the indicated treatment groups were prepared and examined histologically. (A) H&E staining showing inflammation with infiltrating leukocytes. Red arrowheads indicate epithelial cell hyperplasia and subepithelial fibrosis. (B) 100× images of the same H&E-stained slides showing the infiltrating eosinophils more clearly. Yellow arrowheads indicate leukocytes infiltrating across the airways. Leukocyte infiltration was quantified and presented as inflammation score. (C) PAS-stained slides highlighting goblet cell metaplasia (PAS-positive cells). (D) 100× images of the same PAS-stained slides showing mucus plugs and granulated goblet cells. Mucus staining was quantified and presented as mucus score. The results were reproduced at least two times independently; **P < 0.01.
AEC-PPARγ regulates secretion of asthma-associated cytokines and chemokines in BALF by controlling inflammation and mucin mRNA expression in AECs
Airway inflammation promotes secretion of several inflammatory mediators from different cell types including AECs (2, 24). To investigate whether AEC-PPARγ regulates AAD-induced cytokine and chemokine secretion as well as immunoglobulin production, we assessed the levels of the asthma-associated pro-inflammatory mediators IL-5, IL-13, eotaxin, TSLP, IgE, and IgG1 in BALF. We found no difference between the two groups at baseline, however, all six molecules were significantly elevated in AEC-PPARγ−/− compared to WT mice following AAD induction (Fig. 3).
FIGURE 3. AEC-PPARγ knockout increases markers of AAD-induced lung inflammation.
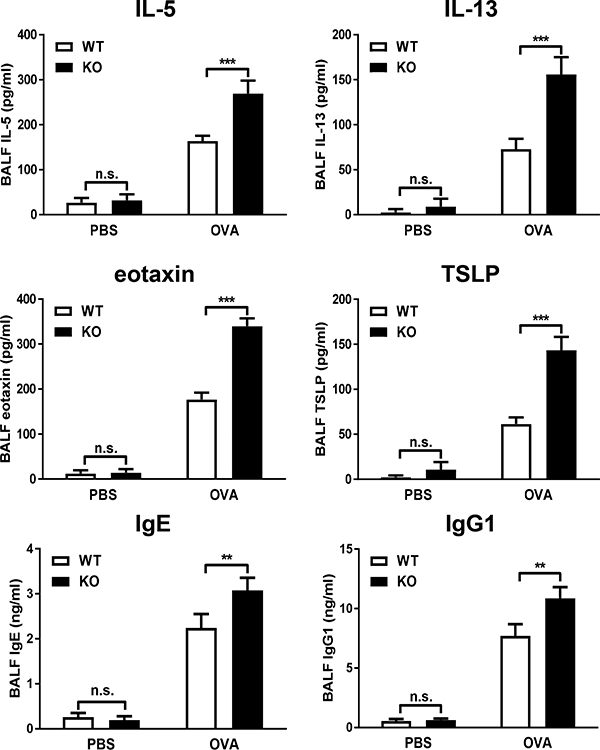
AAD was induced in WT and AEC-PPARγ−/− mice by OVA sensitization on days 0 and 7 and challenge on even-numbered days 14–22. Control mice were similarly treated with PBS. Twenty-four hours later, BALF samples were obtained and indicated cytokine, chemokine, and immunoglobulin levels (IL-5, IL-13, eotaxin, TSLP, IgE, and IgG1) were measured by ELISA. The data are expressed as the mean ± SD with n = 6 mice/group. The results were reproduced at least two times independently; **P < 0.01, ***P < 0.001, n.s. = non-significant.
Next, to determine whether regulation of inflammatory mediators by AEC-PPARγ (Supplemental Fig. S1) was executed at the transcriptional level, we examined the expression of inflammatory genes (Il25, Il33, Tslp, Ccl11 [corresponding to eotaxin], and Rela [corresponding to NF-κB/p65]), as well as the mucin gene Muc5ac, by real-time RT-PCR using samples isolated from AECs. AAD-induced elevation of all the examined genes was further increased in AEC-PPARγ−/− mice compared with WT animals (Fig. 4). These data suggest that AEC-PPARγ deficiency increases inflammation and mucin gene expression in AECs, leading to the observed enhanced secretion of asthma-related cytokines and chemokines in BALF.
FIGURE 4. AEC-PPARγ knockout increases expression of inflammatory and mucin genes.
