

Abstract
S119 was a top hit from an ultrahigh throughput screen performed to identify novel inhibitors of influenza virus replication. It showed a potent antiviral effect (50% inhibitory concentration, IC50 = 20 nM) and no detectable cytotoxicity (50% cytotoxic concentration, CC50 > 500 μM) to yield a selectivity index greater than 25 000. Upon investigation, we found that S119 selected for resistant viruses carrying mutations in the viral nucleoprotein (NP). These resistance mutations highlight a likely S119 binding site overlapping with but not identical to that found for the compound nucleozin. Mechanism of action studies revealed that S119 affects both the oligomerization state and cellular localization of the NP protein which has an impact on viral transcription, replication, and protein expression. Through a hit-to-lead structure−activity relationship (SAR) study, we found an analog of S119, named S119–8, which had increased breadth of inhibition against influenza A and B viruses accompanied by only a small loss in potency. Finally, in vitro viral inhibition assays showed a synergistic relationship between S119–8 and oseltamivir when they were combined, indicating the potential for future drug cocktails.
Keywords: influenza, antiviral, nucleoprotein, aggregation, nucleozin
Influenza virus is an important human pathogen, which accounts for significant morbidity and mortality worldwide in annual epidemics. In addition, through genomic reassortment, influenza A virus has and will continue to cause periodic global pandemics. The worst of these was the 1918 “Spanish flu”, which caused an estimated 50−100 million deaths.32 During the most recent H1N1 pandemic in 2009, efforts to produce and deliver a vaccine to protect the world population were outpaced by the spread of the virus. This failure of the influenza pandemic vaccination program highlights the importance of antiviral therapies under pandemic conditions. Moreover, coverage of seasonal influenza vaccines is estimated at only 33−73%,33 leaving a significant proportion of the population vulnerable to infection every year.
There is an urgent need for new antiviral therapies against influenza virus, as the two current FDA-approved drug classes have both suffered from significant issues of viral resistance, either currently or in the recent past. The CDC no longer recommends the M2 inhibitors (amantadine and rimantadine) for use in the clinic due to high levels of resistance in both H1N1 and H3N2 circulating strains.1 Neuraminidase inhibitors, of which the orally available oseltamivir is the most heavily prescribed, are currently the only FDA approved antiviral drugs recommended for use against influenza virus. Although oseltamivir resistance remains relatively low (0−0.8%) currently, during the 2007−2008 influenza season, the circulating H1N1 strain gained nearly complete resistance.2 This oseltamivir resistant strain was fortunately replaced by the 2009 H1N1 pandemic strain, but the quick spread of this resistance, to our last line of antiviral defense, is highly concerning and highlights the necessity of finding new therapeutics with different targets.
The influenza nucleoprotein (NP) is a critical factor in many stages of the viral life cycle. The NP protein coats the viral genomic RNA to form ribonucleoproteins (RNPs)3 and is involved in transportation of RNPs to the nucleus,4 viral transcription and replication,5,6 and virion assembly.7 Therefore, NP is localized within the cytoplasm during viral entry, within the nucleus during viral replication, and finally in the cytoplasm again during egress.8 Knowledge of the NP crystal structure20 and RNP cryo-EM structure3,9 has significantly advanced our understanding of the multimeric nature of NP and how important its oligomeric state is to viral replication. Recently, the NP protein was shown to be a valid and efficient target for inhibition of influenza virus replication and pathogenesis.10−12 The compound, nucleozin, was found to inhibit influenza via blocking NP import into the nucleus and causing abnormal aggregation within the cytoplasm.11
In a high throughput screen, which we have previously described,13 919, 960 compounds were screened for inhibition of influenza virus replication in a cell-based assay. In this screen, we identified a number of influenza specific inhibitors of viral replication with submicromolar 50% inhibitory concentrations (IC50’s). One of these compounds, designated S119, was found to inhibit replication through alteration of the oligomeric state of the NP protein. Herein, we discuss the mechanism of action of S119 and a preliminary structure−activity relationship (SAR) study to improve the breadth of antiviral activity as proof-of-concept for future drug lead optimization.
RESULTS AND DISCUSSION
S119 Is a Specific Inhibitor of Influenza A Virus.
The compound S119 (Figure 1a) was identified as a potent hit from a previously published cell-based screen of nearly one million compounds utilizing a luciferase-based reporter influenza A/WSN/33 H1N1 (WSN) virus.14 S119 potently inhibited both the reporter virus and wild-type WSN virus in MDCK cells with an IC50 of 60 nM (Figure 1b). When tested in the human lung cell line, A549, S119 caused a 4 log reduction in WSN viral titers resulting in an IC50 of 20 nM (Figure 1c). Cytotoxicity of S119 was tested in A549 cells using the CellTiter-Glo system (Promega), and the compound showed very little toxicity at concentrations as high as 500 μM, yielding a minimum SI of 25 000. When examined for activity against other strains of influenza A virus, we found that S119 caused a 77% reduction in the pandemic strain A/California/2009 (H1N1) infectious titer but was ineffective against A/Puerto Rico/8/1934 (PR8, H1N1), A/Panama/2007/1999 (H3N2), and A/Vietnam/1203/2004 (H5N1) (Table 2).
Figure 1.
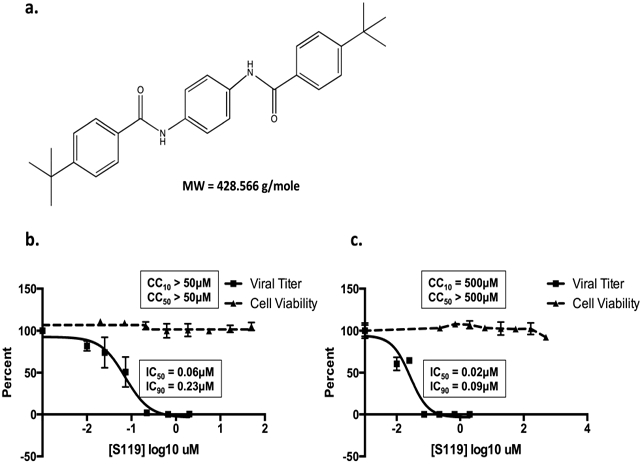
S119 potently inhibits influenza A virus. (a) Chemical structure of compound S119 and its molecular weight (MW). (b) MDCK or (c) A549 cells were infected with influenza A/WSN/33 virus (MOI = 0.01) in the presence of serially diluted S119. Viral titers were determined 24 h post-infection (solid line), and the IC50 and IC90 were calculated. Cell viability was determined in an independent experiment in A549 cells over a 24 h period (dashed line), and the CC50 and CC10 were calculated. Mean of three replicates ± SD are shown.
Table 2.
S119–8 Shows Increased Breadth of Activity against Multiple Influenza A and B Virusesa
| influenza virus | S119 IC50 (μM) | S119–8 IC50 (μM) | S119–8 CC50 (μM) | subtype/lineage | cell line |
|---|---|---|---|---|---|
| A/WSN/1933 | 0.02 | 1.43 | >50 | H1N1 | A549 |
| A/Califomia/04/2009 | 27.43 | 5.32 | >50 | H1N1 | A549 |
| A/Puerto Rico/8/1934* | >50 | 6.05 | >50 | H1N1 | A549 |
| A/Brisbane/59/2007-S* | >50 | 6.68 | 40.66 | H1N1 | A549 |
| A/Brisbane/59/2007-R* | >50 | 3.85 | 40.66 | H1N1 | A549 |
| A/Panama/2007/1999 | >50 | 6.43 | >50 | H3N2 | A549 |
| A/Wyoming/03/2003* | >50 | 11.53 | 40.66 | H3N2 | A549 |
| A/Vietnam/1203/2004 | >50 | 7.94 | >50 | H5N1 | A549 |
| B/Yamagata/16/1988 | >50 | 2.08 | >50 | Yamagata | MDCK |
| B/Brisbane/60/2008* | >50 | 15.153 | >50 | Victoria | MDCK |
S119 and S119–8 were tested against a number of influenza A and B viruses. Average IC50 and CC50 values of triplicate experiments are presented. Each strain indicated with a * was quantified using NP-staining of infected cells 24 h post-infection, and signal was detected using the Celigo Imaging Cytometer (Nexcelcom Biosciences, Lawrence, MA). Infectivity was determined by NP signal over DAPI counterstain of total cells. Otherwise, infections were quantified by standard plaque assay of supernatants from infected cells at 24 h post-infection.
S119 Inhibits a Post-entry Step and Targets the NP protein.
