

Abstract
Physiological responses to short-term environmental stressors, such as infection, can have long-term consequences for fitness, particularly if the responses are inappropriate or nutrient resources are limited. Genetic variation affecting energy acquisition, storage, and usage can limit cellular energy availability and may influence resource-allocation tradeoffs even when environmental nutrients are plentiful. Here, we utilized Drosophila mitochondrial–nuclear genotypes to test whether disrupted mitochondrial function interferes with nutrient-sensing pathways, and whether this disruption has consequences for tradeoffs between immunity and fecundity. We found that an energetically-compromised genotype was relatively resistant to rapamycin—a drug that targets nutrient-sensing pathways and mimics resource limitation. Dietary resource limitation decreased survival of energetically-compromised flies. Furthermore, survival of infection with a natural pathogen was decreased in this genotype, and females of this genotype experienced immunity–fecundity tradeoffs that were not evident in genotypic controls with normal energy metabolism. Together, these results suggest that this genotype may have little excess energetic capacity and fewer cellular nutrients, even when environmental nutrients are not limiting. Genetic variation in energy metabolism may therefore act to limit the resources available for allocation to life-history traits in ways that generate tradeoffs even when environmental resources are not limiting.
Introduction
The energy available to heterotrophic organisms is often determined by nutrients in the environment, and the dynamic allocation of these resources within the lifespan of an individual impacts life-history tradeoffs between organismal maintenance and reproduction. Nutritional stress may be caused by the lack of a single nutrient (Bergland et al. 2008; Jensen et al. 2015), improper nutrient ratios (Skorupa et al. 2008), or reduced overall food availability leading to a decrease in overall calorie consumption. Energetic costs associated with infection are predicted to have a significant impact on survivorship and future reproduction via the allocation of limited resources between reproduction and immunity (Lochmiller and Deerenberg 2000; Harshman and Zera 2007; Schwenke et al. 2016). Energetic costs of infection can be associated with the mechanisms of pathogen resistance (e.g., constitutive and induced immune responses) and tolerance (Rauw 2012), reduced nutrient uptake during infection (Bonfini et al. 2016), or resource consumption by pathogens (Cressler et al. 2014; Kurze et al. 2016).
Despite the prediction that fighting infection will generate a tradeoff with future reproduction, the relationship between infection and reproduction is complex. Under some conditions, adult infection decreases fecundity and the expression of reproduction genes (Short and Lazzaro 2013). However, constitutive immune expression does not always generate life-history tradeoffs (Fellous and Lazzaro 2011), and infection can even increase fecundity (Adamo 1999) and offspring quality (Stahlschmidt et al. 2013; Reavey et al. 2015). Increased reproduction post-infection may occur via parasite manipulation (e.g., Weeks and Stouthamer 2004) or if hosts switch resources toward short-term investment in reproduction (Cressler et al. 2015), a strategy known as terminal investment (Clutton-Brock 1984; Bonneaud et al. 2004). Understanding how host energy metabolism impacts resource allocation and immune function, and the consequences for life-history tradeoffs remain an important area of research, with implications for the field of ecological immunology (Sheldon and Verhulst 1996; Brock et al. 2014).
Investigating how genetic variation in host metabolism impacts immune function and interacts with diet to influence life-history outcomes during periods of environmental stress (e.g., infection) is critical for understanding the evolution of immunity–fecundity tradeoffs. Genetic variation affecting energy metabolism may limit the availability of cellular energy (e.g., Adenosine triphosphate [ATP]) and influence resource-allocation tradeoffs even when environmental nutrients are not limiting. Thus, the extent to which environmental nutrients are limiting is expected to vary among individuals. One regulatory mechanism that integrates information from external (e.g., food availability) and internal (e.g., ATP) inputs is the target of rapamycin (TOR) signaling pathway (Oldham and Hafen 2003). When external and internal nutrient levels are sufficient, TOR upregulates downstream genes to promote protein synthesis and growth. Conversely, poor nutrient levels or treatment with the drug rapamycin decreases protein production and increases recycling of cellular components via autophagy, slowing growth (Zheng et al. 1995; Hahn-Windgassen et al. 2005; Fig. 1). Consistent with these effects, rapamycin delays development, decreases fecundity, and increases lifespan in the fruit fly Drosophila melanogaster (Bjedov et al. 2010).
Fig. 1.
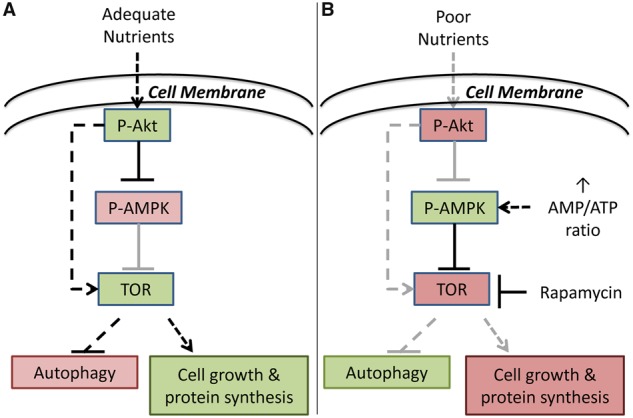
The target of rapamycin (TOR) protein integrates nutrient responses to regulate growth. (A) In the presence of adequate nutrients, TOR is active, which represses recycling of cellular components via autophagy and promotes growth. (B) When nutrients are sensed as being limited either via insulin signaling, an increased AMP/ATP ratio, or artificially by exposure to the drug rapamycin, TOR is repressed which promotes autophagy and inhibits growth.
To investigate how genetic variation in energy metabolism and, specifically, in mitochondrial function affects immune function and immunity–fecundity tradeoffs, we utilized a mitochondrial–nuclear (mito–nuclear) genotype of Drosophila that compromises mitochondrial oxidative phosphorylation (OXPHOS). Compromised OXPHOS in this genotype is caused by an incompatible interaction between a single nucleotide polymorphism in the mitochondrial-encoded mt-tRNATyr and an amino acid polymorphism in the nuclear-encoded mt-tyrosyl-tRNA synthetase that aminoacylates this mt-tRNA (Meiklejohn et al. 2013). Together, these mutations disrupt larval metabolism, delay development, and decrease female fecundity, indicative of inefficient energy metabolism (Hoekstra et al. 2013, 2018; Meiklejohn et al. 2013). Here we measured life-history traits in mito–nuclear genotypes under nutrient- and pathogen-stress conditions to test whether genetic variation that compromises energy metabolism can limit available cellular resources and generate tradeoffs between immunity and fecundity.