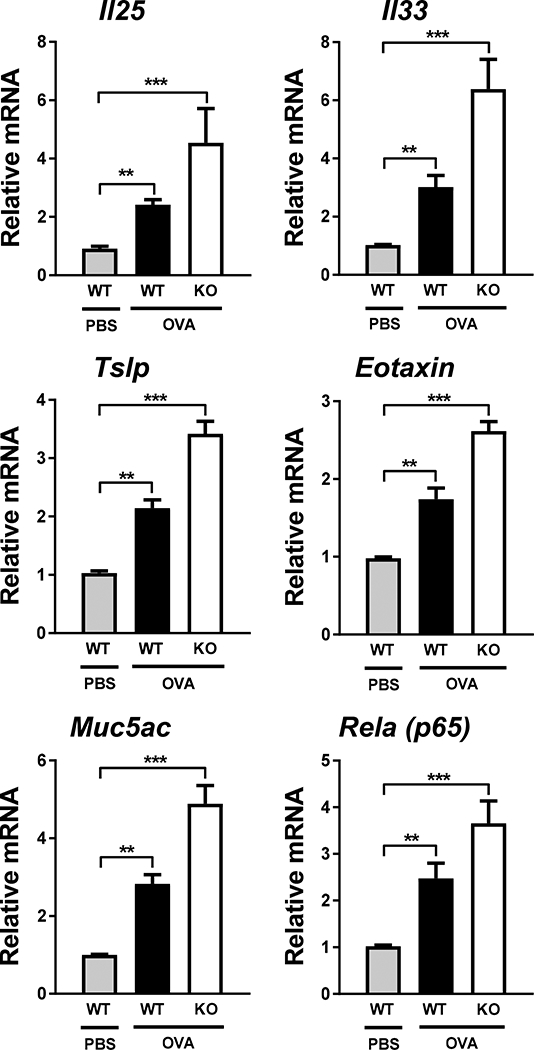
AAD was induced in WT and AEC-PPARγ−/− mice by OVA sensitization on days 0 and 7 and challenge on even-numbered days 14–22. Control mice were similarly treated with PBS. Twenty-four hours later, lung sections were prepared. AECs were collected from lung sections using laser capture microdissection as described in Materials and Methods. mRNA levels of indicated genes (Il25, Il33, Tslp, Ccl11 [eotaxin], Muc5ac, and Rela [NF-κB-p65]) were measured by real-time RT-PCR. The data are expressed as the mean ± SD with n = 3 mice/group. The results were reproduced at least two times independently; **P < 0.01, ***P < 0.001.
The MUC5AC promoter contains novel PPREs
MUC5AC gene modulation by PPARγ (Fig. 4) may be a significant element in AEC-PPARγ’s anti-AAD functions. To investigate the mechanism by which AEC-PPARγ suppresses MUC5AC expression, we analyzed the human MUC5AC promoter for the presence of putative PPARγ transcription factor binding elements. As shown in schematic representation (Fig. 5A), our analysis identified two novel PPREs: one between −1207 and −1194 bp (PPRE1: AGGACAAAGGGCC) and another between −3834 and −3821 bp (PPRE2: TGTTCAGAGGTCAA). To assess the functionality of these MUC5AC PPREs, we performed luciferase assays in NHBE cells transfected with reporter constructs harboring the WT promoter or promoters with one or both of the putative PPREs deleted (ΔPPRE), in the presence or absence of PPARγ. PPARγ significantly decreased luciferase activity of the WT promoter construct (construct 1) (approximately 75% decrease compared with the vector control), whereas deletion of both PPREs (construct 2) completely abolished this effect (Fig. 5B). Reduction in luciferase activity caused by deletion of PPRE2 (construct 3) or PPRE1 (construct 4) was more modest; approximately 65% and 35%, respectively, compared with the control (Fig. 5B). Thus, we identified PPREs in the MUC5AC promoter that may be involved in PPARγ’s suppression of MUC5AC protein expression.
FIGURE 5. The MUC5AC promoter contains novel functional PPREs.