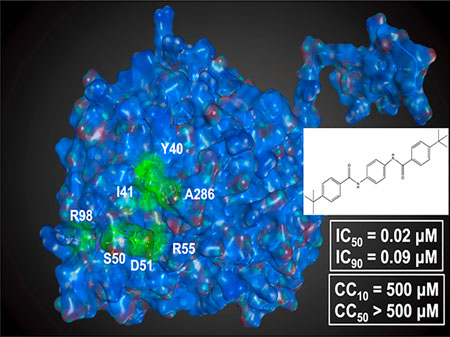
To begin to dissect the mechanism of action of S119, we first sought to determine at which stage of viral infection the compound was inhibiting. We performed a time of addition assay at an MOI of 1 (A/WSN/33) with S119 treatments at −2, 0, 2, 4, 6, or 8 h relative to infection. We found that S119 was able to maximally inhibit virus replication when added before infection and up to 2 h after infection. The compound also showed significant virus inhibition when added 4 h after infection, while still maintaining a 1 log effect at 6 h (Figure 2). This indicated that S119 was exerting its effect at a post-entry step of the viral life cycle, possibly during viral transcription and replication. To determine whether S119 was targeting a viral protein, influenza virus was passaged in the presence of S119 and S119-resistant viruses were selected. Briefly, A549 cells were infected with influenza A/WSN/33 virus at an MOI of 0.01 in the presence of 2 μM S119, which yielded enough viral progeny for subsequent passages at a low MOI as detected by the viral plaque assay. The remaining passages were tracked by the hemagglutination assay to determine if the virus had escaped inhibition by S119. Viral resistance to S119 was detected after 3 to 4 passages, and isolates were obtained from 7 independent experiments, with 3 viruses plaque purified from each experiment and submitted for full genome sequencing. Of the 21 sequenced viral genomes, 11 contained mutations leading to 7 unique amino acid changes all occurring within the nucleoprotein (NP). When these resistance mutations were mapped onto the crystal structure of the NP protein, 6 of the 7 residues were found to form a single pocket on the surface of the protein (Figure 3a). The remaining mutated residue, R98, was found to be within close proximity to this pocket as well. We found this particularly illuminating due to the 246 amino acid distance between the most distal residues (Y40 and A286). This indicated to us that not only was NP the likely target for S119 inhibition but also this pocket highlighted by resistance mutations is also likely the S119 binding site within the NP protein. This putative binding pocket is located in the “body” domain of NP where an important NP−NP interaction occurs in the formation of the ribonucleoprotein (RNP) complex. Quite interestingly, a known inhibitor of NP, nucleozin,11 has been shown to have two binding sites on either side of the putative S119 binding pocket.15 Nucleozin is proposed to cause an atypical NP−NP interaction through the “body” domains,12 which subsequently leads to formation of large, replication deficient NP aggregates. S119 is structurally unrelated to nucleozin, and the binding sites of the two compounds appear to be distinct with a small amount of overlap. Previous publications have shown that mutations at positions Y40, S50, D51, and A286 (which confer S119 resistance) do not cause resistance to nucleozin, while a mutation at position R55 causes resistance to both compounds.12 The NP protein is highly conserved among influenza viruses, so it is surprising that S119 did not have a broad spectrum of inhibition, particularly in the case of the PR8 strain, which is closely related to WSN. The NP proteins of WSN and PR8 share identical amino acids at each of the resistance mutation sites identified; indeed, these sites are also 99.8−100% conserved in currently circulating influenza A viruses. Even currently circulating influenza B viruses show 100% conservation of identical or similar amino acids in the corresponding sites of the influenza B NP protein. Interestingly, there is a proline at position 283 of the PR8 NP protein, whereas there is a serine at this position in WSN. This S283P substitution would have a significant effect on the structure of the α helix in which the A286 resistance site resides and therefore change the conformation of the putative S119 binding pocket. It may be that the extended structure of the S119 molecule is highly intolerant of any change to the size of its binding pocket.
Figure 2.
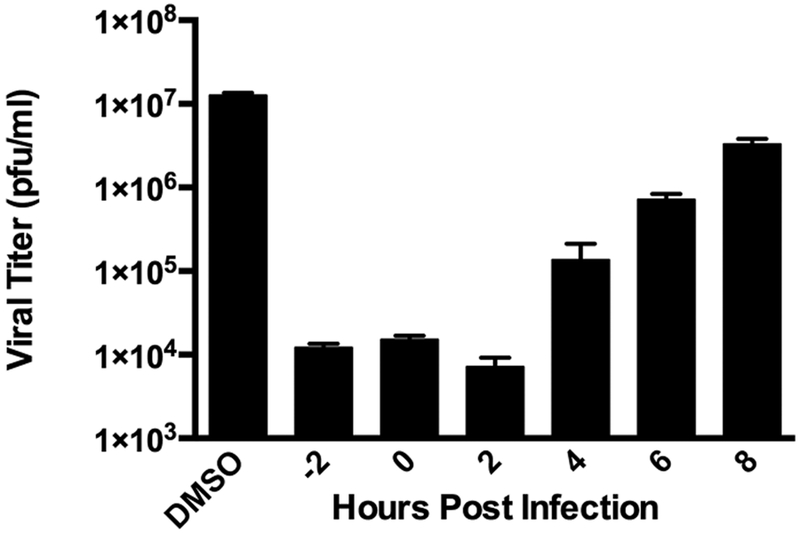
S119 inhibits a post-entry stage of the viral life cycle. Time-of-addition assay for inhibition of influenza A/WSN/33 virus by S119. A549 cells were infected with influenza virus A/WSN/33 (MOI = 1). Compound S119 (10 μM) was present in the culture medium 2 h before infection or added to the medium at the indicated time points post-infection. Viral titers were determined 24 h post-infection by the plaque assay. The assay was performed in triplicate; results are presented as the mean ± SD.
Figure 3.
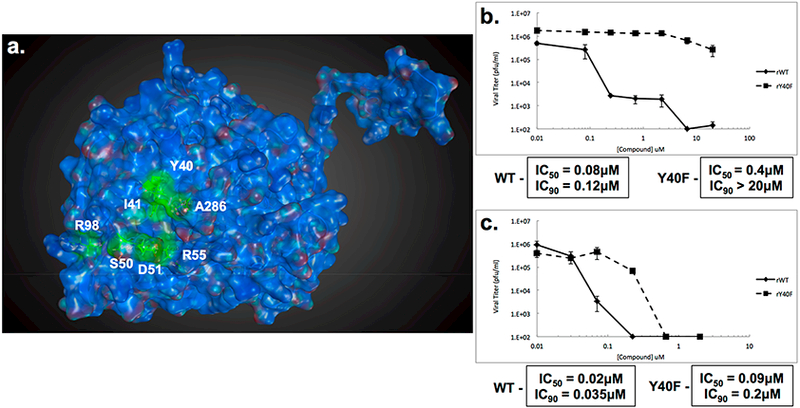
Resistance mutations to S119 occur within the nucleoprotein. (a) Crystal structure of A/WSN/33 NP (PDB 2IQH) monomer. Residue positions where S119 escape mutations occurred are indicated in green. Virus titers from A549 cells infected with either rWSN-WT or rWSN-NP/Y40F viruses (MOI = 0.01) in the presence of increasing concentrations of (b) S119 or (c) nucleozin for 24 h. Curves represent means of triplicate values ± SD. IC50 and IC90 values are indicated.
To further explore the impact of resistance to S119, the Y40F mutation was introduced into a recombinant influenza A/WSN/33 virus using a reverse genetics system.16 This virus, referred to as rWSN-Y40F, was not impaired for growth when compared to a wild-type recombinant virus in a multicycle growth assay (data not shown). When S119 was tested for efficacy against rWSN-Y40F, the mutant virus had a resistant phenotype, with an IC50 50-fold higher than the wild-type counterpart (Figure 3b). Conversely, the IC50 for nucleozin was only 4.5-fold higher with the Y40F NP mutation (Figure 3c). This minor resistance phenotype is in agreement with the literature that this residue is adjacent to, but not part of, the nucleozin binding site in NP.12
S119 Causes Aggregation of the NP Protein.