Methods
Drosophila genotypes and rearing conditions
We employed six mito–nuclear genotypes that combine mtDNAs from Drosophilasimulans—(simw501) and (sm21)—and D. melanogaster (ore) with two wild-type D. melanogaster nuclear genomes—OreR and Aut (Montooth et al. 2010). Of these six genotypes, only the (simw501); OreR mito–nuclear combination generates an incompatible interaction that decreases OXPHOS; the other five genotypes serve as wild-type controls. All genotypes were maintained at 25°C with a 12 h:12 h, light:dark cycle. Three non-isocaloric food types were used in experiments: our standard laboratory food, which is a high-yeast diet (0.88% agar, 8.33% Torula yeast, 10% Cornmeal, 0.33% Tegosept W/V and 4.66% Molasses, 1.66% 95% ethanol, and 0.66% propionic acid V/V dH2O), a low-yeast diet (our standard food with 0.5% Torula Yeast W/V), and a medium-mixed diet (0.93% agar, 2.94% SAF Yeast, 6.12% Cornmeal, 12.94% sugar, 0.28% Tegosept W/V and 1.08% 95% ethanol, and 0.71% propionic acid V/V dH2O).
Rapamycin and diet effects on development
To test whether the energetically-compromised (simw501); OreR genotype has disrupted nutrient-sensing, we developed all six genotypes from egg to adult on the medium-mixed diet containing three concentration of rapamycin concentrations (0, 2, and 10 µM). Fifty females and 30 males of each genotype were mated for 24 h and placed onto grape-agar plates (50 g bacto-agar, 30 mL tegosept in 10% ethanol, 500 mL grape juice, 1500 mL distilled H2O) for collecting cohorts of eggs every 24 h. A total of five replicate vials of 75 eggs per genotype and rapamycin concentration were monitored twice a day to measure the development time of each individual and the number of males and females that eclosed as a measure of sex-specific survival. This assumed a 50:50 sex ratio in the eggs or larvae (see below) placed in each vial.
In order to examine additional rapamycin concentrations, genotypes with the (sm21) mtDNA—which did not behave differently from the (ore) control mtDNA in the initial experiment—were not included in a second experiment. In this experiment, four genotypes were reared on the high-yeast diet for many generations before being reared on food containing 0, 5, 10, or 15 µM rapamycin. Males and females of each genotype were mated, and females were allowed to lay eggs for 12 h on grape-agar plates. Fifty first-instar larvae of each genotype were collected 24 h later. Seven to eight replicate vials of each genotype at each rapamycin concentration were measured for development time and survival as described above.
In order to test the prediction that control genotypes exposed to a low-yeast diet would show a decreased responsiveness to rapamycin, similar to (simw501); OreR (see the “Results” section), we developed all six mito–nuclear genotypes from larvae to adult on either a high-yeast or low-yeast diet, supplemented with 0, 5, or 10 µM rapamycin. Males and females of each genotype were mated, and females were allowed to lay eggs for 4h on high- or low-yeast plates. One hundred first-instar larvae of each genotype were collected 30 h after the egg lay. Five replicate vials of each genotype, yeast, and rapamycin combination were measured for development time and survival as described above.
Bacterial infection and female fecundity
To test whether compromised energy metabolism decreases the ability to survive bacterial–pathogen infection, we infected virgin 1-day old adults of all six mito–nuclear genotypes with the natural pathogen Providencia rettgeri (Juneja and Lazzaro 2009; Short and Lazzaro 2013). Individuals were either sham infected with 1× PBS or infected with P. rettgeri in 1× PBS at a concentration of 1.0 OD (∼5000 bacterial cells) using a 0.1 mm needle (TedPella 13561-50) (Khalil et al. 2015). The infection protocol results in moderate lethality: 40–80% of adults survive depending on the infection method and condition of flies, with infection stabilizing by day 4 (Sackton et al. 2010; Howick and Lazzaro 2014; Duneau et al. 2017a). Flies were then placed in groups of 30 males or females on standard food and survivors were counted twice daily for 10 days. Five replicate groups of each genotype, sex, and infection treatment (sham vs. pathogen) combination were measured for survival. In a parallel infection setup, fecundity was measured using 15–20 females of each genotype–treatment combination that had survived to 5 days post infection. These females were mated with wild-type males that were genetically distinct from the focal genotypes. Mated females were allowed to lay eggs for 72 h, transferring both males and females to a new vial every 24 h.
Statistical analyses
Development time to adult eclosion was analyzed using linear mixed-effects models with mtDNA, nuclear genotype, sex, treatment (rapamycin, diet, infection), and their interactions as fixed effects, and replicate vial as a random variable. Rapamycin concentration was treated as an ordered factor. Tukey’s tests were performed with Holm’s sequential Bonferroni correction. The same fixed effects were included in a generalized linear-model analyses of the proportion of flies surviving treatment in each vial. Cox proportional hazard mixed-effects model estimates of hazard ratios associated with infection were obtained using the coxme function in R (Therneau et al. 2003). Fecundity was analyzed using linear models that included the fixed effects of day, genotype, and treatment. Outliers were identified via the Grubbs test and removed. However, analyses with and without outlier data did not produce qualitatively different results. All analyses were carried out in R version 3.4.2 (R Core Team 2017), and statistical tables are provided in Supplementary Tables. Due to the prevalence of main and interaction effects with sex, as well as extensive evidence of sexual dimorphism for life history and physiology in Drosophila (Millington and Rideout 2018), we plotted female and male data separately.
Results
Individuals with compromised energy metabolism were resistant to rapamycin
The mito–nuclear genotype (simw501); OreR decreases mitochondrial OXPHOS activity with deleterious effects on metabolic rate, development, and female fecundity that are sensitive to energy demand (Hoekstra et al. 2013, 2018; Meiklejohn et al. 2013; Holmbeck et al. 2015; Zhang et al. 2017). Here we tested whether (simw501); OreR flies had altered nutrient sensing due to their predicted low level of cellular energy even when reared on a non-limiting diet. We raised this genotype and genotypic controls that have normal energy metabolism on diets containing rapamycin. This drug represses TOR, an energy-sensing protein downstream of both the insulin receptor and Adenosine monophosphate (AMP)-activated protein kinase (AMPK)—a central regulator of cellular metabolism that responds to the relative abundances of AMP and ATP. Thus, TOR integrates multiple signals of nutrient availability and energetic status to control growth (Fig. 1).
In two independent experiments, we found that rapamycin extended development time of control genotypes in a dose-dependent manner (Fig. 2 and Supplementary Fig. S1), consistent with prior observations in Drosophila (Zhang et al. 2000; Wang et al. 2016). However, the energetically-compromised (simw501); OreR genotype was resistant to the effect of rapamycin on development time and survived rapamycin treatment better than control genotypes (Fig. 2 and Supplementary Fig. S1). An interaction between mtDNA genotype, nuclear genotype, and rapamycin concentration significantly affected development time (mtDNA × nuclear × rapamycin, P < 0.0001), a pattern that was independent of sex (mtDNA × nuclear × rapamycin × sex, P = 0.14) (Supplementary Table S1). In the experiment on the medium-mixed diet, flies with the Aut nuclear genome did not survive at high rapamycin concentrations; in this experiment, an interaction between mtDNA and rapamycin concentration significantly affected development time for individuals with the OreR nuclear genome (mtDNA × rapamycin, P < 0.0001) (Supplementary Fig. S1 and Table S2). In both experiments, the interaction appeared to be driven by an attenuated response of (simw501); OreR development time to rapamycin, relative to the control genotypes (Fig. 2A, B and Supplementary Fig. S1A, B).
