

Abstract
Objective:
To describe low-frequency sensorineural hearing loss (LFSNHL) inherited as a dominant trait in 3 families and in 1 sporadic case.
Design:
Longitudinal clinical study from 1968 to 2001.
Setting:
Tertiary care hospital; field studies conducted by molecular genetic research laboratory.
Participants:
Dominant LFSNHL families.
Interventions:
Questionnaires, serial audiograms, and interviews, correlated with molecular genetic data.
Outcome Measures:
Symptoms, age of onset, serial audiometric data, and hearing aid use.
Results:
Low-frequency sensorineural hearing loss is typically diagnosed in the first decade and slowly progresses over decades; LFSNHL is often asymptomatic in young patients, few of whom use hearing aids. Speech perception becomes affected in later decades when patients develop high-frequency loss. Even children with a strong family history of dominant LFSNHL were not monitoreDroutinely. Penetrance appears complete in that all individuals with a genetic mutation developed hearing loss.
Conclusions:
Dominant LFSNHL is most commonly caused by mutations in the Wolfram syndrome type 1 gene (WFSI). Mutations in WFS1 also cause a rare recessive syndromic form of hearing loss known as Wolfram syndrome or D1DMOAD (diabetes insipidus, diabetes mellitus, optic atrophy, and deafness). Routine newborn hearing screening methods will not typically identify hearing loss affecting frequencies below 2000 Hz; thus, children at risk must be specifically monitored. Genetic counseling and genetic testing may be useful in the management of patients with this type of hearing loss.
Low - frequency sensorineural hearing loss (LFSNHL) is an unusual type of hearing impairment in which frequencies below 2000 Hz are predominantly affected. The EuropeanWorking Group on Genetics of Hearing Impairment has defined such loss as lowto mid-frequency hearing impairment with a difference of at least 15 dB between the poorer lower-frequency and the better higher-frequency thresholds and an air bone gap of less than 15 dB1 There are at least 5 types of LFSNHL: (1) acute low-tone SNHL2; (2) DFNA1 hearing impairment3,4; (3) DFNA6/14 hearing impairment; (4) LFSNHL associated with Meniere disease5; and (5) sporadic isolated LFNSHL. While both DFNA1 and DFNA6/14 are genetic loci linked to nonsyndromic, autosomal dominant LFSNHL, DFNA1 deafness is distinguished by rapid progression that results in profound bilateral deafness by the fourth decade of life. Mutations in the DIAPH1 gene cause LFSNHL linked to DFNA1, which is extremely rare (reported in only 1 family worldwide).
For commentary see page 405
In contrast, DFNA6/14 hearing impairment appears to be composed of a large proportion of hereditary LFSNHL. We have shown that mutations in the Wolfram syndrome type 1 gene (WFSI) underlie DFNA6/14, identifying 5 heterozygous missense mutations in WFSI in 5 dominant LFSNHL families and 1 sporadic case6 One mutation, A716T, was found in 2 unrelated families in our study and also in a third family from Newfoundland7 The purpose of this article is to describe the clinical phenotype of dominant LFSNHL caused by genetic mutations in WFSI in 4 families (families 59, 35, 19, and 21).
METHODS
The family pedigrees have been previously described.6,8,9 Families 59, 35, and 19 have autosomal dominant inheritance, whereas the proband of family 21 has sporadic unilateral low- frequency hearing loss. The institutional review boards of the respective institutions (University of Michigan, Rockefeller University, Vanderbilt University, and the National Institute on Deafness and Other Communication Disorders) approved the study, and informed consent was obtained from each participating subject. Information regarding the history of the hearing loss and assessing risk indicators for acquired hearing loss was obtained through a questionnaire or by personal interviews. Audiometric data were obtained by testing at tertiary care centers and at private audiology offices as well as field testing at subjects’ homes. Prior audiograms were obtained when available, including 10 dating from 1968 for family 59.
The test battery included pure-tone audiometry with high- frequency ipsilateral masking (GSI-16 clinical audiometer [Grason- Stadler Inc, Madison, Wis] and external masker),10 word recognition performance, aural immittance measurement (GSI-33 middleear analyzer; Grason-Stadler Inc), and distortion product oto acoustic emissions (Virtual 330; Virtual Corporation, Portland Ore). For field testing, pure-tone thresholds were obtained using a Beltone 120 audiometer with ER3-A earphones and EAR link foam ear tips (Beltone Electronic Corp, Chicago, 111). Tasco T250 earmuffs (The American Safety Company, Brownwood, Tex) were positioned during air conduction testing. Age of onset was determined by the age at which hearing loss was first documented or by the patient’s recollection, whichever date was earlier.
RESULTS
AUDIOMETRIC ASPECTS
Comparison of audiograms within and across families revealed progression of hearing loss with increasing age and variability in the pattern of hearing loss among affected members within family 59 (Figure 1), family 35 (Figure 2), and families 19 and 21 (Figure 3). Serial audiometric data are presented for selected individuals in family 59 (Figure 4A–B) and family 35 (Figure 5). Overall, the natural history of hearing loss in families 59, 35, and 19 was similar, although variable within families. Several members of family 35 have better hearing at low (250- and 500-Hz) compared with middle (1000- and 2000-Hz) frequencies.
