

Abstract
Long-term monitoring of data of ambient mercury (Hg) on a global scale to assess its emission, transport, atmospheric chemistry, and deposition processes is vital to understanding the impact of Hg pollution on the environment. The Global Mercury Observation System (GMOS) project was funded by the European Commission (http://www.gmos.eu) and started in November 2010 with the overall goal to develop a coordinated global observing system to monitor Hg on a global scale, including a large network of ground-based monitoring stations, ad hoc periodic oceanographic cruises and measurement flights in the lower and upper troposphere as well as in the lower stratosphere. To date, more than 40 ground-based monitoring sites constitute the global network covering many regions where little to no observational data were available before GMOS. This work presents atmospheric Hg concentrations recorded worldwide in the framework of the GMOS project (2010-2015), analyzing Hg measurement results in terms of temporal trends, seasonality and comparability within the network. Major findings highlighted in this paper include a clear gradient of Hg concentrations between the Northern and Southern hemispheres, confirming that the gradient observed is mostly driven by local and regional sources, which can be anthropogenic, natural or a combination of both.
1. Introduction
Mercury (Hg) is found ubiquitously in the atmosphere and is known to deposit to ecosystems, where it can be taken up into food webs and transformed to highly toxic species (i.e., methyl-Hg) which are detrimental to ecosystem and human health. A number of activities have been carried out since the late 1980s in developed countries within European and international strategies and programs (i.e., UNECE-CLRTAP, EU-Mercury Strategy; UNEP Governing Council) to elaborate possible mechanisms to reduce Hg emissions to the atmosphere from industrial facilities, trying to balance the increasing emissions in rapidly industrializing countries of the world (Pirrone et al., 2013, 2008, 2009; Pacyna et al., 2010). Hg displays complex speciation and chemistry in the atmosphere, which influences its transport and deposition on various spatial and temporal scales (Douglas et al., 2012; Good-site et al., 2004, 2012; Lindberg et al., 2007; Soerensen et al., 2010a, b; Sprovieri et al., 2010b; Slemr et al., 2015). Most of Hg is observed in the atmosphere as Gaseous Elemental Mercury (GEM/Hg0), representing 90 to 99 % of the total with a terrestrial background concentration of approximately 1.5-1.7 ng m−3 in the Northern Hemisphere and between 1.0 and 1.3 ngm−3 in the Southern Hemisphere based on research studies published before Global Mercury Observation System (GMOS) (Lindberg et al., 2007; Sprovieri et al., 2010b). The results obtained from newly established GMOS ground-based sites show a background value in the Southern Hemisphere close to 1 ng m−3, which is lower than that obtained in the past. Oxidized Hg species (gaseous oxidized mercury or GOM) and particulate bound mercury (PBM) contribute significantly to dry and wet deposition fluxes to terrestrial and aquatic receptors (Brooks et al., 2006; Goodsite et al., 2004, 2012; Hedgecock et al., 2006; Skov et al., 2006; Gencarelli et al., 2015; De Simone et al., 2015). Although in the past 2 decades a number of Hg monitoring sites have been established (in Europe, Canada, USA and Asia) as part of regional networks and/or European projects (i.e., MAMCS, MOE, MERCYMS) (Munthe et al., 2001, 2003; Wangberg et al., 2001, 2008; Pirrone et al., 2003; Steffen et al., 2008), the need to establish a global network to assess likely southern-northern hemispheric gradients and long-term trends has long been considered a high priority for policy and scientific purposes. The main reason is to make consistent and globally distributed Hg observations available that can be used to validate regional and global-scale models for assessing global patterns of Hg concentrations and deposition and re-emission fluxes. Therefore a coordinated global observational network for atmospheric Hg was established within the framework of the GMOS project (Seventh Framework Program - FP7) in 2010. The aim of GMOS was to provide high-quality Hg datasets in the Northern and Southern hemispheres for a comprehensive assessment of atmospheric Hg concentrations and their dependence on meteorology, long-range atmospheric transport and atmospheric emissions on a global scale (Sprovieri et al., 2013). This network was developed by integrating previously established ground-based atmospheric Hg monitoring stations with newly established GMOS sites in regions of the world where atmospheric Hg observational data were scarce, particularly in the Southern Hemisphere (Sprovieri et al., 2010b). The stations are located at both high altitude and high sea level locations, as well as in climatically diverse regions. The measurements from these sites have been used to validate regional- and global-scale atmospheric Hg models in order to improve our understanding of global Hg transport, deposition and re-emission, as well as to provide a contribution to future international policy development and implementation (Gencarelli et al., 2016; De Simone et al., 2016). The GMOS overarching objective to establish a global Hg monitoring network was achieved having in mind the need to assure high-quality observations in line with international quality assurance/quality control (QA/QC) standards and to fill the gap in terms of spatial coverage of measurements in the Southern Hemisphere where data were lacking or nonexistent. One of the major outcomes of GMOS has been an interoperable e-infrastructure developed following the Group on Earth Observations (GEO) data sharing and interoperability principles which allows us to provide support to UNEP for the implementation of the Minamata Convention (i.e., Article 22 to measure the effectiveness of measures). Within the GMOS network, Hg measurements were in fact carried out using high-quality techniques by harmonizing the GMOS measurement procedures with those already adopted at existing monitoring stations around the world. Standard operating procedures (SOPs) and a QA/QC system were established and implemented at all GMOS sites in order to assure full comparability of network observations. To ensure a fully integrated operation of the GMOS network, a centralized online system (termed GMOS Data Quality Management, G-DQM) was developed for the acquisition of atmospheric Hg data in near real time and providing a harmonized QA/QC protocol. This novel system was developed for integrating data control and is based on a service-oriented approach that facilitates real-time adaptive monitoring procedures, which is essential for producing high-quality data (Cinnirella et al., 2014; D’Amore et al., 2015). GMOS activities are currently part of the GEO strategic plan (2016-2025) within the GEO flagship on “tracking persistent pollutants”. The overall goal of this flagship is to support the development of GEOSS by fostering research and technological development on new advanced sensors for in situ and satellite platforms, in order to lower the management costs of long-term monitoring programs and improve spatial coverage of observations. In this paper we present for the first time a complete global dataset of Hg concentrations at selected ground-based sites in the Southern and Northern hemispheres and highlight its potential to support the validation of global-scale atmospheric models for research and policy scenario analysis.