Fig. 2.

The energetically-compromised genotype (simw501); OreR was relatively resistant to the drug rapamycin. (A, B) The effect of rapamycin to increase development time was attenuated in (simw501); OreR relative to control genotypes in both sexes. (C, D) (simw501); OreR had similar survival to genetic controls in the absence of rapamycin, but had the highest survival in the presence of rapamycin in both sexes. Points are average trait values across seven to eight replicate vials with 95% CI for females (A, C) and males (B, D). Low survivorship of the Aut nuclear background accounts for the increase in variance and lack of error bars for development time at high rapamycin concentrations. Statistical results are in Supplementary Table S1 and the main text.
In addition to delaying development, rapamycin caused significant dose-dependent mortality in all genotypes (Fig. 2C, D and Supplementary Fig. S1C, D). An interaction between mtDNA genotype, nuclear genotype, and rapamycin concentration significantly affected survival (mtDNA × nuclear × rapamycin, P < 0.0003 in both experiments), a pattern that was independent of sex (mtDNA × nuclear × rapamycin × sex, P > 0.39 in both experiments) (Supplementary Tables S1 and S2). Again, this effect was attenuated in (simw501); OreR relative to the control genotypes, with this genotype often having the highest survival in the presence of rapamycin (Fig. 2C, D). This pattern was only observed when first-instar larvae (Fig. 2) rather than embryos (Supplementary Fig. S1) were placed on food containing rapamycin, likely due to high embryonic lethality in this genotype (Zhang et al. 2017). In summary, (simw501); OreR individuals were relatively resistant to the effects of rapamycin on survival to adulthood and development time, suggesting that this genotype may have less responsive TOR signaling as a consequence of a deficient cellular energetic state even when provided a high-nutrient diet.
The effects of diet and rapamycin were genotype and sex specific
Dietary yeast levels affect Drosophila development and ovary size (Bergland et al. 2008; Becher et al. 2012). Yeast is an important source of dietary amino acids, and limiting dietary amino acids slow Drosophila development, possibly via TOR signaling (Colombani et al. 2003; Oldham and Hafen 2003). We reared mito–nuclear genotypes on both high- and low-yeast diets across a range of rapamycin concentrations to test two hypotheses. We first tested whether (simw501); OreR individuals were relatively resistant to the effects of decreased dietary yeast in the absence of rapamycin treatment. While a low-yeast diet extended development in all genotypes in the absence of rapamycin, the effect was dampened in (simw501); OreR (Fig. 3A, B) (Supplementary Table S3). On a high-yeast diet, the development time of this genotype was delayed by nearly 2 days, relative to genotypic controls (Pfemales < 0.05, Pmales < 0.01 for all Tukey’s contrasts). However, in a low-yeast environment the developmental time of (simw501); OreR flies was not significantly different from genotypic controls (Pfemales > 0.38, Pmales > 0.44 for all Tukey’s contrasts). This pattern was also observed on the medium-mixed diet that was intermediate in yeast content (Supplementary Fig. S2B and Table S4). The lack of extended development on a low-yeast diet appeared to come at a cost to female survival to adulthood; female (simw501); OreR larval-to-adult survival was significantly reduced to 50% on a low-yeast diet, relative to control genotypes (P < 0.001 for all Tukey’s contrasts) (Fig. 3C), while males had survival that was similar to OreR genotypic controls under both diets (PHigh-yeast > 0.05, PLow-yeast > 0.05 for all Tukey’s contrasts) (Fig. 3D and Supplementary Table S3).
Fig. 3.

Dietary yeast modified the effects of a mitochondrial–nuclear incompatibility on development time and survival. (A, B) Decreased dietary yeast delayed development of all genotypes, but the response of (simw501); OreR to dietary yeast was less than that of control genotypes. The differences in average development time in hours between (simw501); OreR and OreR nuclear genotypic controls are indicated. (C, D) (simw501); OreR females, but not males, had decreased larval-to-adult survival relative to control genotypes when developed on a low-yeast diet. Points are average trait values across five replicate vials with 95% CI for females (A, C) and males (B, D). Statistical results are in Supplementary Table S3 and the main text.
Second, we aimed to test whether control genotypes developed with decreased dietary nutrients were resistant to rapamycin, in a similar way to (simw501); OreR individuals fed a non-limiting diet. However, flies with the Aut nuclear background had very low survival to adulthood when developed on rapamycin, independent of mtDNA genotype. This effect was enhanced on the low-yeast diet, with very few individuals surviving after greatly extended development in the presence of rapamycin. At 10 µM rapamycin on a low-yeast diet, too few flies of all genotypes survived to provide good estimates of development time (Supplementary Fig. S3). However, we were able to use two compatible mito–nuclear genotypes with the OreR nuclear background—(ore); OreR and (sm21); OreR—to test the prediction that control genotypes fed a low-yeast diet would be less responsive to 5 µM rapamycin, similar to the (simw501); OreR genotype. Consistent with this prediction, (ore); OreR flies developed on a low-yeast diet had a dampened response of development time to 5 µM rapamycin, relative to (ore); OreR flies developed on a high-yeast diet (yeast × rapamycin, P = 0.007), an effect that was independent of sex (yeast × rapamycin × sex, P = 0.11) (Fig. 4 and Supplementary Tables S5 and S6). However, this pattern was not observed in (sm21); OreR (yeast × rapamycin, P = 0.85; yeast × rapamycin × sex, P = 0.45) (Fig. 4 and Supplementary Tables S5 and S6). Together, our results indicate that nutrient limitation—either in the diet or by mutations affecting energy metabolism—can attenuate delays in larval development due to nutrient-signaling via TOR.
Fig. 4.

A low-yeast diet attenuated the response of some mitochondrial–nuclear genotypes to rapamycin. Similar to (simw501); OreR on a high-yeast diet, the (ore); OreR genotype had an attenuated response to rapamycin when fed a low-yeast diet. Points are average trait values across five replicate vials with 95% CI for females (A) and males (B). Statistical results are in Supplementary Table S5 and the main text.