Figure 1.
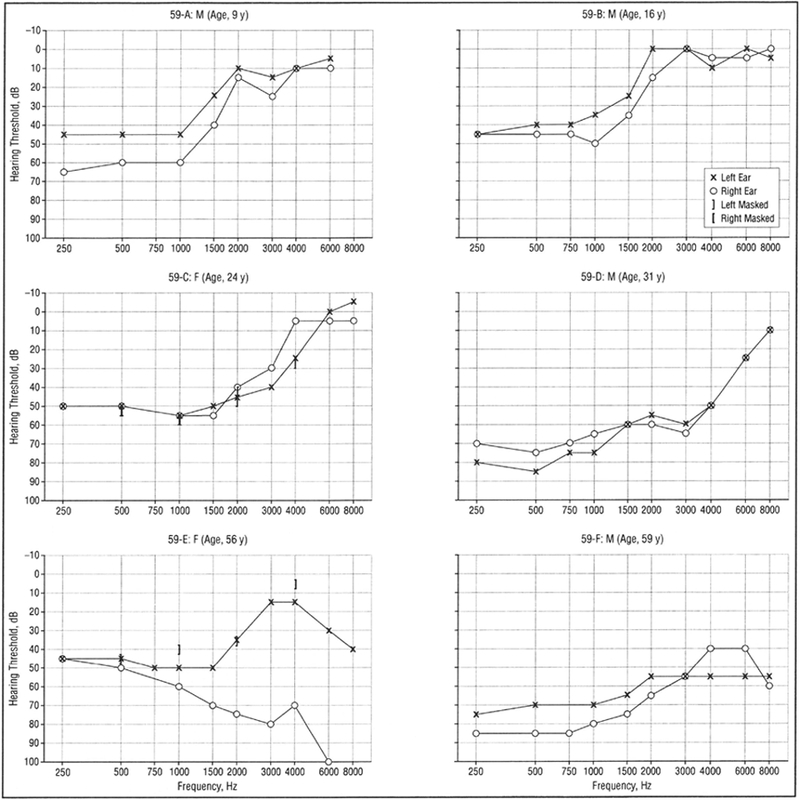
Representative audiograms of affected individuals from family 59 showing pure-tone audiometry results for air conduction bilaterally.
Figure 2.
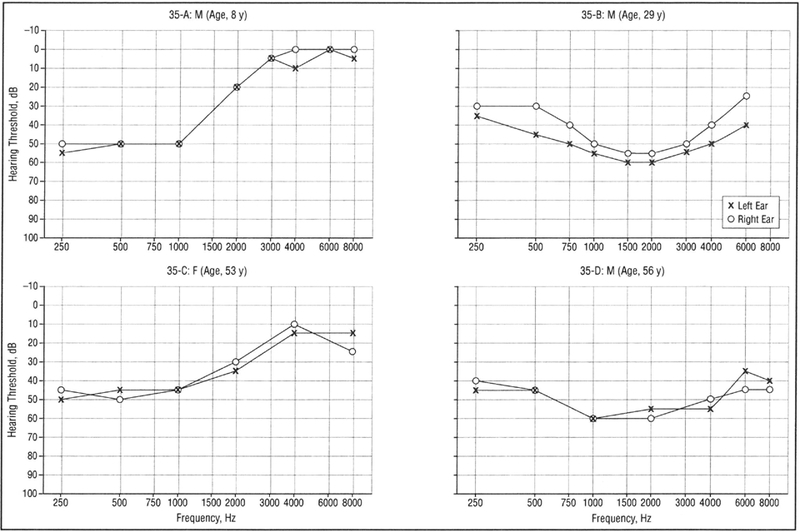
Representative audiograms of affected individuals from family 35 showing pure-tone audiometry results for air conduction bilaterally.
Figure 3.