2. Experimental
GMOS global network
The GMOS network currently consists of 43 globally distributed monitoring stations located both at sea level (i.e., Mace Head, Ireland; Calhau, Cabo Verde; Cape Point, South Africa; Amsterdam Island, southern Indian Ocean) and high-altitude locations, such as the Everest-K2 Pyramid station (Nepal) at 5050 m a.s.l. and the Mt. Walinguan (China) station at 3816 m a.s.1., as well as in climatically diverse regions, including polar areas such as Villum Research Station (VRS), Station Nord (Greenland), Pallas (Finland) and Dome Concordia and Dumont d’Urville stations in Antarctica. It is possible to browse the GMOS monitoring sites at the GMOS Monitoring Services web portal. The monitoring sites are classified as master (M) and secondary (S) with respect to the Hg measurement programs (Table 1). Master stations perform speciated Hg measurements and collect precipitation samples for Hg analysis whereas the secondary stations perform only total gaseous mercury (TGM)/GEM measurements and precipitation samples as well. Table 1 summarize key information about GMOS stations, such as (a) the location, elevation and type of monitoring stations; (b) new sites (master and/or secondary) established as part of GMOS; and (c) existing monitoring sites established by institutions that are part of European and international monitoring programs and managed by GMOS partners and GMOS external partners who have agreed to share their monitoring data and submit them to the central database following the interoperability principles and standards set in GEOSS (Group Earth Observation System of System). The GMOS objective of establishing a global Hg monitoring network was achieved always bearing in mind not only the necessity to provide intercomparable data worldwide but also international standards of intercomparability. In particular, GMOS attempts to comply with the data sharing principles set by the GEO that aim to develop the GEOSS through the use of “observation systems, which include ground-, air-, water- and space-based sensors, field surveys and citizen observatories. GEO works to coordinate the planning, sustainability and operation of these systems, aiming to maximize their added value and use.” Additionally, GMOS makes use of “information and processing systems, which include hardware and software tools needed for handling, processing and delivering data from the observation systems to provide information, knowledge, services and products.” In 2010 the Executive Committee of GEO selected GMOS as a showcase for the work plan (2012-2015) to demonstrate how GEOSS can support the convention and policies as well as pioneering activity in environmental monitoring using highly advanced e-infrastructure. More details about the sites can also be found at http://www.gmos.eu.
Table 1.
Atmospheric ground-based sites locations that are part of the GMOS network and general characteristics of the sites (i.e., code, lat, long), including the type of monitoring station in respect to the Hg measurements carried out as speciated (M) or not (S). In bold, external GMOS partners are indicated.
| Code | Site | Country | Elevation (m a.s.l.) | Lat (°) | Long (°) | GMOS sitea |
|---|---|---|---|---|---|---|
| AMS | Amsterdam Island | Terres Australes et Antarctiques Françaises | 70 | -37.79604 | 77.55095 | M |
| BAR | Bariloche | Argentina | 801 | -41.128728 | -71.420100 | M |
| CAL | Calhau | Cabo Verde | 10 | 16.86402 | -24.86730 | S |
| CHE | Cape Hedo | Japan | 60 | 26.86430 | 128.25141 | M |
| CPT | Cape Point | South Africa | 230 | -34.353479 | 18.489830 | S |
| CST | Celestún | Mexico | 3 | 20.85838 | -90.38309 | S |
| CMA | Col Margherita | Italy | 2545 | 46.36711 | 11.79341 | S |
| DMC | Concordia Station | Antarctica | 3220 | -75.10170 | 123.34895 | S |
| DDU | Dumont d’Urville | Antarctica | 40 | -66.66281 | 140.00292 | S |
| EVK | Ev-K2 | Nepal | 5050 | 27.95861 | 86.81333 | S |
| ISK | Iskrba | Slovenia | 520 | 45.56122 | 14.85805 | M |
| KOD | Kodaicanal | India | 2333 | 10.23170 | 77.46524 | M |
| LSM | La Seyne-sur-Mer | France | 10 | 43.106119 | 5.885250 | S |
| LISb | Listvyanka | Russia | 670 | 51.84670 | 104.89300 | S |
| LON | Longobucco | Italy | 1379 | 39.39408 | 16.61348 | M |
| MHD | Mace Head | Ireland | 8 | 53.32661 | -9.90442 | S |
| MAN | Manaus | Brazil | 110 | -2.89056 | -59.96975 | M |
| MIN | Minamata | Japan | 20 | 32.23056 | 130.40389 | M |
| MAL | Mt. Ailao | China | 2503 | 24.53791 | 101.03024 | S/M |
| MBA | Mt. Bachelor | WA, USA | 2743 | 43.977516 | -121.685968 | M |
| MCH | Mt. Changbai | China | 741 | 42.40028 | 128.11250 | M/S |
| MWA | Mt. Walinguan | China | 3816 | 36.28667 | 100.89797 | M |
| NIKb | Nieuw Nickerie | Suriname | 1 | 5.95679 | -57.03923 | S |
| PAL | Pallas | Finland | 340 | 68.00000 | 24.23972 | S |
| RAO | Rao | Sweden | 5 | 57.39384 | 11.91407 | M |
| SIS | Sisal | Mexico | 7 | 21.16356 | -90.04679 | S |
| VRS | Villum Research Station | Greenland | 30 | 81.58033 | -16.60961 | S |
M indicates master, S indicates secondary.