The importance of the oligomeric state of the influenza virus NP protein has been well-defined through previous biochemical and structural studies,17−19 and the location of the S119 resistance mutations near to those conferring nucleozin resistance indicated that alteration of the NP−NP interaction at this interface may be the mechanism of S119 antiviral activity. To determine whether S119 has an effect on NP oligomeric state, purified baculovirus-expressed NP was incubated with and without compound for 30 min in the presence or absence of purified viral RNA and analyzed by blue native PAGE. In the absence of RNA, the purified NP runs at a low molecular weight, likely representing monomeric NP (Figure 4a, upper panel). The band appears as a doublet because of a slightly truncated form of NP being coexpressed with the full-length protein during baculovirus expression. This was confirmed via Western blot analysis, where the truncated band is recognized by both anti-NP and anti-His antibodies. This indicated a C-terminally truncated NP species, because the His-tag is located on the N-terminus of the recombinant protein. In Figure 4a (lower panel, lane 1), both forms of NP are shown to be capable of oligomerizing on RNA, and therefore, the truncated protein should not affect the experimental outcome. An active analog of the published PB2 inhibitor VX-78720 was used as a negative control. When treated with S119 or nucleozin in the absence of RNA, the low molecular weight NP band is lost relative to controls. For nucleozin, it has been described that NP shifts into an unresolvable higher order complex,11 and this is likely the case for S119 treatment as well. Interestingly, S119 appears to have a greater effect on the low molecular weight NP than nucleozin at the same concentration, despite nucleozin having similar potency in viral inhibition assays. In the presence of influenza vRNA, most of the NP runs at a higher molecular weight (indicative of oligomers), which are clearly visible in the native PAGE gel, but a small amount of low molecular weight NP remains present. In the case of nucleozin treatment, this oligomer ladder becomes a smear on the gel, indicating the formation of an aggregate. When treated with S119, a faint oligomer ladder still remains and the low molecular weight band is completely lost. Our interpretation of this effect is that S119 perturbs the NP oligomerization state both in the presence and in the absence of viral RNA but may be doing so in a manner different from nucleozin. These results could indicate that S119 has a higher affinity for NP in the monomeric state as compared to oligomerized NP within the RNP structure. While these experiments where suggestive of the S119 mechanism of action, the inherent limitations of the in vitro system with a tagged protein outside of the context of an influenza virus-infected cell meant that confirmation of these results would be required from alternative experimental systems.
Figure 4.
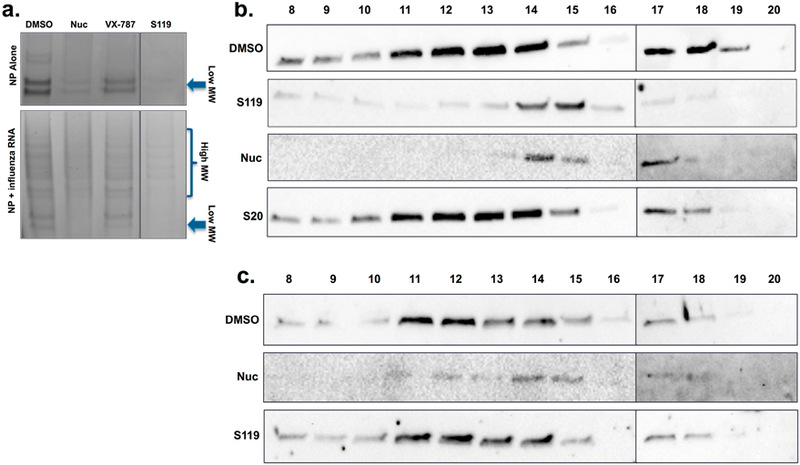
S119 alters the oligomerization state of NP. (a) Visualization and comparison of the effects of S119 and nucleozin on purified recombinant wild-type NP in the absence and presence of RNA; native gradient gel conditions, stained with Coomassie brilliant blue G-250. All samples were loaded on the same gel, and an irrelevant experimental compound originally in the 4th lane was removed resulting in the split figure. Size exclusion chromatography was performed on cell extract from A549 cells infected with (b) A/WSN/33 virus or (c) rY40F WSN virus and treated with either S119 or nucleozin. Extracts were RNase treated and applied to a Superose-6 column, and 1 mL fractions were collected and analyzed by Western blot for NP content.
Due to the inability to resolve higher order aggregations in the native PAGE system, it was difficult to address the exact effect of S119 on NP oligomerization, so size exclusion chromatography was employed to investigate this further. Lysates from WSN infected A549 cells treated with S119 or nucleozin were applied to a Superose-6 column, and 1 mL fractions were collected. S20, a compound that targets HA,13 was used as a negative control. In infected cells, the vast majority of the expressed NP is present in RNP complexes, which are quite large and appeared in the exclusion volume (fraction 8) in the Superose-6 column (Figure S1). However, some of the NP was maintained in a lower molecular weight pool, which is likely to be monomers or small oligomers (fractions 17−19). A large NP aggregate would be expected to appear in the exclusion fraction similar to the RNP structure, making differentiation of these two states difficult in this experimental set up. Therefore, lysates were RNase A treated following drug treatment to disrupt the RNP complexes prior to being applied to the column for subsequent experiments. Once the RNPs were RNase treated, a moderate molecular weight NP oligomer pool was observed in fractions 11 through 14. A clear shift to smaller oligomers (fractions 14−15) was observed in both S119 and nucleozin treated samples along with a distinct loss of the low molecular weight NP pool (Figure 4b). While a reduction in the oligomeric size of NP was unexpected, the fact that the known NP aggregator, nucleozin, showed a similar effect indicated that S119 could be causing a similar formation of abnormal NP complexes. The shift to smaller oligomers might be due to changes in the exposure of viral RNA to RNase A in the presence of drug. For example, if the NP aggregates caused by both S119 and nucleozin were to make the RNA more accessible to enzymatic digestion than the RNA within the tightly packed RNP structure, one would expect the observed reduction in oligomer size after RNase A treatment. Interestingly, S119 appeared to cause a greater reduction in the low molecular weight NP pool than that observed with nucleozin, and this effect occurred in the absence of RNase treatment as well (Figure S1), which aligns well with our assays using purified NP protein (Figure 4a). When we examined NP from the rY40F S119-resistant virus, we found that oligomerization was unaffected by S119 treatment but was altered by nucleozin similarly to wild-type virus (Figure 4c).
Finally, immunofluoresence microscopy was performed on WSN infected A549 cells to determine NP localization at 2, 4, 6, and 24 h after infection. The incoming NP protein (as part of the RNP) is first found in the cytoplasm during viral entry into the cell. Then, at around 4 h post-infection, newly synthesized NP is detected exclusively in the nucleus, and later, it is exported to the cytoplasm for packaging into virions. This temporal pattern of NP localization was observed in the DMSO control experiment (Figure 5). With nucleozin treatment, NP remains exclusively in the cytoplasm and begins to form visible aggregates by 4 h post-infection, consistent with previous publications.11 In the case of S119, while NP translocation to the nucleus was delayed, it was not excluded. At 6 h post-infection, NP was observed in the nucleus, though with much weaker expression. Aggregation of NP was also observed under S119 treatment but not until the 24 h post-infection time point (Figure 5). The S119-induced aggregates also appeared to be larger than those from nucleozin, possibly due to an overall higher expression of NP in the presence of S119 compared to nucleozin. All of these data taken together suggest that S119 is causing an abnormal oligomerization of the NP protein, likely resulting in the formation of large aggregates.
Figure 5.
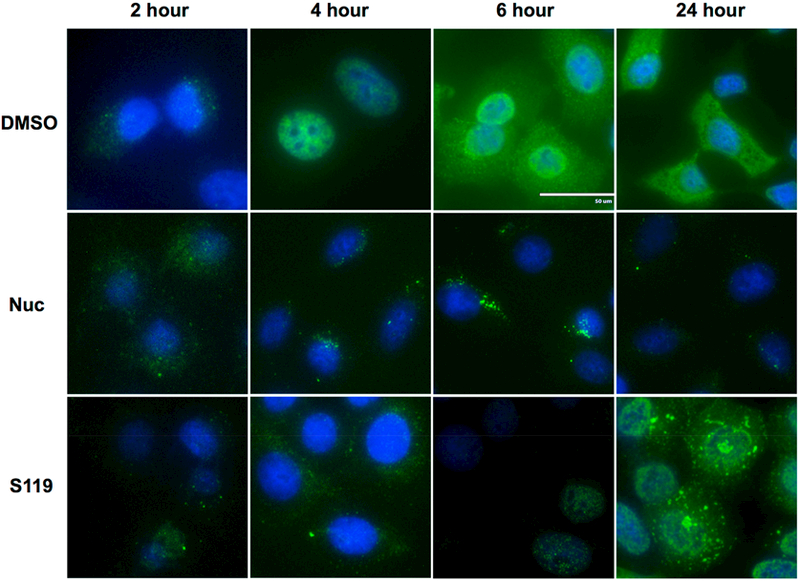
Large aggregates of NP accumulate in the cytoplasm upon S119 treatment. A549 cells infected with A/WSN/33 virus were treated with S119 or nucleozin, and NP localization was tracked at 2, 4, 6, and 24 h. DAPI staining and mouse anti-influenza A NP antibodies were used to define the locations of the nucleus and NP, respectively.