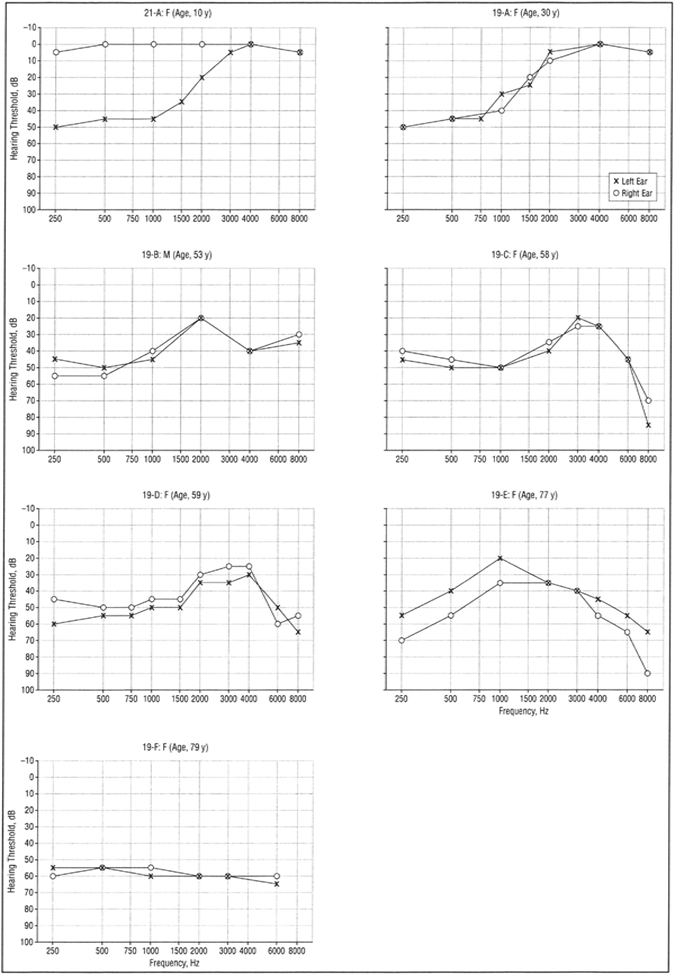
Audiograms of affected individuals from families 19 (19A-F) and 21 (21-A) showing pure-tone audiometry results for air conduction bilaterally.
Figure 4.
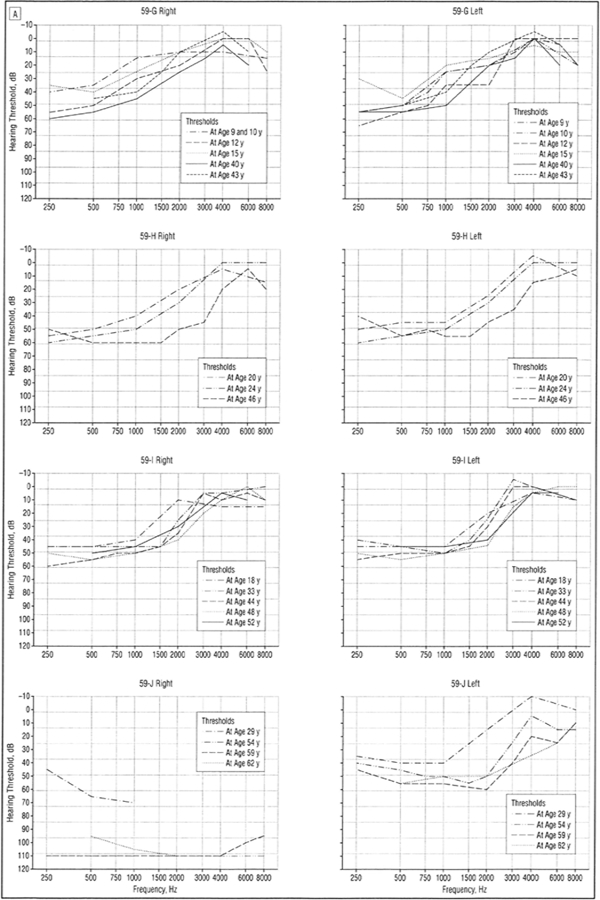
A, Audiograms (divided into right and left ear) of patients 59-G to 59-J with low-frequency hearing loss from family 59 demonstrating progression of hearing loss over decades. All curves represent air conduction thresholds, with the exception of person 59-J, right ear, curve for age 29 years, which represents bone conduction thresholds (considered vibrotactile).
Figure 4. B,
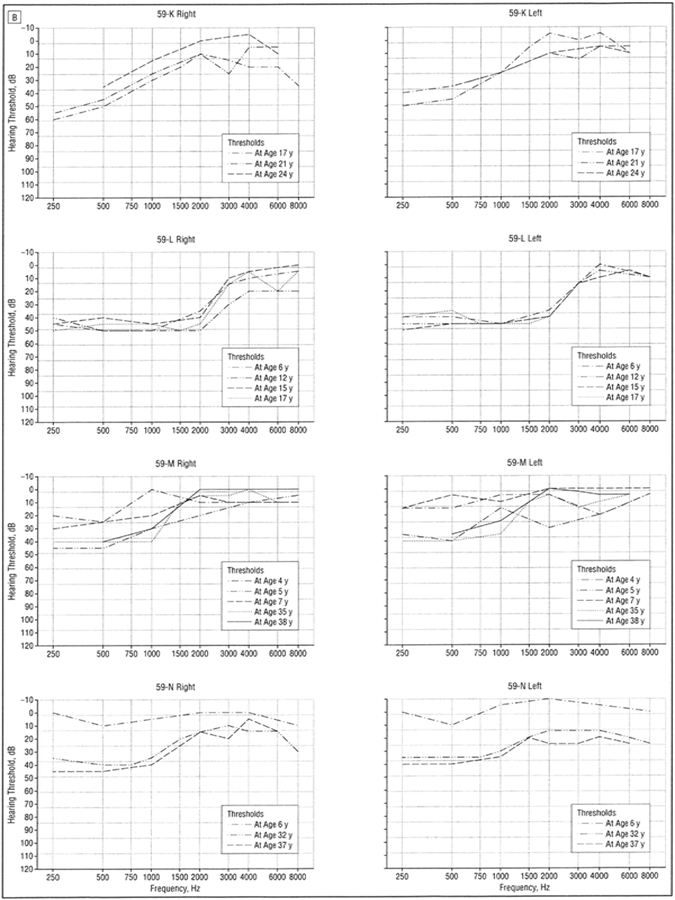
Audiograms (divided into right and left ear) of patients 59-K to 59-N with low-frequency hearing loss from family 59 demonstrating progression of hearing loss over decades. All curves represent air conduction thresholds.