These sites use Lumex, elsewhere Tekran.
Eleven monitoring stations managed by external partners are included within the global network sharing their data with the GMOS central database. These new associated stations follow the “Governance and Data Policy of the Global Mercury Observation System” guidelines established by GMOS (Pirrone, 2012).
From the start of GMOS a small number of monitoring sites have been relocated or have become recently operational, but most of the sites have been fully operational for the entire project period and remain active. These original core group stations consist of 27 monitoring sites. Their spatial coverage is better throughout the Northern Hemisphere with 17 operational monitoring stations, whereas there are 5 sites in the tropical zone (area between the Tropic of Cancer (+23°27´) and the Tropic of Capricorn (−23°27´)), and 5 sites in the Southern Hemisphere. The sites in the Southern Hemisphere include new Hg stations, such as the GMOS site in Bariloche (Patagonia, Argentina), the station in Kodaicanal (South India) and the site on the Amsterdam Island (Terres Australes et Antarctiques Françises, TAAF) in the southern Indian Ocean, and two sites in Antarctica at the Italian-French Dome Concordia station and at the French site Dumont d’Urville.
3. Hg measurements methods
3.1. Field operation
All GMOS secondary sites used the Tekran continuous mercury vapor analyzer, model 2537A/B (Tekran Instruments Corp., Toronto, Ontario, Canada) with the exception of Listvyanka site (LIS), Russia, and Nieuw Nickerie site (NIK), Suriname, which used a Lumex RA-915+ mercury analyzer. The latter provides direct continuous GEM concentrations in air flow without Hg collection on sorbent traps (Sholupov and Ganeyev, 1995; Sholupov et al., 2004). GMOS master sites used the Tekran model 2537A/B mercury vapor analyzer coupled with their speciation system model 1130 for GOM and model 1135 for particulate boundaries mercury (PBM2.5) with fractions less than 2.5 μm in diameter to prevent large particles from depositing on the KCl-coated denuder (Gustin et al., 2015). The principles and operation of the Tekran Hg speciation system are described in Landis et al. (2002). Data were captured using either personal computers or data loggers and were submitted to the GMOS Central database network (http://www.gmos.eu/sdi). During the implementation of the GMOS global network, harmonized SOPs as well as common QA/QC protocols were developed (Munthe et al., 2011; Brown et al., 2010a, b) according to measurement practices followed within existing European and American monitoring networks and based on the most recent literature (Brown et al., 2010b; Steffen et al., 2012; Gay et al., 2013). The GMOS SOPs were reviewed by both GMOS partners and external partners as experts in this issue and finally adopted within the GMOS network (Munthe et al., 2011). Full SOPs are available online (http://www.gmos.eu/sdi) and include sections on site selection, field operations, data management, field maintenance and reporting procedures. All monitoring sites strictly followed the GMOS SOPs to harmonize operations and ensure the comparability of all results obtained worldwide. At the GMOS master sites the Hg analyzers were operated in conjunction with the Tekran 1130/1135 speciation units, and therefore the TGM/GEM data for these sites are explicitly referred to as GEM. GEM concentrations were also provided by the two secondary sites (LIS and NIK) which used the Lumex Hg analyzer (see the Lumex measurements principle in Sect. 2.2.2). Regarding the TGM/GEM at the other GMOS secondary sites, it has been discussed whether the Tekran 2537A/B instruments measure TGM = GEM + GOM or GEM only (Slemr et al., 2011, 2015); considering that previous modeling studies and experimental measurements highlighted that particularly at remote/background monitoring sites the oxidized fraction of the TGM is less than 2% (Gustin et al., 2015), we consider the Tekran 2537A/B data to represent GEM. This is also in line with a study recently published by Slemr et al. (2015) which reports a comparison of Hg concentrations at several GMOS sites in the Southern Hemisphere. Following the SOPs implemented at all GMOS sites, the Hg analyzers used at the secondary sites were operated without the speciation units but using the PTFE (Teflon) filters to protect the instrument from sea salt and other particles intrusion. Slemr et al. (2015) assumed that the surface active GOM in the humid air of the marine boundary layer (MBL) at several GMOS secondary sites, mostly located at the coastline (i.e., Cape Point (South Africa), Cape Grim (Australia) as well as Sisal (Mexico), Nieuw Nickerie (Paramaribo), Calhau (Cabo Verde), etc.), has been filtered out together with particulate matter (PM), partly by the sea salt particles loaded PTFE filter and partly on the walls of the inlet tubing. Consequently, they assumed that measurements at the secondary sites represent GEM only and are thus directly comparable to those at remote master sites. In contrast, the observations made by Temme et al. (2003) at Troll (Antarctica) suggested that at the low temperature and humidity prevailing at this site, GOM passed the inlet tubing and the PTFE filter, thus measuring TGM and not GEM. Taking into account these findings, Slemr et al. (2015) calculated for the GMOS master site on Amsterdam Island (AMS) a value of GOM less than 1 % of TGM compared to the other secondary sites in the Southern Hemisphere, including Troll, therefore highlighting a value which is insignificant when compared with the uncertainties discussed in the available peer-reviewed literature (Slemr et al., 2015). Since we compare results at various stations, in this work we have taken into account analysis of both systematic and random uncertainties associated with the measurements as well as published results of Tekran intercomparison exercises as reported and discussed elsewhere (Slemr et al., 2015, and references there in).