NP Aggregation by S119 Causes a Segment Specific Inhibition of Viral Transcription.
The nucleoprotein is essential for both viral transcription and replication. Therefore, we wanted to determine whether S119-induced NP aggregation caused any downstream effects on the production of mRNA and vRNA from the various influenza genomic segments. We utilized primer extension assays to analyze mRNA and vRNA production and Western blot to detect protein expression from the PB2, NP, and NS segments as representatives of long, medium, and short segments, respectively. Treatment of WT WSN infected A549 cells with nucleozin caused a complete loss of both mRNA, vRNA, and viral protein expression from all three segments, regardless of size (Figure 6a−c). In contrast, S119 inhibition resulted in a segment specific effect. In the presence of S119, mRNA expression from the PB2 segment was reduced to undetectable levels (Figure 6a), although the small amount of PB2 protein indicates that there is a minimal level of mRNA expression. In the case of the NP segment, mRNA expression was severely reduced by S119 but still observable and NP protein levels were moderately reduced (Figure 6b). Finally, mRNA expression from the NS segment was slightly reduced, but NS1 protein expression was unaffected (Figure 6c). vRNA could not be detected from any segment under S119 treatment, indicating a block in replication. In contrast, the rY40F-WSN virus was completely resistant to S119 inhibition of transcription and replication, while nucleozin exhibited efficient inhibition of this virus (Figure 6d−f). The loss of global vRNA expression could be explained through two possible mechanisms. First, the loss of protein expression of the polymerase subunits and the nucleoprotein would make RNP formation impossible, which is the functional unit for both transcription and vRNA replication. Second, the accumulation of the NP monomeric pool has previously been shown to be a key molecular trigger in the switch from transcription of mRNA to replication of vRNA in the viral life cycle.5,21,22 The loss of this monomeric NP pool, evident in the size exclusion chromatography data (Figure 4b), could prevent this switch from occurring and thereby block vRNA synthesis from all segments. These two mechanisms are not mutually exclusive, with each likely playing a role in S119 inhibition of influenza virus replication. The observed differential inhibition of mRNA expression by S119 from the three tested segments indicates a possible length-dependent effect of the compound. An explanation for this phenomenon could again be due to the loss of the monomeric NP pool. It has been well established that the viral polymerase requires an NP-coated template for full processivity and that the lack of NP will cause the polymerase to dissociate prematurely.23 Absence of monomeric NP could cause a reduction in NP-encapsidation of viral vRNAs resulting in reduced processivity of the viral polymerase. It would be reasonable to infer that a loss of processivity would disproportionately affect longer segments, as we observe in our experiments.
Figure 6.
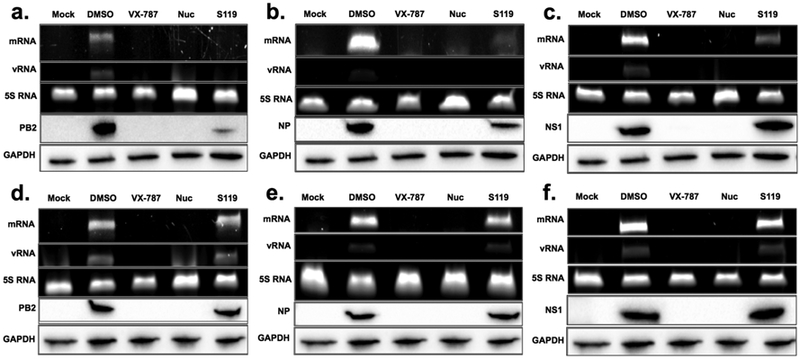
NP aggregation leads to alterations in viral RNA expression. A549 cells infected with either A/WSN/33 (a−c) virus or the mutant rWSNY40F (d−f) virus were treated with S119, nucleozin, or VX-787 control for 24 h. Viral mRNA and vRNA were detected using a primer extension assay with 5S rRNA serving as a loading control. Protein expression was monitored through Western blot for the expression of (a, d) PB2, (b, e) NP, and (c, f) NS1, with GAPDH as a loading control.
Broad Spectrum Influenza A Virus Inhibition by S119 Analogs.
Our initial analysis of the breadth of S119 anti-influenza virus activity indicated that it was only effective against some H1N1 viruses with maximum potency observed against the A/WSN/33 laboratory strain and no effect on another H1N1 laboratory strain A/PR/8/34 (Table 1). Due to this specificity, we performed a structure activity relationship study with a focus on improving the antiviral breadth. Analogs of S119 were synthesized and screened for activity against an influenza A/PuertoRico/8/34 virus expressing GFP,24 and to ensure we were not selecting for H1N1 specific compounds, all analogs were counter screened against an A/Vietnam/1203/2004 (H5N1) HALO GFP virus. Successive rounds of analog production (66 in total), using an iterative process, resulted in increased potency against the PR8 and H5N1 viruses. We found that activity against both the H1N1 and H5N1 viruses was also predictive of the ability to inhibit a representative H3N2 virus (A/Panama/2007/1999). Therefore, we were confident that this screening process could identify S119 analogs that had broad spectrum antiviral activity against influenza A viruses, as listed in Table 1.
Table 1.
S119 Analogs Have Increased Breadth of Inhibitiona
| Name | Structure | PRS IC50 (μM) | H5N1 IC50 (μM) |
|---|---|---|---|
| S119 | >50 | >50 | |
| S119-2 | 13.93 | 44.51 | |
| S119-3 | 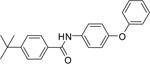 |
11.41 | 17.37 |
| S119-6 | >50 | >50 | |
| S119-8 | 6.05 | 8.42 |
Analogs of S119 were chemically synthesized and screened for antiviral activity against A/Puerto Rico/8/1934 (H1N1) and A/Vietnam/1203/2004 (H5N1-Halo) viruses which express GFP from the NS segment. GFP signal was detected by laser scanning cytometry, and infectivity was determined by GFP signal over counterstain of total cells.
S119 is a symmetrical bis(4-t-butylphenyl)carboxamide of 1,4-diaminobenzene that showed no detectable antiviral activity on the two influenza strains that were screened, and a related symmetrical 1,3-benzenediamine analog S119–6 also showed narrow spectrum activity. It is interesting to note that the unsymmetrical analogs, as in the shortened mono-4-tertbutylphenyl carboxamide S119–2, the diphenyl ether bearing carboxamide S119–3, or the diphenylamino analog S119–8, all possessed broader spectrum anti-influenza activity as compared to S119. The analogs were designed to explore the nature of the influenza activity pharmacophore as well as to improve calculated physical chemical properties of the lipophilic (cLogP: 6.81), poorly soluble S119 parent hit molecule. We plan to expand this SAR and will report on our progress in future manuscripts.
Of these analogs, S119–8 demonstrated that it had acquired enhanced broad-spectrum activity (Figure 7, Table 2) with improved calculated physical properties (i.e., lower cLogP: 5.95), while maintaining significant potency (IC50 = 1.43 μM) at nontoxic (50% cytotoxic concentration, CC50 = 66.10 μM) concentrations, albeit somewhat higher (7-fold) than that of the parent S119 against WSN virus. Most interestingly, S119–8 also showed activity against multiple influenza B viruses and an oseltamivir-resistant influenza A virus (Figure 7d, Table 2) but did not inhibit a noninfluenza virus, vesicular stomatitis virus (VSV) (Figure S2). As evidence that S119–8 was still targeting NP, we showed that the rWSN-Y40F virus maintained resistance to S119–8 (Figure S3).
Figure 7.
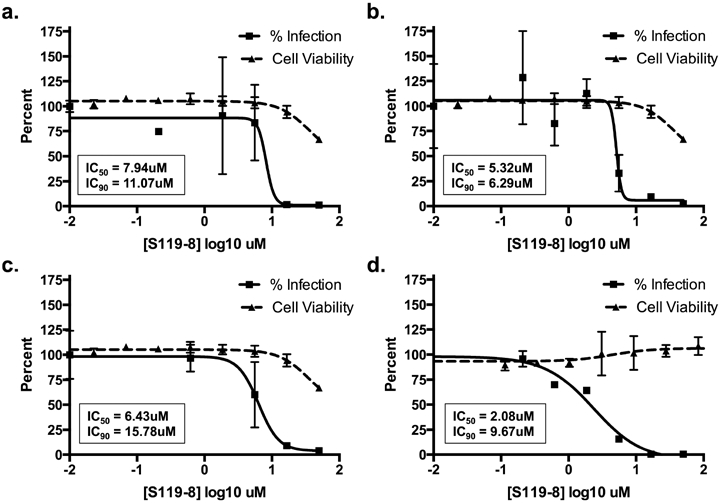
S119–8 has increased antiviral breadth. A549 cells were infected with influenza (a) A/Panama/2007/1999 (H3N2, MOI = 0.01, 24 h), (b) A/California/04/2009 (H1N1, MOI = 0.1, 48 h), or (c) A/Vietnam/1203/2004 (H5N1-HaLo, MOI = 0.01, 24 h) or (d) MDCK cells were infected with B/Yamagata/16/1988 (MOI = 0.1, 48 h) in the presence of serially diluted S119–8. Viral titers were determined and represented as percent infection relative to DMSO control (solid line). The IC50 and IC90 values were calculated. Cell viability was determined in an independent experiment in A549 or MDCK cells over a 24 or 48 h period respective to the infection conditions (dashed line), and the CC50 and CC10 were calculated. Means of three replicates ± SD are shown.