Figure 5.
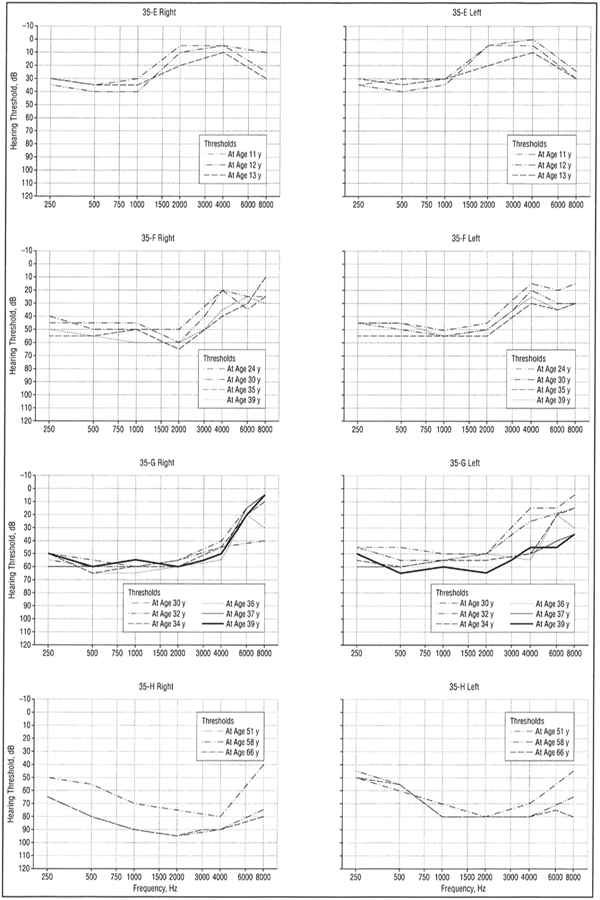
Audiograms (divided into right and left ear) of selected patients with low-frequency hearing loss from family 35 demonstrating progression of hearing loss over decades. All curves represent air conduction thresholds.
The hereditary hearing loss developed in the first and second decades of life as a symmetric bilateral SNHL affecting frequencies below 2000 Hz, with excellent speech discrimination. With advancing age, hearing loss for audiometric frequencies of 250 through 2000 Hz worsened in a low- to high-frequency sequence with thresholds on average exceeding a 50-dB hearing loss by age 50 years. In the fifth and sixth decades, the hearing loss became flatter with hearing loss developing at high frequencies (4000 Hz) with relative preservation at midfrequencies, creating a “peaked” audiogram. Loss of distortion product otoacoustic emissions implicated outer hair cells either directly or indirectly.
Only 2 subjects had profound hearing loss, both in family 59. A 61-year-old woman (Figure 4, person 59-J) had lifelong right profound loss with the more typical low-frequency loss pattern in the left ear. Bone conduction thresholds obtained in the right ear at age 29 years were considered vibrotactile. A second subject was reported to have had a cochlear implant, but the genetic status of this subject is presently unknown.
The proband of family 21 was a 5-year-old girl diagnosed with sporadic unilateral LFSNHL through a routine school screening (Figure 3, person 21-A). She had no risk factors for hearing loss, and the family had suspected hearing loss for at least 2 years.
AGE OF ONSET
A pure-tone hearing loss at 250 and 500 Hz was usually evident by age 10 years (Figure 6). A small number of individuals could not recall when they first became aware of hearing loss and/or had the diagnosis made later in life through participation in our study. One individual had documented normal hearing at age 6 years with a follow-up audiogram at age 22 years documenting LFSNHL (Figure 4, person 59-N). In addition, audiologic data obtained for 10 children at risk, age 10 years or younger, revealed only 1 child with LFSNHL (data not shown).
Figure 6.