3.2. GEM measurements method
Amalgamation with gold is the principle method used to sample Hg0 for atmospheric measurements worldwide (Gustin et al., 2015). The most widely used automated instrument is the Tekran 2537A/B analyzer (Tekran Instrument Corp., Ontario, Canada) which performs amalgamation on dual gold cartridges used alternately and thermal desorption (at 500 °C) to provide continuous GEM measurements. One trap is sampling while the other is heated, releasing Hg0 into an inert carrier gas (usually ultra-high-purity argon); quantification is by cold vapor atomic fluorescence spectroscopy (CVAFS) at 253.7 nm (Landis et al., 2002). Concentrations are expressed in ng m−3 at standard temperature and pressure (STP, 273.15 K, 1013.25 hPa). The sampling interval is between 5 and 15 min based on location logistics and meteorological conditions. Taking into account the elevation of some monitoring sites in the network (i.e., Ev-K2CNR, Nepal (5050 m a.s.l.), Mt. Waliguan, China (3816 m a.s.l.), and Concordia Station (3220 m a.s.l.)), the Tekran 2537A/B analyzers have been operated with a 15 min sample time resolution at a flow rate of 0.8 L min-1. Following the SOPs the Tekran analyzers also perform automatic internal permeation source calibrations every 71 h, and the best estimate of the method detection limit is 0.1 ng m−3 at a flow rate of 1Lmin-1. The alternative automated instrument to measure continuous GEM concentrations is the Lumex RA915AM, which is based on the use of differential atomic absorption spectrometry with direct Zeeman effect, providing a detection limit lower than 1 ng m−3 (Sholupov and Ganeyev, 1995; Sholupov et al., 2004). Comparison studies between the Tekran 2537 and the RA-915AM performed during EN 15852 standard development showed good agreement of the monitoring data obtained with these systems (Brown et al., 2010b).
3.3. GEM/GOM/PBM measurements method
Speciated atmospheric Hg measurements were performed using the Tekran Hg speciation system units (models 1130 and 1135) coupled to a Tekran 2537A/B analyzer. PBM and GOM concentrations are expressed in picograms per cubic meter (pgm−3) at STP (273.15 K, 1013.25 hPa). At most GMOS sites, the speciation units were located on the rooftop of the station and connected to a Tekran 2537A/B analyzer through a heated PTFE line (50 °C, 10 m in length). The sampling time resolution, due to some technical/location issues, was set equal to 5, 10 and 15 min for GEM (see tables in the Supplement) and equal to 1, 2 and 3h for GOM and PBM. Speciation measurements were performed following the GMOS SOPs and procedure as described elsewhere (Landis et al., 2002) using a size-selective impactor inlet (2.5 μm cutoff aerodynamic diameter at 10L min−1), a KCl-coated quartz annular denuder in the 1130 unit and a quartz regenerable particulate filter (RPF) in the 1135 unit.
3.4. Quality assurance and quality control procedures
In terms of network data acquisition, QA/QC implementation procedures and data management, the worldwide configuration of the GMOS network was a challenge for all scientists and site operators involved in GMOS. The traditional approaches to Hg monitoring QA/QC management that were primarily site specific and manually implemented were no longer easily applicable or sustainable when applied to a global network with the number and size of data streams generated from the monitoring stations in near real time. The G-DQM system was designed to automate the QA process, making it available on the web with a user-friendly interface to manage all the QC steps from initial data transmission through final expert validation. From the user’s point of view, G-DQM is a web-based application, developed using an approach based on Software as a Service (SaaS) (D’Amore et al., 2015). G-DQM is part of the GMOS cyberinfrastructure (CI), which is a research environment that supports advanced data acquisition, storage, management, integration, mining and visualization, built on an IT infrastructure (Cinnirella et al., 2014; D’Amore et al., 2015).
4. Results and discussion
4.1. GMOS data coverage and consistency
Almost all GMOS stations provide near-real-time raw data that are archived and managed by GMOS-CI. Figures 1 and 2, over the 2011-2015 period and at some of the ongoing secondary and master GMOS stations, show the elemental and speciated Hg raw data coverage, respectively. For each station the coverage of raw data was generated considering the percentage of the real available raw data in respect to the total potential number of data points on monthly basis. During the first year of the project a number of sites were being established and/or equipped and not enough data were available to support broad network spatial analysis. In 2011 (at the effectively starting of the project) only four monitoring sites produced Hg measurements and, step by step, an increasing number of stations have been established and added to the network in 2012. Therefore, we evaluated the years 2013 and 2014 due to major data coverage (%) of the observations. In fact, our statistical evaluations/calculations are related to this period for all the ground-based sites taken into account within the GMOS network in order to harmonize the discussion and compare the results worldwide.
Figure 1.
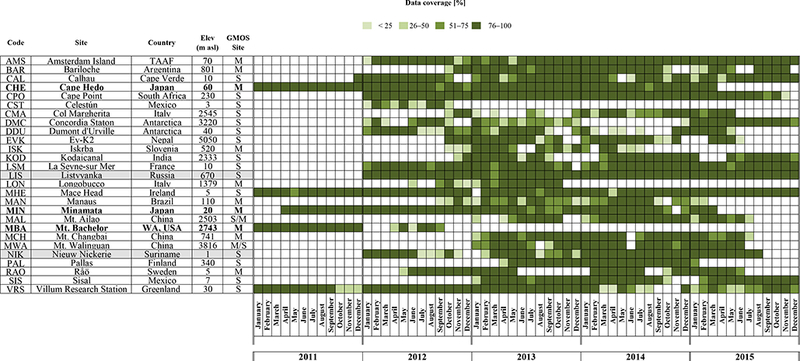
Coverage and consistency (%), on monthly basis, of GEM data collected at some of the ongoing GMOS secondary stations during the period 2011-2015.
Figure 2.
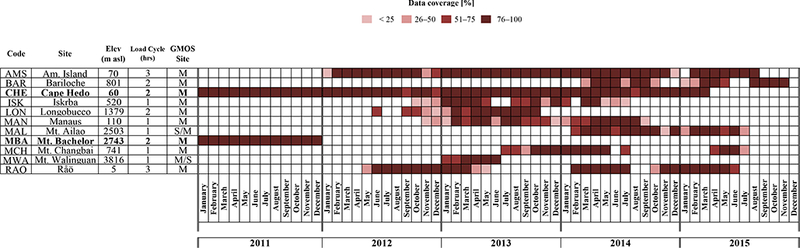
Coverage and consistency, on monthly basis, of GOM/PBM data collected at some of the ongoing GMOS master stations during the period 2011-2015.