S119–8 Synergizes with Oseltamivir.
Finally, future anti-influenza therapies are likely to contain a cocktail of small molecules to deter the development of resistance.25 Therefore, it is important to understand how S119 interacts with currently licensed drugs and potential influenza drugs in development. To this end, we tested the effect of the S119 analog S119–8 in combination with either nucleozin or oseltamivir. A549 cells were infected with influenza PR8-GFP virus in the presence of the two compounds which were titrated against each other in 3-fold serial dilutions starting with a concentration above the 90% inhibitory concentration (IC90) of each compound alone. Isobolograms of viral inhibition were generated (Figure 8), and on the basis of these data, the combination index for the S119–8−nucleozin interaction was determined to be 0.79, suggesting an additive to slightly synergistic interaction. This was somewhat surprising as we anticipated that the close proximity of the resistance mutations of the two compounds could have potentially caused an antagonistic interaction. When we examined the interaction between S119–8 and oseltamivir, we found a combination index of 0.57 which indicates a synergistic relationship between the two compounds. This implies that, if the in vivo PK and safety profile of S119–8 is acceptable, this compound would be a good candidate to include in a potential anti-influenza drug cocktail by enhancing the potency of the current standard of care, oseltamivir, and lowering the chances of developing resistance. Studies to improve the antiviral and drug-like properties26 of the S119 series are ongoing.
Figure 8.
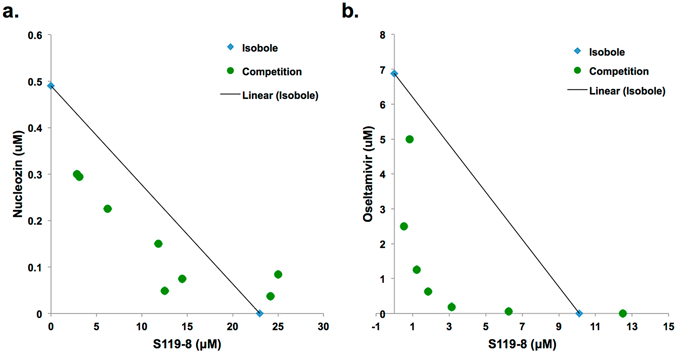
S119 analog, S119–8, synergizes with oseltamivir. Isobolograms showing the effect of fixed ratio combinations of S119–8 and (a) nucleozin or (b) oseltamivir. Each circle represents the IC90 for the fixed ratio of the two inhibitors, while the diamonds indicate the IC90 of each compound alone and the line connecting them is the isobole. Circles underneath the isobole indicate a synergistic relationship, while circles above the isobole show antagonism between the compounds.
CONCLUSION
The discovery of nucleozin has highlighted the viral nucleoprotein as a promising target for novel therapeutics against influenza virus. The highly conserved nature of the nucleoprotein27,28 and its critical importance in multiple stages of the viral life cycle make it an ideal target. Here, we present S119 and its analog S119–8 as antiviral drug leads for the development of a pan-influenza NP inhibitor with a defined mechanism of action. Through a SAR study, S119–8 was shown to extend its breadth to multiple influenza A virus subtypes and even to influenza B viruses, therefore not suffering from the same specificity limitations as nucleozin. Furthermore, the rise in resistance to currently approved influenza antivirals has emphasized the potential benefits of antiviral cocktails for future therapeutics. The observed synergistic relationship between S119–8 and oseltamivir is encouraging for incorporation of such a compound (or improved derivatives) in future influenza antiviral drug cocktails.
METHODS
Cell Culture and Reagents.
Madin-Darby canine kidney (MDCK) epithelial cells, human alveolar epithelial (A549) cells, and human embryonic kidney 293T (293T) cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA). MDCK, A549, and 293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (HyClone, South Logan, UT) and 1% penicillin−streptomycin (P/S) (Gibco). All cells were grown at 37 °C, 5% CO2. Transfection of DNA was performed in Opti-MEM I-reduced serum medium (Opti-MEM) (Gibco) with Lipofectamine LTX (Invitrogen) in A549 cells according to the manufacturer’s specifications.
Expression Plasmids and Cloning.
The influenza virus plasmid pPolI-NP Y40F was generated by exchanging one nucleotide in the parental plasmid pPolI-NP16 using the QuickChange site-directed mutagenesis kit (Agilent Technologies, Wilmington, DE) using specific primers (forward: 5′-GATGGAATTGGACGATTCTTCATCCAAATGTGCACCGAAC-3′; reverse: 5′-GTTCGGTGCACATTTGGATGAAGAATCGTCCAATTCCATC-3′). Presence of the mutation was confirmed by sequencing (Macrogen, Rockville, MD). The mammalian expression vector pCAGGS containing a chicken β-actin promoter has been previously described.29 Proper insertion and presence of the mutation was confirmed by sequencing (Macrogen).
Viruses.
The influenza A/WSN/1933 (H1N1) virus (WSN) was propagated in MDCK cells for 2 days at 37 °C. Influenza A/California/04/2009 (H1N1) virus was propagated in MDCK cells for 3 days at 35 °C. Influenza viruses A/Puerto Rico/8/1934 (H1N1) (PR8), A/Brisbane/59/2007-S, A/Brisbane/59/2007-R, A/Wyoming/03/2003, A/Panama/2007/1999 (H3N2), and A/Vietnam/1203/2004 (H5N1) bearing a mutated polybasic cleavage site in the HA segment (HAlo) were propagated in 10-day old embryonated chicken eggs for 2 days at 37 °C. The A/Brisbane/59/2007-S and A/Brisbane/59/2007-R viruses are oseltamivir sensitive (S) and resistant (R) versions of a clinical isolate generated using a reverse genetics system as previously described.39 The NS segment (A/Vietnam/1203/2004) carrying GFP was generated by overlapping fusion PCR using standard molecular biology techniques. Briefly, the NS1 ORF without the stop codon was fused to the N-terminal of a codon-optimized maxGFP (Amaxa) via a GSGG linker region (NS1-GFP). The maxGFP was followed by a short GSG linker, a 19-aa 2A autoproteolytic site 34 derived from porcine teschovirus 1 and by the NEP ORF. Also, silent mutations in the endogenous splice acceptor site in the NS1 ORF were introduced to prevent splicing.35 The engineered NS-GFP segment was cloned in the pDZ IAV rescue plasmid.36 Influenza B/Yamagata/16/1988 and B/Brisbane/60/2008 viruses were propagated in 8-day old embryonated chicken eggs for 3 days at 33 °C. All influenza viruses were titered by standard plaque assay in MDCK cells. Vesicular stomatitis virus was grown and titered by the plaque assay in VERO cells. Recombinant influenza viruses were generated using the influenza virus rescue protocol as previously described.16 Briefly, 293T cells were transfected with eight pPolI constructs expressing the PB1, PB2, PA, NP (or NP Y40F), HA, NA, M, and NS genomic segments as well as pCAGGS expression plasmids encoding the PB1, PB2, PA, and NP proteins. Twenty four hours post-transfection, MDCK cells were cocultured with the transfected 293Ts for an additional 24−48 h, until cytopathic effects were observed. Newly generated viruses were collected and plaque-purified, and the presence of the mutation was confirmed by sequencing.
Small Molecular Weight Compounds.
Nucleozin was purchased from Sigma-Aldrich (St. Louis, MO). The active VX-787 analog and oseltamivir carboxylate were kindly provided as gifts by Roche (Basel, Switzerland). Compounds were purchased through eMolecules (La Jolla, CA) and dissolved in 100% DMSO. The final concentration of DMSO in the culture medium did not exceed 0.5%.
Cell Viability Assay.