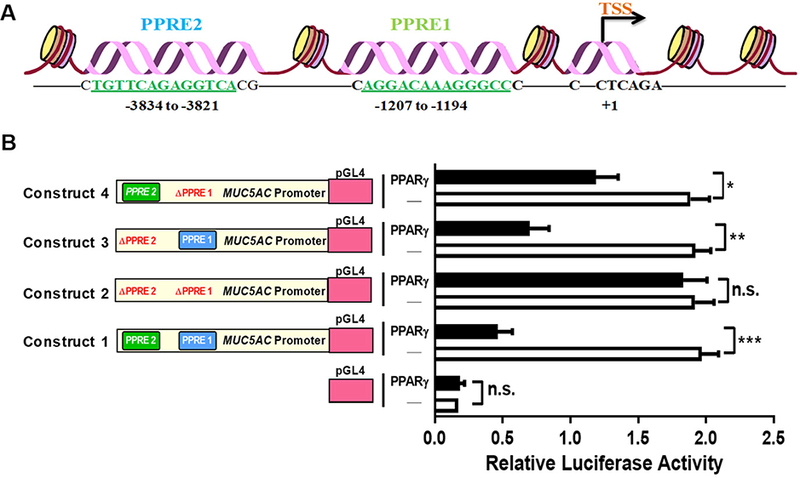
(A) Bioinformatics analysis identified the existence of previously unreported PPREs in the human MUC5AC promoter. The schematic diagram shows the location of those PPREs relative to the transcription start site (TSS); numbers refer to the nucleotide position within the promoter region. (B) NHBE cells were transfected, in the presence or absence of PPARγ expression plasmid, with luciferase constructs containing the WT MUC5AC promoter (construct 1) or promoters with one (constructs 3 and 4) or both (construct 2) of the putative PPREs deleted (ΔPPRE). Empty pGL4 served as a negative control for the assay. The luciferase activities were determined after 24 h. The data are expressed as the mean ± SD with n = 3, and the results were reproduced at least two times independently; *P < 0.05, **P < 0.01, ***P < 0.001, n.s. = non-significant.
PPARγ physically interacts with PPREs in the MUC5AC promoter
To further characterize the novel PPARγ-responsive MUC5AC PPREs and to determine whether MUC5AC is a direct transcriptional target of PPARγ, we examined their interaction. First, we performed DNA affinity precipitation to test whether PPARγ physically interacts with the MUC5AC PPREs. Biotinylated WT and mutated (Mu) double-stranded oligonucleotides corresponding to the PPRE sequences were used to precipitate the proteins to which they bound from nuclear extracts of NHBE cells transfected with or without a PPARγ expression plasmid. Addition of PPARγ caused PPRE-binding proteins from the nuclear extracts to associate with PPARγ, as determined by Western blotting (Fig. 6A). Mutations in the PPRE sequences of the respective oligonucleotides (Supplemental Table 1) eliminated PPARγ from theses complexes (Fig. 6A). As expected, PPRE from CD36 and nonspecific oligonucleotides, used as positive and negative controls respectively, did and did not precipitate PPARγ-containing complexes (Fig. 6A). Notably, Lamin B1, employed as a control for non-specific interaction, was not present in any of the precipitated samples, supporting the specificity of the observed PPARγ-MUC5AC PPRE interactions (Fig. 6A).
FIGURE 6. PPARγ directly binds to PPREs in the MUC5AC promoter.
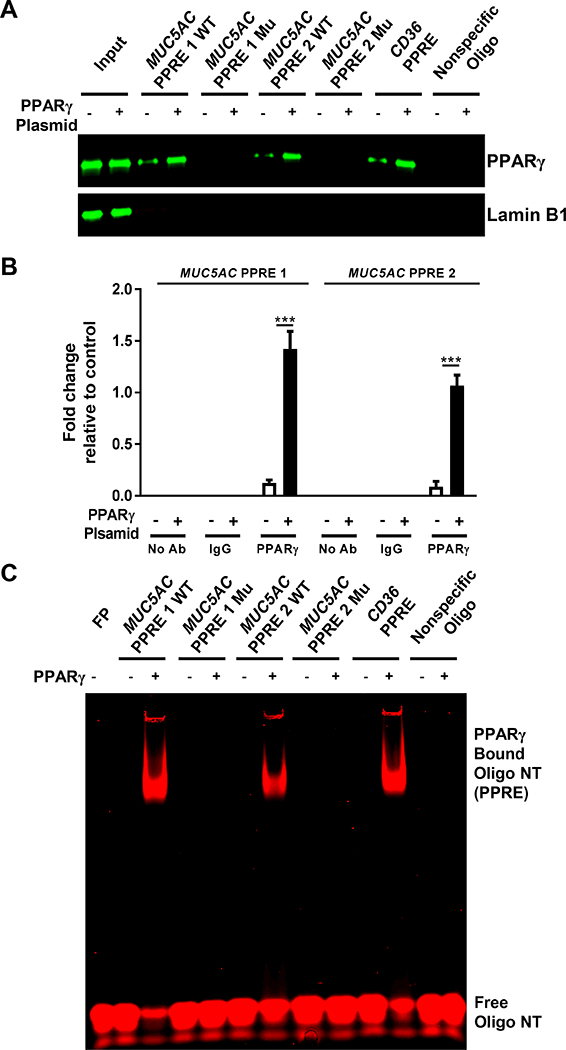
(A, B) NHBE cells were transfected with or without PPARγ expression plasmid. After 24 h, (A) nuclear extracts were obtained and incubated with the indicated biotinylated, double-stranded oligonucleotide-coupled beads. Bead-bound oligonucleotide-protein complexes were eluted and subjected to Western blotting to identify the presence of PPARγ. The PPRE from the CD36 gene and a nonspecific nucleotide sequence were used as positive and negative controls, respectively. Nuclear extracts without added nucleotides were loaded as input. Western blotting for Lamin B1 was performed as a control for non-specific interaction. (B) Chromatin was crosslinked and immunoprecipitated with antibodies against IgG or PPARγ; the antibody-bound DNA-protein complexes were then subjected to real-time RT-PCR with primers that specifically amplified the MUC5AC PPRE1 and PPRE2 regions. (C) Recombinant PPARγ was individually incubated with infrared dye 700 end-labeled double-stranded oligonucleotides for WT or mutated (Mu) MUC5AC PPRE, CD36 PPRE, or nonspecific oligonucleotide. EMSA was performed as described in Materials and Methods. The infrared signal (red) was detected using an Odyssey Infrared Imager. The data are expressed as the mean ± SD with n = 3. The results were reproduced at least two times independently; ***P < 0.001