Energetically-compromised individuals had decreased immune function
We measured the survival of (simw501); OreR adults and genotypic controls after infection with the natural Drosophila bacterial pathogen P. rettgeri, as well as adult flies that were given a sham infection. The majority of deaths occurred 3–4 days post infection, consistent with prior studies using this pathogen (Duneau et al. 2017a). The proportion of flies surviving infection was significantly affected by mito–nuclear genotype (mtDNA × nuclear × infection, P = 0.014), with greater mortality in the energetically-compromised genotype (Fig. 5 and Supplementary Tables S7 and S8). While the four-way interaction with sex was not significant, the magnitude of the effect of infection on (simw501); OreR females was larger than it was in males (Supplementary Tables S7 and S8). Survival analyses also indicated that the hazard ratio associated with infection was larger for individuals with the energetically-compromised genotype, relative to other genotypes, and larger for females of this genotype, relative to males (female hazard ratio = 4.72, male hazard ratio = 3.76) (Supplementary Fig. S4 and Table S9).
Fig. 5.

The energetically-compromised genotype (simw501); OreR had decreased survival of infection with the natural pathogen P. rettgeri, relative to control genotypes, an effect that was greater in females (A) than in males (B). Control refers to sham infection. Points are averages across five to six replicate vials with 95% CI. Survival plots are provided in Supplementary Fig. S4. Statistical results are in Supplementary Table S7 and the main text.
Compromised energy metabolism revealed an immunity–fecundity tradeoff
We measured the offspring produced by females that survived for 5 days following bacterial or sham infection. There was a significant interaction effect between mtDNA, nuclear genotype, and infection treatment on the number of offspring produced by females (mtDNA × nuclear × infection, P = 0.0056). This interaction was only significant when (simw501); OreR females were included in the analysis (Supplementary Table S10). In control genotypes, there was no evidence for a tradeoff between immunity and fecundity; over the course of 3 days, females with control genotypes produced similar numbers of offspring whether they had survived a sham infection or a pathogen infection (infection, P = 0.99), a pattern that was independent of mito–nuclear genotype (mtDNA × nuclear × infection, P = 0.10) (Fig. 6 and Supplementary Fig. S5 and Table S9). However, (simw501); OreR females that survived infection with P. rettgeri had fewer offspring than sham-infected females of the same genotype (Fig. 6) (infection, P = 0.049), an effect that was larger on the second and third days of egg production (Fig. 6 and Supplementary Fig. S5 and Table S10).
Fig. 6.
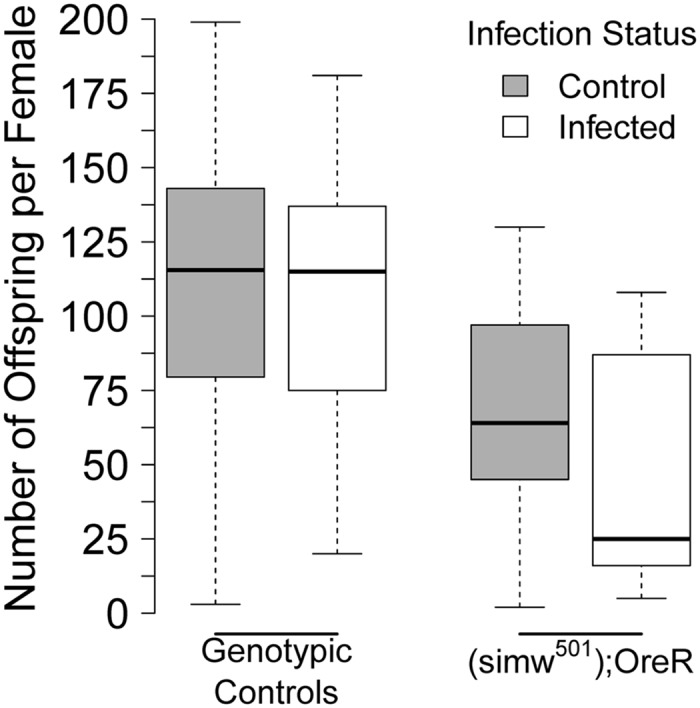
Compromised energy metabolism in (simw501); OreR revealed an immunity–fecundity tradeoff. Surviving infection decreased the total number of offspring produced by (simw501); OreR females, relative to sham-infected females, an effect that was not observed in control genotypes with normal metabolism. Data from 15–20 replicate females for each genotype across 3 days of egg laying are presented in Supplementary Fig. S4. Statistical results are in Supplementary Table S10 and in the main text.
Discussion
Life-history tradeoffs occur due to differential resource allocation to the competing demands of organismal growth, maintenance, performance, and reproduction (Harshman and Zera 2007; King et al. 2011). These tradeoffs can vary among genotypes or within an individual across life stages (Zera and Larsen 2001), and can be modified by environmental stressors, such as temperature (Partridge et al. 1995; Adamo and Lovett 2011), pathogens (Love et al. 2008; McKean et al. 2008; Valtonen and Rantala 2012; Schwenke et al. 2016), and decreased resource availability (Burger et al. 2007). The latter can have particularly strong effects on reproductive fitness that can range from gonadal development (Bergland et al. 2008) to the production of sexual ornaments and signals (Siva-Jothy 2000; Fedorka and Mousseau 2007; Emlen et al. 2012; Gilbert and Uetz 2016; Gilbert et al. 2016). Decreased dietary resources negatively impact ovary development and the number of eggs produced by female Drosophila (Drummond-Barbosa and Spradling 2001; Bergland et al. 2008). In other insects, decreased access to nutritional resources can lower immune activation (Jacot et al. 2005), change gene expression related to immune function (Adamo et al. 2016), and reveal costs of immunity (Moret and Schmid-Hempel 2000). However, immunity–fecundity tradeoffs in insects can also be independent of resource availability (Stahlschmidt et al. 2013). Finally, some insect larvae have diet preferences that maximize the appropriate immune response (Cotter et al. 2011). These observations indicate that energetic–immune interactions are likely important in shaping evolutionary responses to environmental challenges, as well as mediating life-history tradeoffs.
However, nutrient reduction is not always detrimental to immunity (Adamo et al. 2016) or fecundity (May et al. 2015). Short-term starvation can increase survival of infection (Brown et al. 2009), and decreased nutrition can increase generalized immune responses, such as phenyloxidase production (Miller and Cotter 2017a) and encapsulation (Saastamoinen and Rantala 2013), despite the fact that immune responses are energetically expensive (Cutrera et al. 2010; Kvidera et al. 2017). Decreased host cellular resources may also impact pathogen growth independent of changes in host immune function. It is possible that differences between studies are due to differences in the type (generalized vs. specific) of immune response under investigation (Lee 2006), but could also be due to other life-history differences between species (Hawley and Altizer 2011), as well as differences in constitutive versus induced immunity. Our results indicate that genetic variation in mitochondrial and nuclear genomes impacts survival of infection with a natural bacterial pathogen and reveals immunity–fecundity tradeoffs in female Drosophila, likely due to a compromised mitochondrial ability to convert environmental nutrients to cellular resources. While the genotypes in our experiments enable us to infer that the observed effects are due to disrupted mitochondrial protein synthesis, future experiments with additional energetic mutants will be important to test the generality of our findings.