The CellTiterGlo Cell Viability Assay (Promega) was used to detect ATP levels as a function of cell viability, according to the manufacturer’s specifications. A549 cells were seeded into 96-well plates (1250 cells/well) and incubated at 37 °C, 5% CO2 for 24 h. Culture medium was then replaced with 100 μL of fresh medium containing compound (serially diluted), and this was further incubated for 24 h. Cell viability was measured by adding 50 μL of CellTiterGlo reagent to each well, and the luminescence signal was read using a plate reader (Beckman Coulter, Brea, CA). The 10% cytotoxic concentration (CC10) and CC50 for each experiment were determined using the Prism (GraphPad Software) software.
Viral Growth Assays in the Presence of Inhibitors.
100 000 A549 cells were seeded into 24-well plates and incubated for 24 h at 37 °C, 5% CO2. Two hours before infection, the medium was replaced with DMEM containing the compound of interest at the indicated concentrations. Compounds were absent during the 1 h virus incubation but were present in the DMEM post-infection medium. Infections were performed at a low MOI (0.01−0.1) for 24 or 48 h, depending on virus used. For infections with influenza viruses, post-infection medium also contained 1 μg/mL TPCK-treated trypsin (Sigma-Aldrich, St. Louis, MO). The infected cells were incubated at 37 °C with the exception of influenza B virus-infected cells, which were incubated at 33 °C. Viral titers were determined by the standard plaque assay in MDCK cells. The IC50 and IC90 for each experiment were determined using the Prism (GraphPad Software) software.
Selection of S119-Resistant Influenza Viruses.
The concentration of S119 required for maximum virus inhibition (3 logs), while maintaining enough virus production for subsequent passages, was determined (2 μM S119). A549 cells were infected with WSN at an MOI of 0.01 for 24 h at 37 °C under S119 treatment. The supernatant was then collected and titered by the plaque assay. If the recovered S119 treated virus did not show increased viral titer similar to that of the DMSO treated control, the virus was passaged again by the same method. Once increased titers in the presence of S119 were detected for two consecutive passages, the viruses were plaque purified. Following plaque purification, all 8 genome segments were sequenced and compared to DMSO treated control virus to detect escape mutations.
Blue Native PAGE.
Blue Native PAGE was used to examine the effect of S119 on the multimeric state of the NP proteins. Purified recombinant wild-type WSN NP protein was kindly produced by Florian Krammer as previously described,37,38 and 5 μg of purified protein was incubated with 10 μM S119 at 25 °C for 30 min. One μg of purified influenza virus RNA or Nuclease-free water was added, up to 10 μL. After an additional incubation of 15 min at 25 °C, the samples were mixed with 2.5 μL of 5% Coomassie brilliant blue G-250 and loaded into sample wells of nondenaturing 4−20% gradient Mini-Protean TGX gel (Bio-Rad). Samples were separated by electrophoresis at a constant voltage of 150 V for 60 min at 25 °C in a blue native Tris/Glycine buffer system (Coomassie brilliant blue G-250 0.02% Cathode Buffer). Gels were destained with 25% methanol/10% acetic acid solution and visualized using the Bio-Rad (Hercules, CA) ChemiDoc MP Imaging System.
Size Exclusion Chromatography.
To determine the oligomerization state of NP, A549 cells were infected with WSN virus (MOI = 1) in the presence or absence of 10 μM S119 or 1 μM Nucleozin for 24 h. Cells were then lysed in 50 mM Tris-HCl, 100 mM KCl, 5 mM MgCl2, and 0.5% NP40 containing protease and phosphatase inhibitor cocktails. Total cell extract was clarified by centrifugation, treated with 50 μg/mL of RNase A for 2 h at room temperature, and fractionated through a Superose-6 column pre-equilibrated in lysis buffer. Fractions were probed by Western blotting with an anti-NP (HT103) antibody.
Immunofluorescence Microscopy.
A549 cells were grown to 70−80% confluency on coverslips. Cells were infected for 2 h at MOI = 5 in the presence or absence of 10 μM S119 or 1 μM Nucleozin and washed. S119 was maintained in culture throughout the experiment. Infections were stopped at indicated time points by fixation in methanol for 30 min at 4 °C. Cells were blocked with 1% BSA for 30 min and then were incubated for 1 h with primary antibodies against NP (HT103) in PBS (dilution 1:2000), washed, and stained with Alexa Fluor 488-conjugated secondary antibodies (Invitrogen) (dilution 1:1000) for 1 h. Coverslips were then washed and counter-stained with 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI) (Invitrogen) for nucleus localization and mounted on slides using Prolong Gold antifade mounting medium (Invitrogen) before image analysis by fluorescence microscopy.
Fluorescent Primer Extension Assay.
Primer extension of influenza RNA was performed using 5′-end labeled fluorescent primers with either Alexa Fluor 488 for vRNA detection or Alexa Fluor 546 for m/cRNA detection of the PB2 (vRNA: 5′-TGCTAATTGGGCAAGGAGAC-3′ or m/cRNA: 5′-GCCATCATCCATTTCATCCT-3′), NP (vRNA: 5′-TGATGGAAAGTGCAAGACCA-3′ or m/cRNA: 5′-TGATTTCAGTGGCATTCTGG-3′), and NS (vRNA: 5′-TGATTGAAGAAGTGAGACACAG-3′ or m/cRNA: 5′-CGCTCCACTATTTGCTTTCC-3′) segments. An Alexa Fluor 488 5′-labeled primer targeting the human 5S rRNA (5′-TCCCAGGCGGTCTCCCATCC-3′) was used as a loading control. A549 cells were pretreated for 2 h with S119, VX-787, or DMSO and infected with the WSN virus for 6 h (MOI 5) under continued compound treatment. Infected A549 total RNA (100 μg) was added to the fluorescent primer (10 pmol), heated to 90 °C (3 min), and cooled on ice. Reverse transcription (RT) reactions were performed in 50 mM Tris-HCl (pH 8.3), 75 mM KCl, 5 mM MgCl2, and 10 mM DTT with 0.5 mM each dNTP. 50U SuperScript III (Invitrogen) was added, and reactions were incubated at 45 °C for 60 min. cDNA samples were mixed with an equal volume of loading dye solution (formamide, 5 mM EDTA, pH 8, 1% bromophenol blue) and heated to 95 °C for 3 min to stop the reaction. Samples were loaded on a 6% polyacrylamide gel containing 7 M urea in TBE buffer. Gel fluorescence was detected using the Bio-Rad (Hercules, CA) ChemiDoc MP Imaging System.
Antiviral Combination Assay.
The combined antiviral effect of S119–8 (S119 Analog) and nucleozin or oseltamivir carboxylate was tested against the A/Puerto Rico/8/34 strain expressing GFP from segment 8 (PR8-GFP), which has been previously described.24 A549 cells were infected with PR8-GFP (MOI = 0.01) in the presence or absence of multiple drug combinations for 24 h. The concentration of the drugs used was based on their respective 50% inhibitory concentrations (IC50) against PR8-GFP in A549 cells. The assays were performed in 96-well format using a liquid handling robot (Biomek NXP, Beckman) to ensure accuracy. Infected cells were detected through GFP fluorescence, quantified by laser scanning cytometry (Acumen, TTP Labtech). Isobolograms and combination indices were calculated as previous described.30
Synthesis of S119 and Related Analogs.
N,N′-(1,4-Phenylene)bis(4-(tert-butyl)benzamide) (S119).
To a solution of 1,4-diamino-benzene 2c (0.07 g, 1 equiv), triethylamine (3.0 equiv) in CH2Cl2 and 4-(1,1-dimethylethyl)benzoyl chloride 1 (2.2 equiv) were added at 0 °C. The resulted white suspension was stirred at room temperature for 12 h and then filtered. The white cake was washed with CH2Cl2 (10 mL) and EtOH (10 mL) and dried under vacuum to give the desired product 8 as white powder, 0.2 g (72% yield). 1H NMR (600 MHz, DMSO-d6): δ 10.17 (s, 2H), 7.90−7.89 (d, 4H), 7.74 (m, 4H), 7.56− 7.54 (d, 4H), 1.33 (s, 18H). 13C NMR (125 MHz, DMSO-d6): δ 165.2, 154.3, 134.9, 132.3, 127.4, 125.1, 120.5, 34.7, 30.9. LCMS (tof+) for C28H32N2O [M] 428.2464; found 2 [M + H]+ 429.2538
Synthesis of S119–8:4-(tert-butyl)-N-(4-(phenylamino)-phenyl)benzamide (S119–8). Step 1: N-(4-Aminophenyl)-4-(tert-butyl)benzamide.