We also examined this interaction by chromatin immunoprecipitation (ChIP) assay with an anti-PPARγ antibody. Consistent with the DNA affinity precipitation data (Fig. 6A), the ChIP results confirmed the interactions between PPARγ and the MUC5AC PPREs (Fig. 6B). PPARγ’s affinities for the two MUC5AC PPREs were comparable (Fig. 6B). Finally, we performed EMSA to determine whether PPARγ directly binds to the MUC5AC PPREs. Recombinant PPARγ bound to both WT PPRE oligonucleotides, albeit more efficiently to MUC5AC PPRE1 than to MUC5AC PPRE2 (Fig. 6C). Mutations in the PPRE sequences of the respective oligonucleotides abrogated these interactions completely (Fig. 6C). As expected, recombinant PPARγ shifted CD36 PPRE but failed to do so with a nonspecific oligonucleotide (Fig. 6C). Together, these results validate the functionality of the PPARγ-responsive PPREs found in the MUC5AC promoter and provide evidence for their direct interactions.
PPARγ-DBD interacts with MUC5AC PPRE1
PPARγ displays selective and polar half-site binding, which results in the arrangement of the PPARγ-DBD upstream of PPREs. Nucleic acid-protein docking predictions with 5 Å RMSD cutoff and analysis of best pose cluster with higher probability further revealed the interactions of PPARγ-DBD with nucleotides in the MUC5AC PPRE1 and the consensus PPRE. The helix one zinc finger of PPARγ containing P-box was embedded in the major groove of both PPREs in a highly similar and sequence-specific manner. Critical contacts between PPARγ-DBD amino acids and PPRE nucleotides are summarized in Fig. 7. The contact maps shown virtually explain all the conserved DBD amino acids and PPRE nucleotides in the binding site. In the MUC5AC PPRE1 (Fig. 7A) and the consensus PPRE (Fig. 7B) docking complexes, each upstream half-site makes specific contacts through seven and six hydrogen bonds, respectively. Both complexes also harbor seven extensive contacts with the phosphate backbone along the upstream half-site. Consistent with the results from DNA affinity precipitation, ChIP assays, and EMSA, these results indicate that PPARγ-DBD physically interacts with specific nucleotides in the MUC5AC PPRE1. The docking study data also explain why mutations in the MUC5AC PPRE disrupt the PPARγ-MUC5AC interaction (Fig. 6A and 6C).
FIGURE 7. The PPARγ-DBD interacts with nucleotides in the MUC5AC PPRE1.
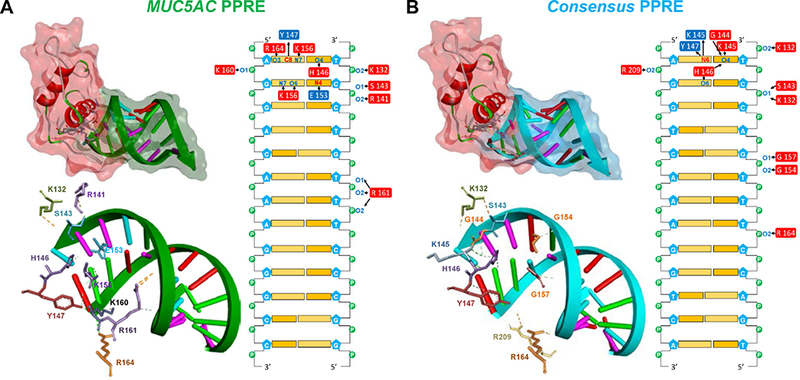
Nucleic acid-protein docking was performed as described in Materials and Methods. Cartoon representation and stereo views of the predicted PPARγ-DBD in complex with (A) MUC5AC PPRE1 and (B) the consensus PPRE. The PPARγ-DBD is depicted in secondary structure with a red surface. The amino acids in contact with the DNA are depicted as colored sticks. The DNA backbone is represented as green (the MUC5AC PPRE1) or sky blue (the consensus PPRE) arrows with semi-transparent surfaces of the same color while nucleotides are drawn as base-colored sticks. Hydrogen bonds are indicated with green dashed lines. Contact maps illustrate interacting amino acids within a 4.0 Å distance from the nucleotides. Donors and acceptors are colored in red and blue, respectively. Arrows indicate hydrogen bonding.