4.2. Northern-southern hemispheric gradients
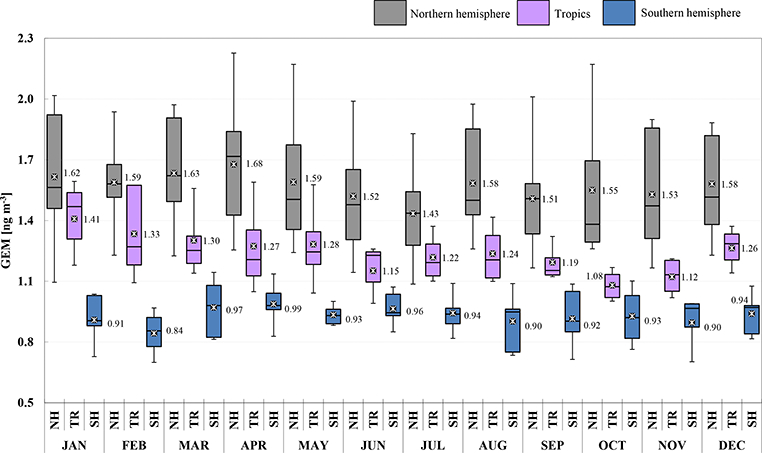
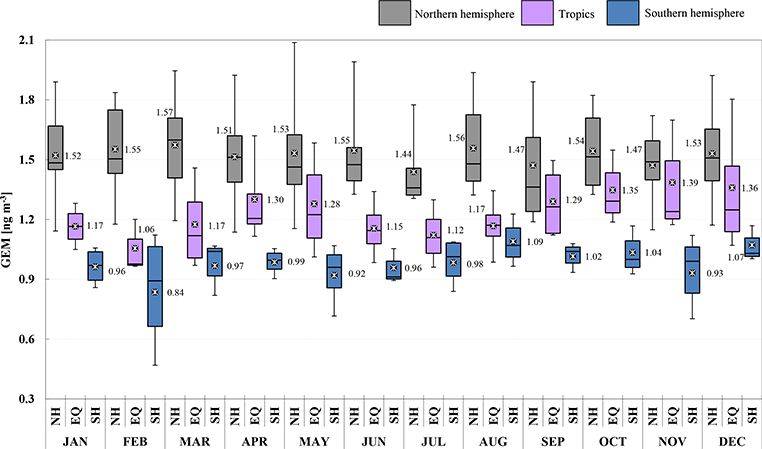
A summary of descriptive statistics based on monthly and annual averages from all GMOS sites is presented in Tables S1 and S2 in the Supplement. The 2013 and 2014 annual mean concentrations of 1.55 and 1.51ngm−3, respectively, for the sites located in the Northern Hemisphere were calculated by averaging the 13 site means for both years. Similar calculations were made for the Southern Hemisphere and the tropics (see Tables S1 and S2). Annual mean concentrations of 1.23 and 1.22 ng m−3 for 2013 and 2014, respectively, were obtained in the tropical zone and 0.93 and 0.97 ng m−3 for the Southern Hemisphere. Figure 3 shows the GEM yearly distribution for 2013 (blue) and 2014 (green). The sites have been organized in the graphic as well as in the tables according to their latitude from those in the Northern Hemisphere to those in the tropics and in the Southern Hemisphere. The data so far do not cover a long enough timespan to investigate temporal trends, but some attempts have been previously made for the more established sites, such as Mace Head (MHD), Ireland (Ebinghaus et al., 2011; Weigelt et al., 2015), and Cape Point (CPT), South Africa (Slemr et al., 2015). At MHD the annual baseline GEM means observed by (Ebinghaus et al., 2011) decreased from 1.82 ng m−3 at the start of the record in 1996 to 1.4 ng m−3 in 2011, showing a downwards trend of 1.4-1.8 % per year. Both a downward trend of 1.6% at MHD from 2013 and 2014 and the slight increase in Hg concentrations seen by Slemr et al. (2015) at CPT from 2007 to 2013 continued through the end of 2014. Some debate remains as to whether anthropogenic emissions are increasing or decreasing (Lindberg et al., 2002; Selin et al., 2008; Pirrone et al., 2013). A clear gradient of GEM concentrations between the Northern and Southern hemispheres is seen in the data for both 2013 and 2014, in line with previous studies (Soerensen et al., 2010a, b; Sommar et al., 2010; Lindberg et al., 2007; Sprovieri et al., 2010b).
Figure 3.
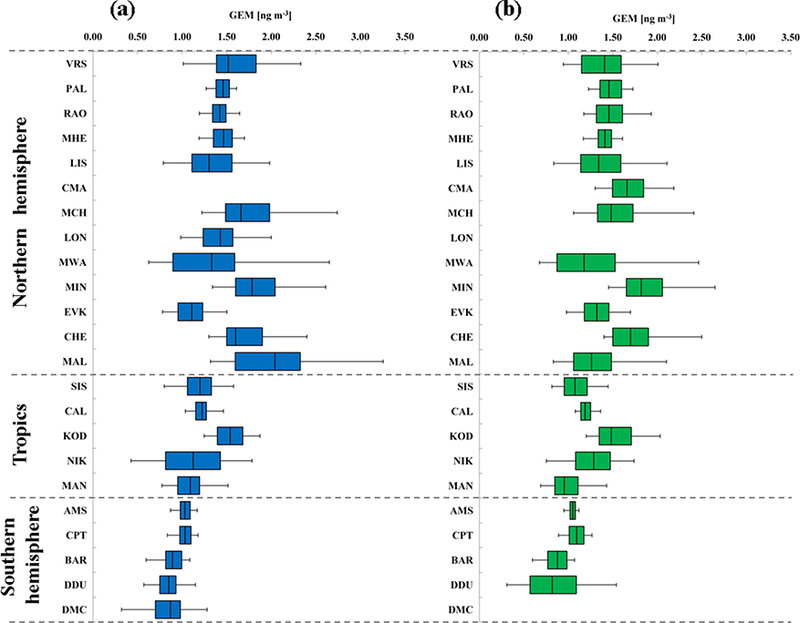
Box-and-whisker plots of gaseous elemental mercury yearly distribution (GEM, ng m−3) at all GMOS stations for (a) 2013 and (b) 2014. The sites are organized according to their latitude from the northern to the southern locations. Each box includes the median (midline) and 25th and 75th percentiles (box edges), 5th and 95th percentiles (whiskers).