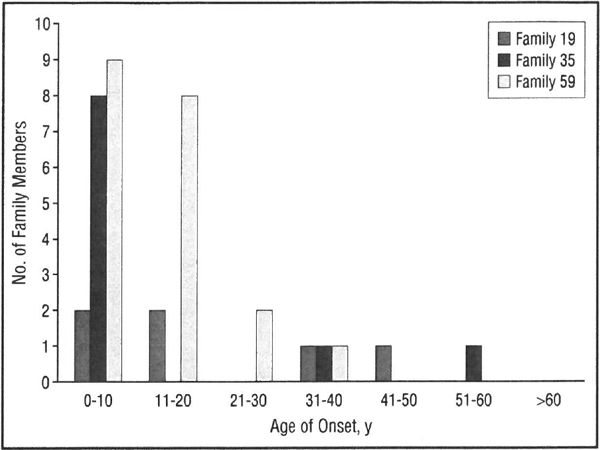
Age of onset of hearing loss in families 59, 35, and 19. Bars indicate number of individuals noting onset of hearing loss in each decade of life.
USE OF AMPLIFICATION
Most of the affected family members were not successful hearing aid users. Of the affected members in family 59, 5 (24%) of 21 had hearing aids and even fewer were regular users. Of the affected members in families 35 and 19,5 (50%) of 10 and 3 (50%) of 6 wore hearing aids, respectively.
ASSOCIATED SYMPTOMS
On questioning, nearly all subjects with low-frequency hearing loss reported tinnitus. However, none found it disabling, and there were no periods of exacerbations or Meniere-like episodes. None reported vestibular complaints. Based on a comprehensive medical examination in 1968 as well as a health questionnaire in 1993, no other clinical features were found to segregate with the hearing loss in family 598 Review of family questionnaires in families 19, 35, and 21 was also consistent with nonsyndromic hearing loss.
MOLECULAR GENETICS
The hearing loss was completely penetrant in an age-dependent fashion, since all subjects with a confirmed WFS1 deafness-causing mutation generally developed hearing loss by age 20 years, and there were no affected children without an affected parent. However, presymptomatic genetic testing was not performed on children with normal hearing. In family 59, a 27-year-old woman (Figure 7, person 59–0) with minimal LFSNHL lacked the L829P mutation in WFSI common to the affected members of the family and was considered a phenocopy.
Figure 7.
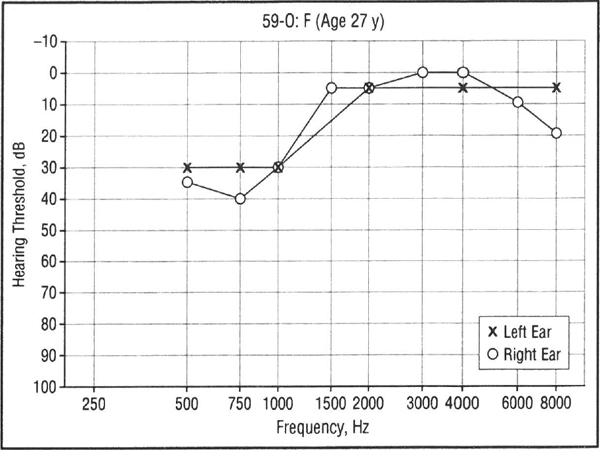
Audiogram of an individual from family 59 with an atypical pattern of hearing loss that is not caused by WFS1 mutation (phenocopy).
COMMENT
Low-frequency sensorineural hearing loss was formerly called bass perceptive deafness, although some doubted whether isolated LFSNHL even existed11 Because inadequate methods for masking made it difficult to obtain true bone conduction levels, a rising audiometric configuration was often assumed to herald a conductive hearing loss.11–13 These authors also noted that patients with isolated LFSNHL had excellent speech discrimination, which tends to delay diagnosis.
Family 59 was the subject of the first description of dominant LFSNHL, reported as kindred I by the Vanderbilt Hereditary Deafness Study Group.8 At that time, 15 family members were identified as affected, with 4 children considered “borderline” affected. The hearing loss was interpreted as cochlear in nature based on Bekesy audiometry and short increment sensitivity index scores. In 1969, Konigsmark14 published a seminal article describing 8 major types of hereditary hearing loss. He suggested that dominant low-frequency hearing loss “is not an infrequent cause of deafness,” since 3 such families from their patient population were ascertained.15 However, most other studies agree that LFSNHL is rare,12,16 with an incidence of only 0.6% in Denmark.17 Hereditary LFSNHL has been described in affecteDrelative pairs13,18 and in other large families with autosomal dominant inheritance.19,20
Overall, hearing loss is one of the most genetically heterogeneous disorders described, with over 40 genetic loci identified for dominant deafness alone.21 The audiometric pattern may vary even among members of a family with the same genetic mutation.22 Modifier genes23 or environmental factors may modulate the effect of the primary genetic mutation.24 In general, the responsible gene cannot be predicted from the audiogram; for example, congenital profound deafness may be caused by GJB2 or Connexin 26 (CX26). In contrast, autosom dominant LFSNHL appears highly predictive of a WFSI mutation.6,25
Other causes of LFSNHL include retrocochlear lesions and cochlear otosclerosis.26 Acquired LFSNHL has been reported as a rare complication of spinal anesthesia, with loss of perilymph hypothesized to cause a relative increase in endolymphatic pressure analogous to hydrops.27 WFS1 mutations do not appear to be a major cause of sporadic LFSNHL (unpublished data, 2001).25 Further genetic studies may help elucidate whether sporadic LFSNHL without vertigo is a variant of Meniere disease or a separate clinical entity.