PPARγ modulates expression of inflammatory and mucin proteins in primary human bronchial epithelial cells
The above findings substantiate PPARγ’s role as a key regulator of AECs’ functions in a murine AAD model. To determine whether PPARγ may have the same capacity in HBE cells, we compared the expression of key mediators involved in airway inflammation (NF-κB-p65 and TSLP) and mucus production (MUC5AC) in NHBE and DHBE cells by Western blotting. At baseline, compared with DHBE cells, NHBE cells expressed significantly higher levels of PPARγ and lower levels of inflammatory and mucus proteins (Fig. 8A). siRNA-mediated PPARγ silencing, however, abolished this trend in NHBE cells, causing them to mimic the expression profile of DHBE cells at baseline (Fig. 8A, B). Conversely, DHBE cells treated with the natural PPARγ agonist OA-NO2 showed expression of PPARγ, NF-κB-p65, TSLP, and MUC5AC that was comparable to that in NHBE cells at baseline (Fig. 8A, C). Scrambled siRNA or vehicle control treatment did not affect these proteins’ expression in NHBE or DHBE cells (Fig. 8). Thus, in agreement with data from the mouse studies, these results imply that PPARγ is capable of modulating mediators of airway inflammation and mucus hypersecretion in bronchial epithelial cells of human origin.
FIGURE 8. PPARγ plays a key role in regulating mediators of inflammation and mucus production in human bronchial epithelial cells.
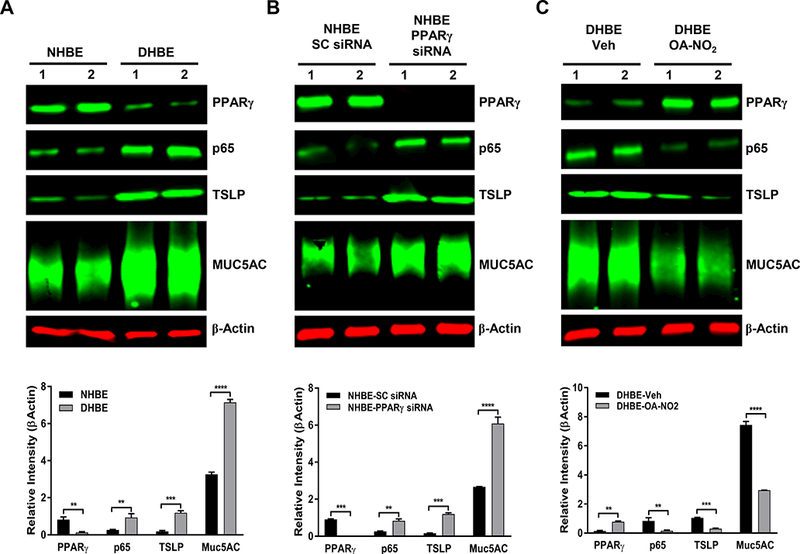
NHBE and DHBE cells were cultured as indicated in Materials and Methods. PPARγ was silenced with siRNA in NHBE cells or activated by OA-NO2 in DHBE cells. Western blotting was performed to determine the levels of indicated proteins (PPARγ, NF-κB/p65, TSLP, and MUC5AC) in (A) untreated NHBE and DHBE cells, (B) NHBE cells treated with scrambled (SC) siRNA or PPARγ-specific siRNA, or (C) DHBE cells treated with vehicle (Veh) or 1 μM OA-NO2 for 6 h. β-Actin served as a loading control. The results were reproduced at least two times independently. Image quantification was performed with MATLAB Image Processing Toolbox. The data are expressed as the mean ± SD; **P < 0.01, ***P < 0.001.
Discussion
The airway epithelium is a physical barrier that separates the lungs from the external environment (25). It protects the underlying tissue from a variety of inhaled gases and particles and initiates immune responses to such challenges (25). In addition to secreting inflammatory cytokines and chemokines and activating innate immune cells, AECs can initiate signaling that may lead to prevention or resolution of inflammation by promoting production of anti-inflammatory molecules such as prostaglandin E2, heparan sulfate proteoglycan syndecan-1, and IL-37 (3). With these biological effects, AECs are central to asthma pathogenesis; therapeutically targeting their functions is thus a key element in asthma therapy.