The 13 northern sites had significantly higher median concentrations than the southern sites did. The north-south gradient is clearly evident in Fig. 4 where the probability density functions (PDFs) of the data are reported. The datasets have been divided into three principal groups related to the latitude: north samples, tropical samples and south samples. The histograms, normalized to the unit area, have been constructed following the Scott rule for the bin width , where σ represents the standard deviation and n the number of samples. This choice is optimal when dealing with normal distributed samples since it minimizes the integrated mean squared error of the density estimate and is then fitted through a normal distribution (full line in Fig. 4), obtained through the classical maximum likelihood estimation method. Since a clear overlap can be observed between the three datasets presented in Fig. 4, in order to make the distinction between the distributions clear we perform the standard Student t test against the null hypothesis (h0) that the three distributions come from the same mother distribution with the same mean (μ0) and unknown standard deviation (σ0).
Figure 4.
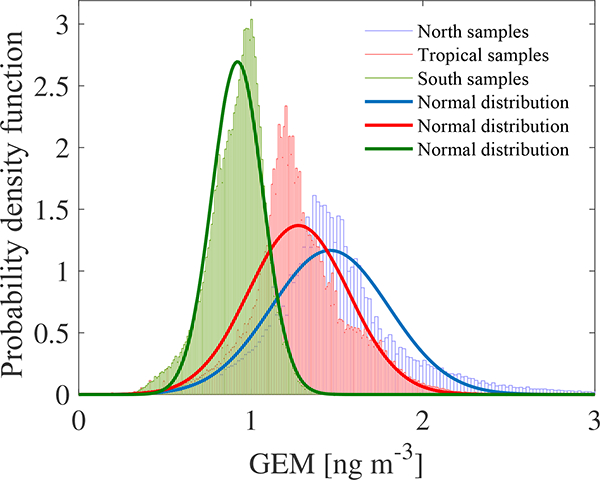
Probability density functions (PDFs) of the GEM data (ng m−3) for the northern, southern and tropical sample groups (dash dotted lines). Full lines the normal distribution fit of the samples.
For every case the null hypothesis (h0) can be rejected, as the means of the three distribution are significantly different, with a 99 % confidence level. If XN, XS and XT are the mean of the experimental measures, respectively, for the northern, southern and tropical groups, the confidence intervals evaluated from the t test are reported in Table 2. The interpretations of the results clearly demonstrate that XN > XT > XS (Table 2), so that a significant gradient exists in the GEM concentrations from the Northern Hemisphere to the Southern Hemisphere. Due to the significant difference in the PDFs, the probability p (p value) of observing a test statistic as extreme as, or more extreme than, the observed value under the null hypothesis is close to zero. Thus the validity of the null hypothesis should be rejected. The spatial gradient observed from northern to southern regions is highlighted in both Figs. 5 and 6, which also report the statistical monthly distribution of GEM values obtained for 2013 and 2014, respectively, at all GMOS sites in the Northern and Southern hemispheres as well as in the tropical area.
Table 2.
The mean (X) of the experimental measures for the northern (XN), southern (XS) and tropical (XT) groups and the confidence intervals evaluated from the Student t test among them.
| Difference between means |
Minimum of the confidence interval |
Maximum of the confidence interval |
|---|---|---|
| XN – XS | 0.590 | 0.592 |
| XN – XT | 0.225 | 0.229 |
| XT – XS | 0.362 | 0.365 |
Figure 5.
Monthly statistical distribution and spatial gradient for 2013 year from Northern Hemisphere to Southern Hemisphere.
Figure 6.
Monthly statistical distribution and spatial gradient for 2014 from Northern Hemisphere to Southern Hemisphere.