In response to the natural bacterial pathogen P. rettgeri, Drosophila activate the Toll, IMD, and JAK/STAT pathways in the first day of infection and the degree of activation is predictive of survivorship (Sackton et al. 2010; Duneau et al. 2017a). However, natural populations harbor significant genetic variation for surviving infection by P. rettgeri and these genetic effects are modified by diet (Howick and Lazzaro 2014). Our results suggest that mutations that impact mitochondrial function may be an important source of genetic variation for immune function in natural populations. Mitochondria have been linked to innate and adaptive immune responses (West et al. 2011; Pourcelot and Arnoult 2014; Weinberg et al. 2015), although mitochondrial genotype does not always affect post-infection reproduction (Nystrand et al. 2017). While we infer that reduced survival and fecundity of infected (simw501); OreR females is due to a compromised energy supply that cannot meet the competing demands of immune function and reproduction, we did not directly measure immune responses in this study. Mitochondria have other roles that may contribute to our observations, including reactive oxygen species production, mitochondrial antiviral signaling, and cellular damage responses (West et al. 2011; Pourcelot and Arnoult 2014; Weinberg et al. 2015). Furthermore, changes in host cellular energetics may have effects on pathogen growth that are independent of host immune function.
Our results suggest that TOR signaling may be less responsive in energetically inefficient genotypes. External and internal energy sensing is integrated by TOR (Xu et al. 2012; Rider 2016) to regulate growth (Zhang et al. 2000; Kavitha et al. 2014), fecundity (Zhai et al. 2015), and autophagy (Neufeld 2010), and there is some indication of a role for TOR signaling in immunity (Cobbold 2013; Allen et al. 2016). TOR signaling is sensitive to many factors, including decreased nutrition (Nagarajan and Grewal 2014), mitochondrial dysfunction (Kemppainen et al. 2016), and overnutrition (Jia et al. 2014), and populations of D.melanogaster harbor genetic variation, including mitochondrial, that influences energy sensing via TOR (Villa-Cuesta et al. 2014b; Stanley et al. 2017). Thus, TOR signaling is an important pathway integrating external and internal energetic and immunity status that may influence the evolution of life-history traits in response to the environment. Our results are consistent with other studies that indicate that this pathway may be limited in the extent to which the addition of multiple inputs can continue to cause increased signaling via TOR. Both simulated low nutrition via rapamycin (Villa-Cuesta et al. 2014a) and genetic manipulation of TOR (Nagarajan and Grewal 2014) fail to generate the expected phenotypic effects of nutrient limitation. Together, these observations indicate that there may be a threshold for nutrient sensing that, once crossed, prevents further repression of TOR. An alternative hypothesis is that mitochondrial protein synthesis, which is the target of this genetic incompatibility, may act downstream of TOR signaling; in Drosophila, cytoplasmic tRNA synthesis and subsequent protein synthesis are downstream of TOR and are necessary for nutrient-dependent growth regulation via this nutrient-sensing pathway (Rideout et al. 2012).
In our study, infection reduced (simw501); OreR survival more strongly in females than in males. In general, male Drosophila survive infection better than do females (Short and Lazzaro 2010; Vincent and Sharp 2014; Duneau et al. 2017b), a pattern that we also observed. The higher survival of males could result from sex-specific differences in immune expression due to Y-linked regulation (Fedorka and Kutch 2015), differences in antimicrobial peptide production (Jacobs et al. 2016; Duneau et al. 2017b), or potentially from differential suppression of the immune system by juvenile hormone, which has been shown to underlie differences in immune function between mated and un-mated females (Schwenke and Lazzaro 2017). An energetic explanation may be that females have less excess supply to invest in immune function, due to differential costs of gamete production (Bateman 1948; Rolff 2002; McKean et al. 2008; Hayward and Gillooly 2011; Schwenke et al. 2016). Consistent with this idea, mated females have lower antimicrobial peptide production than non-mated females (Short and Lazzaro 2010), and our prior results indicate that compromising cellular energy metabolism has greater effects on female reproduction, relative to male reproduction (Hoekstra et al. 2018).
These patterns are counter to the expectation that female Drosophila might mount stronger immune responses, because the resulting increase in longevity would provide greater lifetime opportunity for reproduction (McKean and Nunney 2005), a pattern that has been observed in many species (Klein 2004; Nunn et al. 2009; Miller and Cotter 2017b). In fact, investment in immunity has been shown to be greater in the sex that has higher investment in offspring, regardless of sex (Roth et al. 2011). However, this pattern may not be observed across all conditions, as environmental effects, such as stress, can decrease immune responses (Husak et al. 2017). Furthermore, in a study where female Drosophila appeared to invest more in immune function than did males, the effects were influenced by the presence of Wolbachia (Gupta et al. 2017). While none of our genotypes are infected with Wolbachia, understanding the interactions between this endosymbiont and mitochondrial effects on host energetics, immunity, and reproduction would provide important insight on the spread of Wolbachia in natural populations. An energetic framework that considers how external environmental conditions and internal conditions, such as sex, endosymbiont status, and tissue (e.g., ovary vs. testes) affect the balance of energy supply and demand (Hoekstra et al. 2018), may be a powerful framework for predicting under what conditions sexes may differ in their immune investment and when genetic variation in mitochondrial function will have sex-specific effects on immune function and tradeoffs between reproduction and immunity (Cressler et al. 2014; Tate and Graham 2015).
Author contributions
J.L.B., C.D.M., and K.L.M. conceived and designed the study and analyzed the data. J.L.B. and C.D.M. carried out the experiments. J.L.B. and K.L.M. drafted the initial version of the manuscript, and all authors revised and gave the final approval for publication.
Supplementary Material
Acknowledgments
We would like to thank Brian Lazzaro for P. rettgeri and intellectual contributions to this study. We are grateful for the technical help of Katie Gordon, Rudy Villegas, Abhilesh Dhawanjewar, Cole Julick, and Omera Matoo. We acknowledge the unwavering support of David Rand.
Funding
This study was supported by National Science Foundation awards [IOS-1149178 and DEB-1701876] and funds from the University of Nebraska–Lincoln. Some data were collected by C.D.M. when he was supported by National Institutes of Health NIGMS [R01GM067862] to David Rand (Brown University).
Supplementary data
Supplementary data are available at ICB online.
Data available from the Dryad Digital Repository at https://datadryad.org/resource/doi:10.5061/dryad.88mk4dh.