Our study using AEC-PPARγ−/− mice now adds PPARγ to the list of anti-inflammatory mechanisms activated by AECs. Our observations show that AEC-PPARγ deficiency significantly intensifies airway epithelial pathology in our murine AAD model: In OVA-sensitized and -challenged mice, airway inflammation, cytokine and chemokine expression, and tissue remodeling, were all exaggerated in the absence of AEC-PPARγ. These data agree with previous studies showing that PPARγ agonists ameliorate several AAD features (12, 13, 15, 19, 26) and substantiate the idea that targeting AEC functions via PPARγ may be a suitable therapeutic strategy for asthma.
Type 2 innate lymphoid cells (ILC2) modulate Th2 cell responses as a key source of type 2 cytokines (27). They have been shown to contribute to asthma-associated airway hyperresponseivess, inflammation, mucus hypersecretion, and airway remodeling, at least in mice (27). Because IL-25, IL-33, and TSLP, whose expression was enhanced in our AEC-PPARγ knockout mice, are potent inducers of ILC2 (27), it would be intriguing to determine whether PPARγ deficiency also indirectly exacerbates OVA-induced AAD by enhancing ILC2 activation in addition to its direct effect on AECs.
Moreover, we revealed a novel interaction between PPARγ and the MUC5AC promoter that accounts for effects on MUC5AC repression. Specifically, two novel PPREs in the MUC5AC promoter were identified and demonstrated to directly interact with the PPARγ-DBD. Production of MUC5AC by goblet cells is known to be regulated at the transcriptional level (10). Yet data on such regulation are limited to demonstration that dexamethasone suppresses MUC5AC mRNA expression in multiple cell types (28) and a study implicating FOXA2 as a MUC5AC transcriptional repressor (29). Moreover, knowledge regarding PPARγ’s precise role in regulation of AEC mucus secretion has so far been extremely limited. Thus, our study assigning PPARγ, a well-characterized anti-inflammatory molecule, an additional role as a negative transcriptional regulator of MUC5AC brings new insight to the field of AAD/asthma.
Several studies using various in vivo models of airway inflammation support our conclusion; different PPARγ agonists are capable of blocking airway mucus hypersecretion, as evidenced by decreased PAS staining in goblet cells, airway epithelium, or the lungs (16–18). The PPARγ agonist rosiglitazone also inhibits upregulation of MUC5AC expression in the airway epithelium and its secretion in BALF, as well as Muc5ac mRNA expression in the lungs of rats exposed to acrolein, a cigarette smoke toxin (16). Pioglitazone has also demonstrated a similar effect (26). As Muc5ac mRNA expression closely correlates with the presence of goblet cell metaplasia in the airway epithelium (30) and as airway mucus hypersecretion is associated with morbidity and mortality in asthma patients (8), PPARγ’s ability to suppress MUC5AC provides another layer of support for the therapeutic application of PPARγ agonists in asthma.
Airway mucus hypersecretion is observed in other diseases. Chronic obstructive pulmonary disease and cystic fibrosis display aberrant mucin expression (9, 10). Inflammation in other organs is likewise associated with mucin overexpression (31). Considering that the more than 20 mucin genes that have been identified to date are differentially expressed, either at an mRNA or protein level, in organs of respiratory, digestive, and reproductive systems where mucus serves a protective biofilm (9), it is possible that other mucin genes contain PPREs and respond to PPARγ activation. It would thus be intriguing to investigate whether such PPARγ-mucin gene interactions are absent or dysregulated in other diseases.
Greater amounts of mucin, primarily due to an increase in MUC5AC (mRNA and protein levels), have been observed in bronchial biopsies from asthmatic patients compared with controls (32). A significantly larger amount of intraluminal mucus was also found in samples from patients with severe asthma and was linked to asthma-associated mortality (33, 34). Because mucus hypersecretion leads to airflow obstruction and impaired pulmonary function, which is a major cause of asthma morbidity and mortality (35, 36), it should be a focal point of therapeutic interventions for this disease. Yet current standard therapies, namely corticosteroids and β2-adrenergic receptor agonists, focus on airway inflammation control without directly targeting mucus hypersecretion, thus rationalizing a search for a more effective approach. PPARγ agonists, whose benefits as anti-inflammatory agents for asthma have been recognized by several clinical studies (37), have also been proposed as a treatment for airway mucus hypersecretion (6). This study corroborates this idea by providing evidence for AEC-PPARγ’s function as a transcriptional regulator of MUC5AC.