The WFSI gene was first identified as the cause Wolfram syndrome, also known as DIDMOAD (diabetes insipidus, diabetes mellitus, optic atrophy, anddeafness).28,29 Wolfram syndrome is a rare autosomal recessive syndrome with the minimal diagnostic criteria juvenile-onset diabetes mellitus and optic atrophy. The hearing loss associated with Wolfram syndrome is most commonly delayed onset and progressive, and surprisingly, is usually described as affecting the high frequencies.30,31 Patients with Wolfram syndrome generally lack any functional wolframin protein, while the deafness-associated WFSI mutations are heterozygous missense mutations that affect only 1 copy of the gene and typically change only 1 amino acid.
In the 3 dominant families in our study, the natural history of the hearing loss varied among family members, beginning in childhood and progressing slowly over time. With increasing age, affected family members developed high-frequency SNHL (HFSNHL) with relative preservation of hearing in the mid-frequencies. The like lihood of hearing aid use depended on the person’s age and probably also on socioeconomic status. A similar “double-notch audiogram” has been reported in rare patients with noise exposure.32 The HFSNHL in the older affected members in our study could be caused by the WFSI mutation or may be merely superimposed on the LFSNHL secondary to noise or presbycusis. Because presbycusis and noise-induced hearing loss usually do not segregate in families as autosomal dominant traits and WFSI mutations cause HFSNHL in patients with Wolfram syndrome, we hypothesize that the HFSNHL is part of the WFSJ phenotype. Genetic analysis of the WFSI gene in additional patients with LFSNHL as well as other types of hearing loss such as presbycusis will be necessary to further define genotype-phenotype correlations.
The molecular mechanisms explaining the different phenotypes caused by WFSJ mutations are currently unknown. WFSi encodes a unique protein, wolframin, that has no homology to any known protein. The WFSI gene is expressed in the endoplasmic reticulum of cells, and therefore a role in protein sorting, protein trafficking, or calcium homeostasis has been hypothesized.33 There are currently no animal models of human low-frequency hearing loss, since few nonhuman mammals have useful hearing below 2000 Hz. Furthermore, experiments attempting to create such a model found that animals with apical cochlear lesions are still able to respond to low-frequency stimuli.34 This phenomenon results from the response of the basal cochlea to the harmonics of low-frequency tones, thus masking at frequencies higher than the stimulus frequency is necessary.35,36 Transgenic mice may be helpful in the future to elucidate the role of WFS1 in the pathophysiology of LFSNHL.
The final question is whether patients with WF51 mutations have normal hearing at birth or congenital LFSNHL that is not detected until years later. Determining the exact age of onset is hampered by the lack of audiologic data at early ages, the relative lack of symptoms with isolated LFSNHL, and the tendency for people to delay getting a hearing test even when hearing loss is suspected. Often, 10 years or more elapsed between the time that hearing loss was suspected and when it was formally diagnosed by audiometry. In our study, several members had not noted hearing loss until they had a routine hearing test at school, at work, or through participation in our study. Indeed, participation in our study increased awareness in the families that their hearing loss was hereditary. One patient (Figure 1, person 59-C) attributed her hearing loss to noise exposure, since she first noticed it after attending a rock concert at age 13 years. Most likely, she experienced a temporary threshold shift in the high frequencies, which in combination with hereditary LFSNHL left her severely disabled for a period, whereas others experiencing a temporary threshold shift still had useful hearing in the low frequencies.
In family 59, the incidence of LFSNHL in children with an affected parent was far lower than 50% as would be expected with a fully penetrant dominant trait. Ascertainment bias may explain why all children tested were found to have normal hearing, if parents who suspect hearing loss in their child were reluctant to participate. Alternatively, it is possible that only 1 in 10 children inherited the dominant trait by chance alone. However, considering the case of person 59-N, with documented normal hearing prior to developing LFSNHL, we favor the explanation that WFS1 hearing loss develops later in children born with normal hearing. Evaluating children for hearing loss at an early age will be the only way to answer the question, but to date, almost none of the affected family members or their physicians sought newborn hearing screening for children at risk.
Routine methods of newborn hearing screening, whether by distortion product otoacoustic emissions or auditory brainstem response, will not typically assess frequencies lower than 2000 Hz. A family history should be obtained in patients with LFSNHL to allow audiologic monitoring for children at risk. For such children, visual reinforcement audiometry at age 6 months, or preferably, auditory brainstem response prior to 6 months, is advised. Because the sound energy of a click stimulus is concentrated at 2000 to 4000 Hz,37 the physician should specifically request 500- and 1000-Hz tone bursts to rule out LFSNHL. An awareness of the hereditary nature of LFSNHL among clinicians will facilitate identification of families who might benefit from genetic counseling or perhaps genetic testing.