4.2.1. Seasonal pattern analysis in the Northern Hemisphere
Statistics describing the spatial and temporal distribution of GEM concentrations at all GMOS sites for 2013 and 2014 are summarized in Fig. 3 whereas Figs. 5 and 6 show the monthly statistical GEM distribution for both years considered. The GEM concentrations highlight that the mean GEM values of most of the GMOS sites were between 1.3 and 1.6 ng m−3, with a typical interquartile range of about 0.25 ng m-3. Only a few sites have shown a mean values above 1.6 ng m−3, such as MCH, MIN and MAL, and only the EVK site, located at 5050 m a.s.l. in the Eastern Himalaya of Nepal, reported mean values below 1.3 ng m-3. This value is comparable with free tropospheric concentrations measured in August 2013 over Europe (Weigelt et al., 2016). The mean GEM concentration observed at EVK is less than the reported background GEM concentration for the Northern Hemisphere (1.5-1.7 ng m−3) and more similar to expected background levels of GEM in the Southern Hemisphere (1.1-1.3 ng m−3) (Lindberg et al., 2007; Pirrone, 2016). The values between 1.3 and 1.6 ng m−3 observed at the other GMOS sites in the Northern Hemisphere are comparable to the concentrations measured at the long-term monitoring stations at Mace Head, Ireland (Ebinghaus et al., 2011; Slemr et al., 2011; Weigelt et al., 2015), and Zingst, Germany (Kock et al., 2005). GEM concentration means are also in good agreement with the overall mean concentrations observed at multiple sites in the Canadian Atmospheric Mercury Measurement Network (CAMNet) (1.58 ng m−3) reported by Temme et al. (2007) and those reported from Arctic stations in this paper (VRS, PAL). Seasonal variations of GEM concentrations have also been observed at all GMOS sites in the Northern Hemisphere. Most sites show higher concentrations during the winter and spring and lower concentrations in summer and fall seasons (Figs. 5 and 6). However, few sites such as VRS, Station Nord (northeastern Greenland: 81°36’ N, 16°40’ W), show a slightly different seasonal variation. In winter this high Arctic site (VRS) is sporadically impacted by episodic transport of pollution mainly due to high atmospheric pressure systems over Siberia and low pressure systems over the North Atlantic (Skov et al., 2004; Nguyen et al., 2013). During the spring (April-May) and summer (August-September) seasons GEM concentrations show a higher variability with low concentrations near the instrumental detection limit due to episodic atmospheric Hg depletion events (AMDEs) that occur in the spring (Skov et al., 2004; Sprovieri et al., 2005a, b; Hedgecock et al., 2008; Steffen et al., 2008; Dommergue et al., 2010a) and high GEM concentrations (2 ng m−3) in June and July, probably due to GEM emissions from snow and ice surfaces (Poulain et al., 2004; Sprovieri et al., 2005a, b, 2010b; Dommergue et al., 2010b; Douglas et al., 2012) and Hg evasion from the Arctic Ocean (Fisher et al., 2012; Dastoor and Dumford, 2014). Models of the MBL that simulate the temporal variations of Hg species (Hedgecock and Pirrone, 2005, 2004; Holmes et al., 2009; Soerensen et al., 2010b) show that photo-induced oxidation of GEM by Br can reproduce the diurnal variation of GOM observed in the MBL during cruise measurements better than other oxidation candidates (Hedgecock and Pirrone, 2005; Sprovieri et al., 2010a) and also the seasonal variation (Soerensen et al., 2010b). Although Br is currently considered to be the globally most important oxidant for determining the lifetime of GEM in the atmosphere, there are also other possible candidates that can enhance Hg oxidation (Hynes et al., 2009; Ariya et al., 2008; Subir et al., 2011, 2012). The lack of a full understanding of the reaction kinetics and fate of atmospheric Hg highlights the need to have a global observation system as presented here in order to calibrate and constrain atmospheric box and global/regional-scale models (Hedgecock and Pirrone, 2005; Dastoor et al., 2008).
4.2.2. GMOS sites in Asia
As can be seen in Fig. 3, the group with the highest GEM median variability and maximum concentrations is in Asia, which includes the following sites: Mt. Ailao (MAL), Mt. Changbai (MCH), Mt. Waliguan (MWA) and Minamata (MIN), where 95th percentile values ranged from 3.26 to 2.74 ng m−3 in 2013 (Table S2). These sites are often impacted by air masses that have crossed emission source regions (AMAP/UNEP, 2013). GEM concentrations recorded at all remote Chinese sites (MAL, MCH and MWA) are elevated compared to that observed at background/remote areas in Europe and North America, and at others sites in the Northern Hemisphere (Fu et al., 2012a, b, 2015). A previous study by Fu et al. (2012a) at MWA suggested that long-range atmospheric transport of GEM from industrial and urbanized areas in northwestern China and northwestern India contributed significantly to the elevated GEM at MWA. MAL station is located in Southwest China, at the summit of Ailao Mountain National Nature Reserve, in central Yunnan province. It is a remote station, isolated from industrial sources and populated regions in China. Kunming, one of the largest cities in Southwest China, is located 180 km to the northeast of the MAL site. The winds are dominated by the Indian summer monsoon (ISM) in warm seasons (May to October), and the site is mainly impacted by Hg emission from eastern Yunnan, western Guizhou and southern Sichuan of China and the northern part of the Indochinese Peninsula. In cold seasons the impact of emissions from India and northwestern part of the Indochinese Peninsula increased and played an important role in elevated GEM observed at MAL (Zhang et al., 2016). However, most of the important Chinese anthropogenic sources of Hg and other air pollutants are located to the north and east of the station, whereas anthropogenic emissions from southern and western Yunnan province are fairly low (Wu et al., 2006; Kurokawa et al., 2013; Zhang et al., 2016). Average atmospheric GEM concentrations during this study calculated for MWA and MAL during 2013 and 2014 are in good agreement with those observed during previous measurements at both sites from October 2007 to September 2009 at MWA and from September 2011 to March 2013 at MAL (Fu et al., 2015; Zhang et al., 2016). Also the overall mean GEM concentration observed in 2013 and 2014 at MCH background air pollution site (1.66±0.48 ng m−3 in 2013 and 1.48±0.42 ng m−3 in 2014, respectively) is in good agreement with the overall mean value recorded earlier from 24 October 2008 to 31 October 2010 (1.60±0.51ng m−3, Fu et al., 2012b). Fu et al. (2012a) highlighted a higher mean TGM concentration of 3.58 ± 1.78 ng m−3 observed from August 2005 to July 2006 that was probably due to surface winds circulation with effect of regional emission sources, such as the large iron mining district in the northern part of North Korea and two large power plants and urban areas to the southwest of the sampling site.
In summary, the observed concentrations are a function of site location relative to both natural and anthropogenic sources, elevation and local conditions (i.e., meteorological parameters), often showing links to the patterns of regional air movements and long-range transport. Seasonal variations at ground-based remote sites in China have been observed. At MCH GEM was significantly higher during cold seasons compared to that recorded in warm seasons (from April to September) whereas the reverse has been observed at the other two Chinese GMOS sites.
In order to statistically check the difference of GEM concentrations among the three Chinese sites an alternative statistical test has been performed, since in this case the distributions are strongly non-normal.