Supplementary Material
Acknowledgments
We thank Professor Francesco J. DeMayo for providing the Cc10Cre mice.
Grant Support: This work was supported by a Merit Review award from the U.S. Department of Veterans Affairs and National Institutes of Health grants HL093196, AI125338 and HL137842 (R.C.R).
Abbreviations used in this article:
- AAD
allergic airway disease
- AEC
airway epithelial cell
- B&W
binding and washing
- BALF
bronchoalveolar lavage fluid
- CHARMM
chemistry at HARvard Macromolecular Mechanics
- ChIP
chromatin immunoprecipitation
- DBD
DNA binding domain
- DHBE
diseased (asthmatic) human bronchial epithelial
- HBE
human bronchial epithelial
- ILC2
type 2 innate lymphoid cells
- NHBE
normal human bronchial epithelial
- OA-NO2
10-nitro-oleic acid
- PAS
periodic acid–Schiff
- Poly(dI-dC)
poly(deoxyinosinic-deoxycytidylic)
- PPAR
peroxisome proliferator-activated receptor
- PPRE
PPAR response element
- RMSD
root-mean-square deviation
- TSLP
thymic stromal lymphopoietin
Footnotes
Disclaimer: The contents in this article do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Conflict of Interest: The authors declare that they have no conflicts of interest.
References
- 1.To T, Stanojevic S, Moores G, Gershon AS, Bateman ED, Cruz AA, and Boulet LP 2012. Global asthma prevalence in adults: findings from the cross-sectional world health survey. BMC Public Health 12: 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Erle DJ, and Sheppard D 2014. The cell biology of asthma. J Cell Biol 205: 621–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lambrecht BN, and Hammad H 2012. The airway epithelium in asthma. Nat Med 18: 684–692. [DOI] [PubMed] [Google Scholar]
- 4.Thai P, Loukoianov A, Wachi S, and Wu R 2008. Regulation of airway mucin gene expression. Annu Rev Physiol 70: 405–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donovan C, Tan X, and Bourke JE 2012. PPARgamma Ligands Regulate Noncontractile and Contractile Functions of Airway Smooth Muscle: Implications for Asthma Therapy. PPAR Res 2012: 809164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen Y, Chen L, Wang T, and Wen F 2012. PPARgamma as a Potential Target to Treat Airway Mucus Hypersecretion in Chronic Airway Inflammatory Diseases. PPAR Res 2012: 256874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hiemstra PS, McCray PB Jr., and Bals R 2015. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur Respir J 45: 1150–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans CM, Kim K, Tuvim MJ, and Dickey BF 2009. Mucus hypersecretion in asthma: causes and effects. Curr Opin Pulm Med 15: 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rose MC, and Voynow JA 2006. Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol Rev 86: 245–278. [DOI] [PubMed] [Google Scholar]
- 10.Fahy JV, and Dickey BF 2010. Airway mucus function and dysfunction. N Engl J Med 363: 2233–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holgate ST 2007. Epithelium dysfunction in asthma. J Allergy Clin Immunol 120: 1233–1244; quiz 1245–1236. [DOI] [PubMed] [Google Scholar]
- 12.Belvisi MG, and Mitchell JA 2009. Targeting PPAR receptors in the airway for the treatment of inflammatory lung disease. Br J Pharmacol 158: 994–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ward JE, and Tan X 2007. Peroxisome proliferator activated receptor ligands as regulators of airway inflammation and remodelling in chronic lung disease. PPAR Res 2007: 14983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daynes RA, and Jones DC 2002. Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol 2: 748–759. [DOI] [PubMed] [Google Scholar]
- 15.Becker J, Delayre-Orthez C, Frossard N, and Pons F 2006. Regulation of inflammation by PPARs: a future approach to treat lung inflammatory diseases? Fundam Clin Pharmacol 20: 429–447. [DOI] [PubMed] [Google Scholar]
- 16.Liu DS, Liu WJ, Chen L, Ou XM, Wang T, Feng YL, Zhang SF, Xu D, Chen YJ, and Wen FQ 2009. Rosiglitazone, a peroxisome proliferator-activated receptor-gamma agonist, attenuates acrolein-induced airway mucus hypersecretion in rats. Toxicology 260: 112–119. [DOI] [PubMed] [Google Scholar]
- 17.Lee KS, Park SJ, Kim SR, Min KH, Jin SM, Lee HK, and Lee YC 2006. Modulation of airway remodeling and airway inflammation by peroxisome proliferator-activated receptor gamma in a murine model of toluene diisocyanate-induced asthma. J Immunol 177: 5248–5257. [DOI] [PubMed] [Google Scholar]