To a solution of 1,4-diamino-benzene (0.42 g, 1.2 equiv), triethylamine (1.5 equiv) in CH2Cl2 and 4-(1,1-dimethylethyl)benzoyl chloride 1 (1.0 equiv) were added at 0 °C. The resulted white suspension was stirred at room temperature for 12 h and then filtered. Product was purified via flash chromatography (gradient elution from 40% EtOAc in hexanes to 60% EtOAc in hexanes) to give product as light yellow powder, 0.6 g (70% yield). 1H NMR (600 MHz, CDCl3): δ 7.97 (s, 1H), 7.80−7.79 (d, 2H), 7.46−7.45 (d, 2H), 7.40−7.39 (d, 2H), 6.67−6.66 (d, 2H), 3.58 (s, 2H), 1.35 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 165.2, 154.5, 142.8, 131.8, 129.0, 126.4, 125.1, 121.9, 115.1, 34.5, 30.7. LCMS (tof +) for C17H20N2O [M] 268.1576; found [M + H]+ 269.1654.
Step 2.
To a solution of N-(4-aminophenyl)-4-(tert-butyl)-benzamide (0.115 g, 1.0 equiv), BiPh3 (0.19 g, 1.01 equiv) and Cu(OAc)2 (0.08 g, 1.0 equiv) in CH2Cl2 (20 mL) were added with triethylamine (0.07 mL, 0.48 mmol, 1.1 equiv) at room temperature.31 The resulted suspension was stirred at room temperature for 16 h before being diluted with CH2Cl2 (20 mL), followed by addition of 1 N HCl (12 mL) and kept stirring for 1 h. The organic layer was separated, washed with 1 N HCl (20 mL), and dried over K2CO3. After filtration, the filtrate was concentrated and purified via flash chromatography (gradient elution from 10% EtOAc in hexanes to 25% EtOAc in hexanes) to give product as brown solid, 0.1 g (71% yield). 1HNMR (600 MHz, CDCl3): δ 8.00 (s, 1H), 7.84−7.83 (d, 2H), 7.55−7.54 (d, 2H), 7.23 (t, 2H), 7.08−7.04 (dd, 4H), 6.94 (t, 1H), 1.37 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 165.3, 154.8, 143.0, 139.2, 131.6, 131.2, 128.9, 126.4, 125.2, 121.5, 120.2, 118.4, 116.7, 34.5, 30.7. LCMS (tof+) for C23H24N2O [M] 344.1889; found [M + H]+ 345.1956.
Similar procedures as for S119 were used to prepared related analogs.
S119–2: 4-(tert-butyl)-N-(4-(tert-butyl)phenyl)benzamide (S119–2).
To a solution of 4-tert-butylaniline (0.23 g, 1.54 mmol, 1.0 equiv) and triethylamine (0.16 g, 1.54 mmol, 1.0 equiv) in CH2Cl2 (20 mL) was added 4-(1,1-dimethylethyl)-benzoyl chloride (0.28 g, 1.54 mmol, 1.0 equiv) at 0 °C. The resulted white suspension was stirred at room temperature for 12 h and then filtered. The filtrate was concentrated and purified via flash chromatography (gradient elution from 0% EtOAc in hexanes to 4% EtOAc in hexanes) to give product as white powder, 0.46 g (95% yield). 1H NMR (600 MHz, CDCl3): δ 7.98 (s, 1H), 7.83−7.82 (d, 2H), 7.60−59 (d, 2H), 7.49−7.48 (d, 2H), 7.40−7.39 (d, 2H), 1.36 (s, 9H), 1.35 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 165.2, 154.8, 146.9, 135.0, 131.7, 126.4, 125.4, 125.2, 119.5, 34.5, 33.9, 30.9, 30.7. LCMS (tof+) for C21H27NO [M] 309.2093; found [M + H]+ 310.2158
S119–3: 4-(tert-butyl)-N-(4-phenoxyphenyl)benzamide (3b).
S119–3 was prepared as above from 4-phenoxy-phenylamine 2b (0.10 g, 1.0 equiv), triethylamine (1.0 equiv), and 4-(1,1-dimethylethyl)benzoyl chloride 1 (1.0 equiv). Product was purified via flash chromatography (gradient elution from 0% EtOAc in hexanes to 8% EtOAc in hexanes) to give a white powder, 0.18 g (96% yield). 1H NMR (600 MHz, CDCl3): δ 8.05 (s, 1H), 7.84−7.82 (d, 2H), 7.63−7.62 (d, 2H), 7.50−7.48 (d, 2H), 7.35 (t, 1H), 7.11 (t, 1H), 7.03−7.01 (m, 3H), 1.37 (s, 9H). 13C NMR (125 MHz, CDCl3): δ 165.3, 157.1, 154.9, 153.1, 133.1, 131.5, 129.3, 126.5, 125.2, 122.6, 121.6, 119.2, 117.9, 34.5, 30.7. LCMS (tof+) for C23H23NO2 [M] 345.1729; found [M + H]+ 346.1787.
S119–6: N,N′-(1,3-phenylene)bis(4-(tert-butyl)benzamide) (S119–6).
S119–6 was prepared as above from 1,3-diamino-benzene (0.10 g, 1.0 equiv), triethylamine (3.0 equiv), and 4-(1,1-dimethylethyl)benzoyl chloride 1 (2.2 equiv). Product was purified via flash chromatography (gradient elution from 0% EtOAc in Hexanes to 25% EtOAc in Hexanes) to give a white powder, 0.3 g (77% yield). 1H NMR (600 MHz, CDCl3): δ 8.13 (s, 1H), 8.02−7.94 (m, 2H), 7.82−7.81 (m, 4H), 7.51− 7.48 (m, 6H), 7.38−7.34 (m, 1H), 1.38−1.38 (d, 18H). 13C NMR (125 MHz, CDCl3): δ 165.3, 155.1, 138.3, 131.4, 129.2, 126.4, 125.3, 115.3, 110.9, 34.5, 30.7. LCMS (tof+) for C28H32N2O2 [M] 428.2464; found [M + H]+ 429.2523.
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Florian Krammer for producing the purified NP proteins. JChem for Excel was used for structure database management, search, and prediction, JChem for Excel 6.1.1, 2013, ChemAxon (http://www.chemaxon.com). This work was supported in part by National Institutes of Health (NIH) Grants U01AI1074539, R21AI102169, and U19AI106754 and a Mount Sinai seed fund to the DeVita laboratory. This work was also partially supported by CRIP (Center for Research on Influenza Pathogenesis), an NIAID-funded Center of Excellence for Influenza Research and Surveillance (CEIRS, HHSN272201400008C).
ABBREVIATIONS
- VSV
vesicular stomatitis virus
- SAR
structure−activity relationship
- IC50
50% inhibitory concentration
- IC90
90% inhibitory concentration
- CC50
50% cytotoxic concentration
- CC10
10% cytotoxic concentration
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.7b00120.