References
- Adamo SA. 1999. Evidence for adaptive changes in egg laying in crickets exposed to bacteria and parasites. Anim Behav 57:117–24. [DOI] [PubMed] [Google Scholar]
- Adamo SA, Davies G, Easy R, Kovalko I, Turnbull KF.. 2016. Reconfiguration of the immune system network during food limitation in the caterpillar Manduca sexta. J Exp Biol 219:706–18. [DOI] [PubMed] [Google Scholar]
- Adamo SA, Lovett MME.. 2011. Some like it hot: the effects of climate change on reproduction, immune function and disease resistance in the cricket Gryllus texensis. J Exp Biol 214:1997–2004. [DOI] [PubMed] [Google Scholar]
- Allen VW, O’Connor RM, Ulgherait M, Zhou CG, Stone EF, Hill VM, Murphy KR, Canman JC, Ja WW, Shirasu-Hiza MM.. 2016. Period-regulated feeding behavior and TOR signaling modulate survival of infection. Curr Biol 26:184–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman AJ. 1948. Intra-sexual selection in Drosophila. Heredity 2:349–68. [DOI] [PubMed] [Google Scholar]
- Becher PG, Flick G, Rozpędowska E, Schmidt A, Hagman A, Lebreton S, Larsson MC, Hansson BS, Piškur J, Witzgall P, et al. 2012. Yeast, not fruit volatiles mediate Drosophila melanogaster attraction, oviposition and development. Funct Ecol 26:822–8. [Google Scholar]
- Bergland AO, Genissel A, Nuzhdin SV, Tatar M.. 2008. Quantitative trait loci affecting phenotypic plasticity and the allometric relationship of ovariole number and thorax length in Drosophila melanogaster. Genetics 180:567–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A, Partridge L.. 2010. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab 11:35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfini A, Liu X, Buchon N.. 2016. From pathogens to microbiota: how Drosophila intestinal stem cells react to gut microbes. Dev Comp Immunol 64:22–38. [DOI] [PubMed] [Google Scholar]
- Bonneaud C, Mazuc J, Chastel O, Westerdahl H, Sorci G, Poulin R.. 2004. Terminal investment induced by immune challenge and fitness traits associated with major histocompatibility complex in the house sparrow. Evolution 58:2823–30. [DOI] [PubMed] [Google Scholar]
- Brown AE, Baumbach J, Cook PE, Ligoxygakis P.. 2009. Short-term starvation of immune deficient Drosophila improves survival to Gram-negative bacterial infections. PLoS One 4:e4490.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger JM, Hwangbo DS, Corby-Harris V, Promislow DE.. 2007. The functional costs and benefits of dietary restriction in Drosophila. Aging Cell 6:63–71. [DOI] [PubMed] [Google Scholar]
- Brock PM, Murdock CC, Martin LB.. 2014. The history of ecoimmunology and its integration with disease ecology. Integr Comp Biol 54:353–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clutton-Brock TH. 1984. Reproductive effort and terminal investment in iteroparous animals. Am Nat 123:212–29. [Google Scholar]
- Cobbold SP. 2013. The mTOR pathway and integrating immune regulation. Immunology 140:391–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombani J, Raisin S, Pantalacci S, Radimerski T, Montagne J, Léopold P.. 2003. A nutrient sensor mechanism controls Drosophila growth. Cell 114:739–49. [DOI] [PubMed] [Google Scholar]
- Cotter SC, Simpson SJ, Raubenheimer D, Wilson K.. 2011. Macronutrient balance mediates trade-offs between immune function and life history traits. Funct Ecol 25:186–98. [Google Scholar]
- Cressler CE, Graham AL, Day T.. 2015. Evolution of hosts paying manifold costs of defence. Proc Biol Sci 282:20150065.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cressler CE, Nelson WA, Day T, McCauley E.. 2014. Disentangling the interaction among host resources, the immune system and pathogens. Ecol Lett 17:284–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutrera AP, Zenuto RR, Luna F, Antenucci CD.. 2010. Mounting a specific immune response increases energy expenditure of the subterranean rodent Ctenomys talarum (tuco-tuco): implications for intraspecific and interspecific variation in immunological traits. J Exp Biol 213:715–24. [DOI] [PubMed] [Google Scholar]
- Drummond-Barbosa D, Spradling AC.. 2001. Stem cells and their progeny respond to nutritional changes during Drosophila oogenesis. Dev Biol 231:265–78. [DOI] [PubMed] [Google Scholar]
- Duneau D, Ferdy J-B, Revah J, Kondolf HC, Ortiz GA, Lazzaro BP, Buchon N.. 2017a. Stochastic variation in the initial phase of bacterial infection predicts the probability of survival in D. melanogaster. Elife 6:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duneau DF, Kondolf HC, Im JH, Ortiz GA, Chow C, Fox MA, Eugenio AT, Revah J, Buchon N, Lazzaro BP.. 2017b. The Toll pathway underlies sexual dimorphism in resistance to both Gram-negative and positive-bacteria in Drosophila. BMC Biol 1:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emlen DJ, Warren IA, Johns A, Dworkin I, Lavine LC.. 2012. A mechanism of extreme growth and reliable signaling in sexually selected ornaments and weapons. Science 337:860–4. [DOI] [PubMed] [Google Scholar]
- Fedorka KM, Kutch IC.. 2015. Y-linked variation for autosomal immune gene regulation has the potential to shape sexually dimorphic immunity. Proc Biol Sci 282:20151301.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorka KM, Mousseau TA.. 2007. Immune system activation affects male sexual signal and reproductive potential in crickets. Behav Ecol 18:231–5. [Google Scholar]
- Fellous S, Lazzaro BP.. 2011. Potential for evolutionary coupling and decoupling of larval and adult immune gene expression. Mol Ecol 20:1558–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert R, Karp RD, Uetz GW.. 2016. Effects of juvenile infection on adult immunity and secondary sexual characters in a wolf spider. Behav Ecol 27:946–54. [Google Scholar]
- Gilbert R, Uetz GW.. 2016. Courtship and male ornaments as honest indicators of immune function. Anim Behav 117:97–103. [Google Scholar]
- Gupta V, Vasanthakrishnan RB, Siva-Jothy J, Monteith KM, Brown SP, Vale PF.. 2017. The route of infection determines Wolbachia antibacterial protection in Drosophila. Proc Biol Sci 284:20170809.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn-Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N.. 2005. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J Biol Chem 280:32081–9. [DOI] [PubMed] [Google Scholar]
- Harshman LG, Zera AJ.. 2007. The cost of reproduction: the devil in the details. Trends Ecol Evol 22:80–6. [DOI] [PubMed] [Google Scholar]
- Hawley DM, Altizer SM.. 2011. Disease ecology meets ecological immunology: understanding the links between organismal immunity and infection dynamics in natural populations. Funct Ecol 25:48–60. [Google Scholar]