- 18.Honda K, Marquillies P, Capron M, and Dombrowicz D 2004. Peroxisome proliferator-activated receptor gamma is expressed in airways and inhibits features of airway remodeling in a mouse asthma model. J Allergy Clin Immunol 113: 882–888. [DOI] [PubMed] [Google Scholar]
- 19.Reddy AT, Lakshmi SP, Dornadula S, Pinni S, Rampa DR, and Reddy RC 2013. The nitrated fatty acid 10-nitro-oleate attenuates allergic airway disease. J Immunol 191: 2053–2063. [DOI] [PubMed] [Google Scholar]
- 20.Li H, Cho SN, Evans CM, Dickey BF, Jeong JW, and DeMayo FJ 2008. Cre-mediated recombination in mouse Clara cells. Genesis 46: 300–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reddy AT, Lakshmi SP, and Reddy RC 2012. Murine model of allergen induced asthma. J Vis Exp: e3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reddy AT, Lakshmi SP, Zhang Y, and Reddy RC 2014. Nitrated fatty acids reverse pulmonary fibrosis by dedifferentiating myofibroblasts and promoting collagen uptake by alveolar macrophages. FASEB J 28: 5299–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tuszynska I, Magnus M, Jonak K, Dawson W, and Bujnicki JM 2015. NPDock: a web server for protein-nucleic acid docking. Nucleic acids research 43: W425–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barnes PJ 2008. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol 8: 183–192. [DOI] [PubMed] [Google Scholar]
- 25.Gohy ST, Hupin C, Pilette C, and Ladjemi MZ 2016. Chronic inflammatory airway diseases: the central role of the epithelium revisited. Clin Exp Allergy 46: 529–542. [DOI] [PubMed] [Google Scholar]
- 26.Narala VR, Ranga R, Smith MR, Berlin AA, Standiford TJ, Lukacs NW, and Reddy RC 2007. Pioglitazone is as effective as dexamethasone in a cockroach allergen-induced murine model of asthma. Respir Res 8: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lund S, Walford HH, and Doherty TA 2013. Type 2 Innate Lymphoid Cells in Allergic Disease. Curr Immunol Rev 9: 214–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Voynow JA, Gendler SJ, and Rose MC 2006. Regulation of mucin genes in chronic inflammatory airway diseases. Am J Respir Cell Mol Biol 34: 661–665. [DOI] [PubMed] [Google Scholar]
- 29.Wan H, Kaestner KH, Ang SL, Ikegami M, Finkelman FD, Stahlman MT, Fulkerson PC, Rothenberg ME, and Whitsett JA 2004. Foxa2 regulates alveolarization and goblet cell hyperplasia. Development 131: 953–964. [DOI] [PubMed] [Google Scholar]
- 30.Zuhdi Alimam M, Piazza FM, Selby DM, Letwin N, Huang L, and Rose MC 2000. Muc-5/5ac mucin messenger RNA and protein expression is a marker of goblet cell metaplasia in murine airways. Am J Respir Cell Mol Biol 22: 253–260. [DOI] [PubMed] [Google Scholar]
- 31.Kufe DW 2009. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer 9: 874–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ordonez CL, Khashayar R, Wong HH, Ferrando R, Wu R, Hyde DM, Hotchkiss JA, Zhang Y, Novikov A, Dolganov G, and Fahy JV 2001. Mild and moderate asthma is associated with airway goblet cell hyperplasia and abnormalities in mucin gene expression. Am J Respir Crit Care Med 163: 517–523. [DOI] [PubMed] [Google Scholar]
- 33.Aikawa T, Shimura S, Sasaki H, Ebina M, and Takishima T 1992. Marked goblet cell hyperplasia with mucus accumulation in the airways of patients who died of severe acute asthma attack. Chest 101: 916–921. [DOI] [PubMed] [Google Scholar]
- 34.Shimura S, Andoh Y, Haraguchi M, and Shirato K 1996. Continuity of airway goblet cells and intraluminal mucus in the airways of patients with bronchial asthma. Eur Respir J 9: 1395–1401. [DOI] [PubMed] [Google Scholar]
- 35.Cohn L, Elias JA, and Chupp GL 2004. Asthma: mechanisms of disease persistence and progression. Annu Rev Immunol 22: 789–815. [DOI] [PubMed] [Google Scholar]
- 36.Rogers DF 2002. Airway goblet cell hyperplasia in asthma: hypersecretory and anti-inflammatory? Clin Exp Allergy 32: 1124–1127. [DOI] [PubMed] [Google Scholar]
- 37.Banno A, Reddy AT, Lakshmi SP, and Reddy RC 2018. PPARs: Key Regulators of Airway Inflammation and Potential Therapeutic Targets in Asthma. Nucl Receptor Res 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.