Figure S1, size exclusion chromatography data from infected cell extracts treated with S119 or nucleozin in the absence of RNase treatment; Figure S2, showing that S119–8 does not inhibit VSV and is a specific inhibitor of influenza viruses; Figure S3, showing that S119–8 retains the same Y40F resistant phenotype as the parental S119 (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Dong G, Peng C, Luo J, Wang C, Han L, Wu B, Ji G, and He H (2015) Adamantane-resistant influenza a viruses in the world (1902−2013): frequency and distribution of M2 gene mutations. PLoS One 10, e0119115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Whitley RJ, Boucher CA, Lina B, Nguyen-Van-Tam JS, Osterhaus A, Schutten M, and Monto AS (2013) Global assessment of resistance to neuraminidase inhibitors, 2008−2011: the Influenza Resistance Information Study (IRIS). Clin. Infect. Dis 56, 1197–1205. [DOI] [PubMed] [Google Scholar]
- (3).Arranz R, Coloma R, Chichon FJ, Conesa JJ, Carrascosa JL, Valpuesta JM, Ortin J, and Martin-Benito J (2012) The structure of native influenza virion ribonucleoproteins. Science 338, 1634–1637. [DOI] [PubMed] [Google Scholar]
- (4).Amorim MJ, Bruce EA, Read EK, Foeglein A, Mahen R, Stuart AD, and Digard P (2011) A Rab11- and microtubule-dependent mechanism for cytoplasmic transport of influenza A virus viral RNA. J. Virol 85, 4143–4156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Newcomb LL, Kuo RL, Ye Q, Jiang Y, Tao YJ, and Krug RM (2009) Interaction of the influenza a virus nucleocapsid protein with the viral RNA polymerase potentiates unprimed viral RNA replication. J. Virol 83, 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Turrell L, Lyall JW, Tiley LS, Fodor E, and Vreede FT (2013) The role and assembly mechanism of nucleoprotein in influenza A virus ribonucleoprotein complexes. Nat. Commun 4, 1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Noton SL, Medcalf E, Fisher D, Mullin AE, Elton D, and Digard P (2007) Identification of the domains of the influenza A virus M1 matrix protein required for NP binding, oligomerization and incorporation into virions. J. Gen. Virol 88, 2280–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ashour J, Schmidt FI, Hanke L, Cragnolini J, Cavallari M, Altenburg A, Brewer R, Ingram J, Shoemaker C, and Ploegh HL (2015) Intracellular expression of camelid single-domain antibodies specific for influenza virus nucleoprotein uncovers distinct features of its nuclear localization. J. Virol 89, 2792–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Moeller A, Kirchdoerfer RN, Potter CS, Carragher B, and Wilson IA (2012) Organization of the influenza virus replication machinery. Science 338, 1631–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kakisaka M, Sasaki Y, Yamada K, Kondoh Y, Hikono H, Osada H, Tomii K, Saito T, and Aida Y (2015) A Novel Antiviral Target Structure Involved in the RNA Binding, Dimerization, and Nuclear Export Functions of the Influenza A Virus Nucleoprotein. PLoS Pathog. 11, e1005062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kao RY, Yang D, Lau LS, Tsui WH, Hu L, Dai J, Chan MP, Chan CM, Wang P, Zheng BJ, et al. (2010) Identification of influenza A nucleoprotein as an antiviral target. Nat. Biotechnol 28, 600–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Gerritz SW, Cianci C, Kim S, Pearce BC, Deminie C, Discotto L, McAuliffe B, Minassian BF, Shi S, Zhu S, et al. (2011) Inhibition of influenza virus replication via small molecules that induce the formation of higher-order nucleoprotein oligomers. Proc. Natl. Acad. Sci. U. S. A 108, 15366–15371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).White KM, De Jesus P, Chen Z, Abreu P Jr., Barile E, Mak PA, Anderson P, Nguyen QT, Inoue A, Stertz S, et al. (2015) A Potent Anti-influenza Compound Blocks Fusion through Stabilization of the Prefusion Conformation of the Hemagglutinin Protein. ACS Infect. Dis 1, 98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Konig R, Stertz S, Zhou Y, Inoue A, Hoffmann HH, Bhattacharyya S, Alamares JG, Tscherne DM, Ortigoza MB, Liang Y, et al. (2010) Human host factors required for influenza virus replication. Nature 463, 813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Pang B, Cheung NN, Zhang W, Dai J, Kao RY, Zhang H, and Hao Q (2016) Structural Characterization of H1N1 Nucleoprotein-Nucleozin Binding Sites. Sci. Rep 6, 29684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Martinez-Sobrido L, and Garcia-Sastre A (2010) Generation of recombinant influenza virus from plasmid DNA. J. Visualized Exp, DOI: 10.3791/2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ng AK, Zhang H, Tan K, Li Z, Liu JH, Chan PK, Li SM, Chan WY, Au SW, Joachimiak A, et al. (2008) Structure of the influenza virus A H5N1 nucleoprotein: implications for RNA binding, oligomerization, and vaccine design. FASEB J. 22, 3638–3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Tarus B, Bakowiez O, Chenavas S, Duchemin L, Estrozi LF, Bourdieu C, Lejal N, Bernard J, Moudjou M, Chevalier C, et al. (2012) Oligomerization paths of the nucleoprotein of influenza A virus. Biochimie 94, 776–785. [DOI] [PubMed] [Google Scholar]
- (19).Ye Q, Krug RM, and Tao YJ (2006) The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 444, 1078–1082. [DOI] [PubMed] [Google Scholar]
- (20).Byrn RA, Jones SM, Bennett HB, Bral C, Clark MP, Jacobs MD, Kwong AD, Ledeboer MW, Leeman JR, McNeil CF, et al. (2015) Preclinical activity of VX-787, a first-in-class, orally bioavailable inhibitor of the influenza virus polymerase PB2 subunit. Antimicrob. Agents Chemother 59, 1569–1582.\ [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Portela A, and Digard P (2002) The influenza virus nucleoprotein: a multifunctional RNA-binding protein pivotal to virus replication. J. Gen. Virol 83, 723–734. [DOI] [PubMed] [Google Scholar]
- (22).Vreede FT, Jung TE, and Brownlee GG (2004) Model suggesting that replication of influenza virus is regulated by stabilization of replicative intermediates. J. Virol 78, 9568–9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Honda A, Ueda K, Nagata K, and Ishihama A (1988) RNA polymerase of influenza virus: role of NP in RNA chain elongation. J. Biochem 104, 1021–1026. [DOI] [PubMed] [Google Scholar]
- (24).Manicassamy B, Manicassamy S, Belicha-Villanueva A, Pisanelli G, Pulendran B, and Garcia-Sastre A (2010) Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc. Natl. Acad. Sci. U. S. A 107, 11531–11536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).van der Vries E, Schutten M, Fraaij P, Boucher C, and Osterhaus A (2013) Influenza virus resistance to antiviral therapy. Adv. Pharmacol 67, 217–246. [DOI] [PubMed] [Google Scholar]
- (26).Meanwell NA (2011) Improving drug candidates by design: a focus on physicochemical properties as a means of improving compound disposition and safety. Chem. Res. Toxicol 24, 1420–1456. [DOI] [PubMed] [Google Scholar]
- (27).Kukol A, and Hughes DJ (2014) Large-scale analysis of influenza A virus nucleoprotein sequence conservation reveals potential drug-target sites. Virology 454–455, 40–47. [DOI] [PubMed] [Google Scholar]
- (28).Babar MM, and Zaidi NU (2015) Protein sequence conservation and stable molecular evolution reveals influenza virus nucleoprotein as a universal druggable target. Infect., Genet. Evol 34, 200–210. [DOI] [PubMed] [Google Scholar]
- (29).Niwa H, Yamamura K, and Miyazaki J (1991) Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108, 193–199. [DOI] [PubMed] [Google Scholar]
- (30).Chou TC (2006) Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 58, 621–681. [DOI] [PubMed] [Google Scholar]
- (31).Sorenson RJ (2000) Selective N-arylation of amino-benzanilides under mild conditions using triarylbismuthanes. J. Org. Chem 65, 7747–7749. [DOI] [PubMed] [Google Scholar]
- (32).Morens DM, Taubenberger JK, Harvey HA, and Memoli MJ (2010) The 1918 influenza pandemic: lessons for 2009 and the future. Crit. Care Med 38, e10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Belongia EA, Simpson MD, King JP, Sundaram ME, Kelley NS, Osterholm MT, and McLean HQ (2016) Variable influenza vaccine effectiveness by subtype: a systematic review and meta-analysis of test-negative design studies. Lancet Infect. Dis 16, 942–951. [DOI] [PubMed] [Google Scholar]
- (34).Basler CF, Reid AH, Dybing JK, Janczewski TA, Fanning TG, Zheng H, Salvatore M, Perdue ML, Swayne DE, Garcia-Sastre A, et al. (2001) Sequence of the 1918 pandemic influenza virus nonstructural gene (NS) segment and characterization of recombinant viruses bearing the 1918 NS genes. Proc. Natl. Acad. Sci. U. S. A 98, 2746–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Donnelly ML, Hughes LE, Luke G, Mendoza H, ten Dam E, Gani D, and Ryan MD (2001) The ‘cleavage’ activities of foot-and-mouth disease virus 2A site-directed mutants and naturally occurring ‘2A-like’ sequences. J. Gen. Virol 82, 1027–1041. [DOI] [PubMed] [Google Scholar]
- (36).Quinlivan M, Zamarin D, Garcia-Sastre A, Cullinane A, Chambers T, and Palese P (2005) Attenuation of equine influenza viruses through truncations of the NS1 protein. J. Virol 79, 8431–8439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Krammer F, Schinko T, Palmberger D, Tauer C, Messner P, and Grabherr R (2010) Trichoplusia ni cells (High Five) are highly efficient for the production of influenza A virus-like particles: a comparison of two insect cell lines as production platforms for influenza vaccines. Mol. Biotechnol 45, 226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Pica N, Hai R, Krammer F, Wang TT, Maamary J, Eggink D, Tan GS, Krause JC, Moran T, Stein CR, et al. (2012) Hemagglutinin stalk antibodies elicited by the 2009 pandemic influenza virus as a mechanism for the extinction of seasonal H1N1 viruses. Proc. Natl. Acad. Sci. U. S. A 109, 2573–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Bouvier NM, Rahmat S, and Pica N (2012) Enhanced mammalian transmissibility of seasonal influenza A/H1N1 viruses encoding an oseltamivir-resistant neuraminidase. J. Virol 86, 7268–7279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.