- Hayward A, Gillooly JF.. 2011. The cost of sex: quantifying energetic investment in gamete production by males and females. PLoS One 6:e16557.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra LA, Cole R, Julick Mika KM, Montooth KL.. 2018. Energy demand and the context-dependent effects of genetic interactions underlying metabolism. Evol Lett 2:102–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra LA, Siddiq MA, Montooth KL.. 2013. Pleiotropic effects of a mitochondrial–nuclear incompatibility depend upon the accelerating effect of temperature in Drosophila. Genetics 195:1129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmbeck MA, Donner JR, Villa-Cuesta E, Rand DM.. 2015. A Drosophila model for mito–nuclear diseases generated by an incompatible interaction between tRNA and tRNA synthetase. Dis Model Mech 8:843–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howick VM, Lazzaro BP.. 2014. Genotype and diet shape resistance and tolerance across distinct phases of bacterial infection. BMC Evol Biol 14:56.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husak JF, Roy JC, Lovern MB.. 2017. Exercise training reveals trade-offs among endurance performance and immune function, but not growth, in juvenile lizards. J Exp Biol 220:1497–502. [DOI] [PubMed] [Google Scholar]
- Jacobs CGC, Steiger S, Heckel DG, Wielsch N, Vilcinskas A, Vogel H.. 2016. Sex, offspring and carcass determine antimicrobial peptide expression in the burying beetle. Sci Rep 6:25409.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacot A, Scheuber H, Kurtz J, Brinkhof MW.. 2005. Juvenile immune system activation induces a costly upregulation of adult immunity in field crickets Gryllus campestris. Proc Biol Sci 272:63–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K, McClure C, Priest NK, Hunt J.. 2015. Sex-specific effects of protein and carbohydrate intake on reproduction but not lifespan in Drosophila melanogaster. Aging Cell 14:605–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Aroor AR, Martinez-Lemus LA, Sowers JR.. 2014. Overnutrition, mTOR signaling, and cardiovascular diseases. Am J Physiol Regul Integr Comp Physiol 307:R1198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juneja P, Lazzaro BP.. 2009. Providencia sneebia sp. nov. and Providencia burhodogranariea sp. nov., isolated from wild Drosophila melanogaster. Int J Syst Evol Microbiol 59:1108–11. [DOI] [PubMed] [Google Scholar]
- Kavitha JV, Rosario FJ, Nijland MJ, McDonald TJ, Wu G, Kanai Y, Powell TL, Nathanielsz PW, Jansson T.. 2014. Down-regulation of placental mTOR, insulin/IGF-I signaling, and nutrient transporters in response to maternal nutrient restriction in the baboon. FASEB J 28:1294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemppainen E, George J, Garipler G, Tuomela T, Kiviranta E, Soga T, Dunn CD, Jacobs HT.. 2016. Mitochondrial dysfunction plus high-sugar diet provokes a metabolic crisis that inhibits growth. PLoS One 11:e0145836.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil S, Jacobson E, Chambers MC, Lazzaro BP.. 2015. Systemic bacterial infection and immune defense phenotypes in Drosophila melanogaster. J Vis Exp 99:52613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King EG, Roff DA, Fairbairn DJ.. 2011. Trade-off acquisition and allocation in Gryllus firmus: a test of the Y model. J Evol Biol 24:256–64. [DOI] [PubMed] [Google Scholar]
- Klein SL. 2004. Hormonal and immunological mechanisms mediating sex differences in parasite infection. Parasite Immunol 26:247–64. [DOI] [PubMed] [Google Scholar]
- Kurze C, Mayack C, Hirche F, Stangl GI, Le Conte Y, Kryger P, Moritz RFA.. 2016. Nosema spp. infections cause no energetic stress in tolerant honeybees. Parasitol Res 115: 2381–8. [DOI] [PubMed] [Google Scholar]
- Kvidera SK, Horst EA, Abuajamieh M, Mayorga EJ, Fernandez MVS, Baumgard LH.. 2017. Glucose requirements of an activated immune system in lactating Holstein cows. J Dairy Sci 100:2360–74. [DOI] [PubMed] [Google Scholar]
- Lee KA. 2006. Linking immune defenses and life history at the levels of the individual and the species. Integr Comp Biol 46:1000–15. [DOI] [PubMed] [Google Scholar]
- Lochmiller RL, Deerenberg C.. 2000. Trade-offs in evolutionary immunology: just what is the cost of immunity? Oikos 88:87–98. [Google Scholar]
- Love OP, Salvante KG, Dale J, Williams TD.. 2008. Sex-specific variability in the immune system across life-history stages. Am Nat 172:E99–112. [DOI] [PubMed] [Google Scholar]
- May CM, Doroszuk A, Zwaan BJ.. 2015. The effect of developmental nutrition on life span and fecundity depends on the adult reproductive environment in Drosophila melanogaster. Ecol Evol 5:1156–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKean KA, Nunney L.. 2005. Bateman’s principle and immunity: phenotypically plastic reproductive strategies predict changes in immunological sex differences. Evolution 59:1510–7. [PubMed] [Google Scholar]
- McKean KA, Yourth CP, Lazzaro BP, Clark AG.. 2008. The evolutionary costs of immunological maintenance and deployment. BMC Evol Biol 8:76.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn CD, Holmbeck MA, Siddiq MA, Abt DN, Rand DM, Montooth KL.. 2013. An incompatibility between a mitochondrial tRNA and its nuclear-encoded tRNA synthetase compromises development and fitness in Drosophila. PLoS Genet 9:e1003238.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CVL, Cotter SC.. 2017a. Resistance and tolerance: the role of nutrients on pathogen dynamics and infection outcomes in an insect host. J Anim Ecol 87:500–10. [DOI] [PubMed] [Google Scholar]
- Miller CVL, Cotter SC.. 2017b. Pathogen and immune dynamics during maturation are explained by Bateman’s principle. Ecol Entomol 42:28–38. [Google Scholar]
- Millington JW, Rideout EJ.. 2018. Sex differences in Drosophila development and physiology. Curr Opin Physiol 6:46. [Google Scholar]
- Montooth KL, Meiklejohn CD, Abt DN, Rand DM.. 2010. Mitochondrial–nuclear epistasis affects fitness within species but does not contribute to fixed incompatibilities between species of Drosophila. Evolution 64:3364–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moret Y, Schmid-Hempel P.. 2000. Survival for immunity: the price of immune system activation for bumblebee workers. Science 290:1166–8. [DOI] [PubMed] [Google Scholar]
- Nagarajan S, Grewal SS.. 2014. An investigation of nutrient-dependent mRNA translation in Drosophila larvae. Biol Open 3:1020–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld TP. 2010. TOR-dependent control of autophagy: biting the hand that feeds. Curr Opin Cell Biol 22:157–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunn CL, Lindenfors P, Pursall ER, Rolff J.. 2009. On sexual dimorphism in immune function. Philos Trans R Soc Lond B Biol Sci 364:61–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystrand M, Cassidy EJ, Dowling DK.. 2017. No effect of mitochondrial genotype on reproductive plasticity following exposure to a non-infectious pathogen challenge in female or male Drosophila. Sci Rep 7:42009.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham S, Hafen E.. 2003. Insulin/IGF and target of rapamycin signaling: a TOR de force in growth control. Trends Cell Biol 13:79–85. [DOI] [PubMed] [Google Scholar]