In summary, we describe a sporadic case and 3 families with autosomal dominant nonsyndromic progressive low-frequency cochlear SNHL, all found to have mutations in the WF51 gene. In the future, molecular genetic studies may be increasingly useful to allow clinicians to distinguish between similar clinical entities.
Acknowledgments
This work was supported by grants DC00161 (Dr Les- perance) and DC03594 (Dr Leal) from the National Institutes of Health, Bethesda, Md; Professional Service Contract 92-DC-0234 from the National Institute on Deafness and Other Communication Disorders, Bethesda (Dr Hall); the Starr Center for Human Genetics, New York, NY (Dr Leal), and the American Hearing Research Foundation, Chicago, III (Dr Leal).
This study was presented at the 17th Annual Meeting of the American Society of Pediatric Otolaryngology, Boca Raton, Fla, May 13, 2002.
We thank the families for their participation; Laura Miller and Angelique Boerst, MA, CCC-A, for assistance with preparation of the manuscript; and Pamella McMillan, MA, CCC-A, Susan Barker, MA, CCC-A, David Brown, MD, and Theresa Kim, BA, for assistance with data collection.
REFERENCES
- 1.European Work Group on Genetics of Hearing Impairment. lnfoletter2 Vol 2 1996. [Google Scholar]
- 2.Imamura S, Nozawa I, Imamura M, Murakami Y. Clinical observations on acute low-tone sensorineural hearing loss: survey and analysis of 137 patients. Ann Otol Rhinol Laryngol 1997;106:746–750. [DOI] [PubMed] [Google Scholar]
- 3.Lynch ED, Lee MK, Morrow JE, Welcsh PL, Leon PE, King MC. Nonsyndromic deafness DFNA1 associated with mutation of a human homolog of the Drosophila gene diaphanous. Science 1997;278:1315–1318. [PubMed] [Google Scholar]
- 4.Lalwani AK, Jackler RK, Sweetow RW, et al. Further characterization of the DFNA1 audiovestibular phenotype. Arch Otolaryngol Head NeckSurg 1998;124:699–702. [DOI] [PubMed] [Google Scholar]
- 5.van de Heyning PH, Wuyts FL, Claes J, Koekelkoren E, Van Laer C, Valcke H. Definition, classification anDreporting of Meniere’s disease and its symptoms. Acta Otolaryngol Suppi 1997;526:5–9. [DOI] [PubMed] [Google Scholar]
- 6.Bespalova IN, Van Camp G, Bom SJH, et al. Mutations in the Wolfram syndrome 1 gene (WFS1) are a common cause of low frequency sensorineural hearing loss. Hum Mol Genet 2001; 10:2501–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Young TL, Ives E, Lynch E, et al. Nonsyndromic progressive hearing loss DFNA38 is caused by heterozygous missense mutation in the Wolfram syndrome gene W FS1. Hum Mol Genet 2001; 10:2509–2514. [DOI] [PubMed] [Google Scholar]
- 8.Vanderbilt Hereditary Deafness Study Group. Dominantly inherited low- frequency hearing loss. Arch Otolaryngol 1968;88:242–250. [PubMed] [Google Scholar]
- 9.Lesperance MM, Hall JW III, Bess FH, et al. A gene for autosomal dominant nonsyndromic hereditary hearing impairment maps to 4p16.3. Hum Mol Genet 1995; 4:1967–1972. [DOI] [PubMed] [Google Scholar]
- 10.Humes LE, Tharpe AM, Bratt GW. Validity of hearing thresholds obtained from the rising portion of the audiogram in sensorineural hearing loss. J Speech Hear Res 1984;27:206–211. [DOI] [PubMed] [Google Scholar]
- 11.Gravendeel DW, Plomp R. Perceptive bass deafness. Acta Otolaryngol 1960; 64:548–560. [DOI] [PubMed] [Google Scholar]
- 12.Ross M, Matkin ND. The rising audiometric configuration. J Speech Hear Disord 1967;32:377–382. [DOI] [PubMed] [Google Scholar]
- 13.Parving A, Bak-Pedersen K. Clinical findings and diagnostic problems in sensorineural low frequency hearing loss. Acta Otolaryngol 1978;85:184–190. [DOI] [PubMed] [Google Scholar]
- 14.Konigsmark BW. Hereditary deafness in man. N EnglJ Med 1969;281:827–832. [DOI] [PubMed] [Google Scholar]
- 15.Konigsmark BW, Mengel M, Berlin Cl. Familial low frequency hearing loss. Laryngoscope 1971; 81:759–771. [DOI] [PubMed] [Google Scholar]
- 16.Lundborg T Nerve deafness for low tones. Acta Otolaryngol 1955;45:215–225. [DOI] [PubMed] [Google Scholar]
- 17.Parving A, Sakihara Y, Christensen B. Inherited sensorineural low-frequency hearing impairment: some aspects of phenotype and epidemiology. Audiology 2000; 39:50–60. [DOI] [PubMed] [Google Scholar]
- 18.linuma T, Shitara T, Hoshino T, Kirikae I. Sensorineural hearing loss for low tones. Arch Otolaryngol 1967;86:110–116. [DOI] [PubMed] [Google Scholar]
- 19.Parving A, Johnsen NJ, Holm-Jensen S. Dominantly inherited low-frequency hearing loss. Audiology 1978;17:165–172. [DOI] [PubMed] [Google Scholar]
- 20.Kunst H, Marres H, Huygen P, Van Camp G, Joosten F, Cremers C. Autosomal dominant non-syndromal low-frequency sensorineural hearing impairment linked to chromosome 4p16 (DFNA14): statistical analysis of hearing threshold in relation to age and evaluation of vestibulo-ocular functions. Audiology 1999;38: 165–173. [DOI] [PubMed] [Google Scholar]
- 21.Smith RJH, Van Camp G, eds. Hereditary Hearing Loss Homepage Available at: http://www.uia.ac.be/dnalab/hhh/. Accessed 2002.