- Partridge L, Barrie B, Barton NH, Fowler K, French V.. 1995. Rapid laboratory evolution of adult life-history traits in Drosophila melanogaster in response to temperature. Evolution 49:538–44. [DOI] [PubMed] [Google Scholar]
- Pourcelot M, Arnoult D.. 2014. Mitochondrial dynamics and the innate antiviral immune response. FEBS J 281:3791–802. [DOI] [PubMed] [Google Scholar]
- R Core Team. 2017. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing (https://www.R-project.org/). [Google Scholar]
- Rauw WM. 2012. Immune response from a resource allocation perspective. Front Genet 3:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reavey CE, Silva FWS, Cotter SC.. 2015. Bacterial infection increases reproductive investment in burying beetles. Insects 6:926–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rideout EJ, Marshall L, Grewal SS.. 2012. Drosophila RNA polymerase III repressor Maf1 controls body size and developmental timing by modulating tRNAiMet synthesis and systemic insulin signaling. Proc Natl Acad Sci U S A 109:1139–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rider MH. 2016. Role of AMP-activated protein kinase in metabolic depression in animals. J Comp Physiol B 186:1–16. [DOI] [PubMed] [Google Scholar]
- Rolff J. 2002. Bateman’s principle and immunity. Proc Biol Sci 269:867–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth O, Scharsack JP, Keller I, Reusch TBH.. 2011. Bateman’s principle and immunity in a sex-role reversed pipefish. J Evol Biol 24:1410–20. [DOI] [PubMed] [Google Scholar]
- Saastamoinen M, Rantala MJ.. 2013. Influence of developmental conditions on immune function and dispersal-related traits in the glanville fritillary (Melitaea cinxia) butterfly. PLoS One 8:e81289.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackton TB, Lazzaro BP, Clark AG.. 2010. Genotype and gene expression associations with immune function in Drosophila. PLoS Genet 6:e1000797.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenke RA, Lazzaro BP.. 2017. Juvenile hormone suppresses resistance to infection in mated female Drosophila melanogaster. Curr Biol 27:596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenke RA, Lazzaro BP, Wolfner MF.. 2016. Reproduction–immunity trade-offs in insects. Annu Rev Entomol 61:239–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon BC, Verhulst S.. 1996. Ecological immunology: costly parasite defences and trade-offs in evolutionary ecology. Trends Ecol Evol 11:317–21. [DOI] [PubMed] [Google Scholar]
- Short SM, Lazzaro BP.. 2010. Female and male genetic contributions to post-mating immune defence in female Drosophila melanogaster. Proc Biol Sci 277:3649–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short SM, Lazzaro BP.. 2013. Reproductive status alters transcriptomic response to infection in female Drosophila melanogaster. G3 3:827–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siva-Jothy MT. 2000. A mechanistic link between parasite resistance and expression of a sexually selected trait in a damselfly. Proc Biol Sci 267:2523–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorupa DA, Dervisefendic A, Zwiener J, Pletcher SD.. 2008. Dietary composition specifies consumption, obesity, and lifespan in Drosophila melanogaster. Aging Cell 7:478–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahlschmidt ZR, Rollinson N, Acker M, Adamo SA.. 2013. Are all eggs created equal? Food availability and the fitness trade-off between reproduction and immunity. Funct Ecol 27:800–6. [Google Scholar]
- Stanley PD, Ng′oma E, O′Day S, King EG. 2017. Genetic dissection of nutrition-induced plasticity in insulin/insulin-like growth factor signaling and median life span in a Drosophila multiparent population. Genetics 206:587–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate AT, Graham AL.. 2015. Dynamic patterns of parasitism and immunity across host development influence optimal strategies of resource allocation. Am Nat 186:495–512. [DOI] [PubMed] [Google Scholar]
- Therneau TM, Grambsch PM, Pankratz VS.. 2003. Penalized survival models and frailty. J Comput Graph Stat 12:156–75. [Google Scholar]
- Valtonen TM, Rantala MJ.. 2012. Poor early nutrition reveals the trade-off between immune defense and mating success. Ecol Parasitol Immunol 1:1–7. [Google Scholar]
- Villa-Cuesta E, Fan F, Rand DM.. 2014a. Rapamycin reduces Drosophila longevity under low nutrition. IOSR J Pharm 4:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa-Cuesta E, Holmbeck M, Rand D.. 2014b. Rapamycin increases mitochondrial efficiency by mtDNA-dependent reprogramming of mitochondrial metabolism in Drosophila. J Cell Sci 127:2282–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent CM, Sharp NP.. 2014. Sexual antagonism for resistance and tolerance to infection in Drosophila melanogaster. Proc Biol Sci 281:20140987.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Mouser J, Pitt J, Promislow D, Kaeberlein M.. 2016. Rapamycin enhances survival in a Drosophila model of mitochondrial disease. Oncotarget 7:80131–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks AR, Stouthamer R.. 2004. Increased fecundity associated with infection by a Cytophaga-like intracellular bacterium in the predatory mite, Metaseiulus occidentalis. Proc Biol Sci 271:S193–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg SE, Sena LA, Chandel NS.. 2015. Mitochondria in the regulation of innate and adaptive immunity. Immunity 42:406–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Shadel GS, Ghosh S.. 2011. Mitochondria in innate immune responses. Nat Rev Immunol 11:389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Ji J, Yan X-H.. 2012. Cross-talk between AMPK and mTOR in regulating energy balance. Crit Rev Food Sci Nutr 52:373–81. [DOI] [PubMed] [Google Scholar]
- Zera AJ, Larsen A.. 2001. The metabolic basis of life history variation: genetic and phenotypic differences in lipid reserves among life history morphs of the wing-polymorphic cricket, Gryllus firmus. J Insect Physiol 47:1147–60. [DOI] [PubMed] [Google Scholar]
- Zhai Y, Sun Z, Zhang J, Kang K, Chen J, Zhang W.. 2015. Activation of the TOR signalling pathway by glutamine regulates insect fecundity. Sci Rep 5:10694.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Montooth KL, Calvi BR.. 2017. Incompatibility between mitochondrial and nuclear genomes during oogenesis results in ovarian failure and embryonic lethality. Development 144:2490–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Stallock JP, Ng JC, Reinhard C, Neufeld TP.. 2000. Regulation of cellular growth by the dTOR. Genes Dev 14:2712–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng XF, Florentino D, Chen J, Crabtree GR, Schreiber SL.. 1995. TOR kinase domains are required for two distinct functions, only one of which is inhibited by rapamycin. Cell 82:121–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.