- 22.Van Camp G, Willems PJ, Smith RJ. Nonsyndromic hearing impairment: unparalleled heterogeneity. Am J Hum Genet 1997;60:758–764. [PMC free article] [PubMed] [Google Scholar]
- 23.Riazuddin S, Castelein CM, Ahmed ZM, et al. Dominant modifier DFNM1 suppresses recessive deafness DFNB26. Nat Genet 2000;26:431–434. [DOI] [PubMed] [Google Scholar]
- 24.Prezant TR, Agapian JV, Bohlman MC, et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat Genet 1993;4:289–294. [DOI] [PubMed] [Google Scholar]
- 25.Cryns K, Pfister M, Pennings RJE, et al. Mutations in the WFS1 gene that cause low-frequency sensorineural hearing loss are small non-inactivating mutations. Hum Genet 2002;110:389–394. [DOI] [PubMed] [Google Scholar]
- 26.Carhart R Atypical audiometric configurations associated with otosclerosis. Ann Otol 1962;71:744–758. [DOI] [PubMed] [Google Scholar]
- 27.Walsted A, Salomon G, Olsen KS. Low-frequency hearing loss after spinal anesthesia. Perilymphatic hypotonia? Scand Audio! 1991; 20:211 –215. [DOI] [PubMed] [Google Scholar]
- 28.Strom TM, Hortnagel K, Hofmann S, et al. Diabetes insipidus, diabetes mellitus, optic atrophy and deafness (DIDMOAD) caused by mutations in a novel gene (wolframin) coding for a predicted transmembrane protein. Hum Mol Genet 1998; 7:2021–2028. [DOI] [PubMed] [Google Scholar]
- 29.Inoue H, Tanizawa Y, Wasson J, et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat Genet 1998;20:143–148. [DOI] [PubMed] [Google Scholar]
- 30.Higashi K Otologic findings of DIDMOAD syndrome. Am J Otol 1991; 12:57–60. [PubMed] [Google Scholar]
- 31.Cremers CW, Wijdeveld PG, Pinckers AJ. Juvenile diabetes mellitus, optic atrophy, hearing loss, diabetes insipidus, atonia of the urinary tract and bladder, and other abnormalities (Wolfram syndrome): a review of 88 cases from the literature with personal observations on 3 new patients. Acta Paediatr Scand Suppl 1977;264:1–16. [DOI] [PubMed] [Google Scholar]
- 32.Chung DY. Meanings of a double-notch audiogram. Scand Audio! 1980;9:29–32. [DOI] [PubMed] [Google Scholar]
- 33.Takeda K, Inoue H, Tanizawa Y, et al. WFS1 (Wolfram syndrome 1) gene product: predominant subcellular localization to endoplasmic reticulum in cultured cells and neuronal expression in rat brain. Hum Mol Genet 2001;10:477–484. [DOI] [PubMed] [Google Scholar]
- 34.Prosen CA, Moody DB, Stebbins WC, et al. Apical hair cells and hearing. Hear Res 1990;44:179–193. [DOI] [PubMed] [Google Scholar]
- 35.Turner C, Burns EM, Nelson DA. Pure tone pitch perception and low-frequency hearing loss. JAcoust Soc Am 1983;73:966–975. [DOI] [PubMed] [Google Scholar]
- 36.Prosen CA, Moody DB. Low-frequency detection and discrimination following apical hair cell destruction. Hear Res 1991;57:142–152. [DOI] [PubMed] [Google Scholar]
- 37.Gorga MP, Worthington DW, Reiland JK, Beauchaine KA, Goldgar DE. Some comparisons between auditory brain stem response thresholds, latencies, and the pure-tone audiogram. Ear Hear 1985;6:105–112. [DOI] [PubMed] [Google Scholar]