As in the previous case we construct the unit-area histogram, then we fit with a log-normal distribution. It is worth noting that in this case the histograms has been constructed by manually setting the bin width ᐃW. With this choice the total number of bins can be evaluated as
| (1) |
By looking at Fig. 7, is easy to notice that the skewness (μ3/σ3 ~ 2 where μ3 is third-order moment of the distribution and σ is the standard deviation) and the kurtosis ( where μi is the i th-order moment of the distribution) are far from being zero. In the following the alternative is briefly described. Let us consider a pair of our three time series, namely Xi (i = 1,2), which corresponds to independent random samples described by the log-normal distributions. Then the random variables Yi = ln(Xi) are close to normal distribution with means μi and variances , namely .
Figure 7.
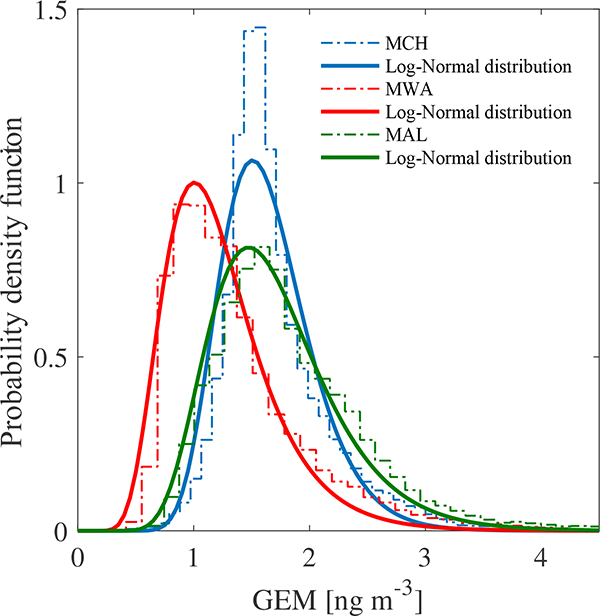
Probability density functions (PDFs) of the GEM data (ng m−3) for the Chinese sample groups (dash dotted lines). Full lines the log-normal distribution fit of the samples.
Since is the expectation value for Xi, the problem of our interest is then to test the null hy-pothesis on η2 – η1 More formally, we test H0 : θ < 0, where θ = η2 – η1. In other words, we test the null hypothesis to see if there is a significant difference in the sample means. Using the algorithm described in Krishnamoorthya and Mathewb (2003) and Abdollahnezhad et al. (2012), specifically designed to perform the inference on difference of means of two log-normal distributions, we obtain the estimates for the p-values which are close to 1 and the confidence intervals, calculated at a confidence level of 95 % (reported in Table 3).
Table 3.
Differences between the ηi obtained for MCH, MWA and MAL, confidence intervals and associated p values.
| Difference of ηi |
Minimum of the confidence interval |
Maximum of the confidence interval |
P |
|---|---|---|---|
| ηMCH–ηMWA | 0.285 | 0.286 | 1 |
| ηMCH–ηMAL | 0.043 | 0.043 | 1 |
| ηMWA–ηMAL | 0.328 | 0.329 | 1 |
From the statistical results we can conclude that a clear distinction exists between the MWA site and the other two (MCH, MAL) as shown from the values in Table 3. However, despite the large overlap in the samples distributions of MCH and MAL the difference in their ηi (ηMCH and ηMAL, respectively) is also significant, with a smaller confidence interval.
Several hypothesis have been made to explain the seasonal variations of GEM in China, including seasonal changes in anthropogenic GEM emissions and natural emissions. The seasonal emission changes mainly resulted from coal combustion for urban and residential heating during cold seasons. This source lacks emission control devices and releases large amounts of Hg, leading to elevated GEM concentrations in the area and thus at MCH (Feng et al., 2004; Fu et al., 2008a, b, 2010). Conversely, GEM at MAL and MWA was higher in warm seasons than in cold seasons. These findings highlight that emissions from domestic heating during the winter could not explain the lower winter GEM concentrations observed at MWA and MAL, but there might be other not-yet-understood factors that played a key role in the observed GEM seasonal variations at these sites, such as the monsoonal winds influence which can change the source-receptor relationship at observational sites and subsequently the seasonal GEM trends (An, 2000; Fu et al., 2015). Among the remote Chinese sites, MAL started as secondary site and in 2014 was upgraded to a master site; conversely, MWA started as a master site and then became a secondary site whereas MCH operated continuously as a master site. Therefore, PBM and GOM concentrations have been measured during the years 2013 and 2014 at all Chinese sites even if not continuously (see Fig. 2 for Hg speciation data coverage). The GOM and PBM concentrations measured at these sites were substantially elevated compared to the background values in the Northern Hemisphere, from 1.8 to 42.8 pg m−3 and from 40.4 to 167.4 pg m−3 at the MCH and MWA, respectively, in 2013. The 2014 PBM maxima were 44.2 and 45.0 pg m−3 at MCH and MAL, respectively. Regional anthropogenic emissions and long-range transport from domestic source regions are likely to be the primary causes of these elevated values (Sheu et al., 2013). Seasonal variations of PBM observed at the Chinese master sites mostly showed lower concentrations in summer and higher concentrations (up to 1 order of magnitude higher) in winter and fall (Wang et al., 2006, 2007; Fu et al., 2008b; Zhu et al., 2014; Xu et al., 2015; Xiu et al., 2009; Zhang et al., 2013). The higher PBM in winter was likely caused by direct PBM emissions, formation of secondary particulate Hg via gas-particle partitioning and a lack of wet scavenging processes (Wang et al., 2006; Fu et al., 2008b; Zhu et al., 2014). PBM has an atmospheric residence time ranging from a few hours to several days and can therefore be transported to the remote sites when conditions are favorable (Sheu et al., 2013). Atmospheric PM pollution is of special concern in China due to the spatial distribution of anthropogenic emission concentrations of PM2.5 in heavily populated areas of eastern and northern China, which are among the highest in the world (van Donkelaar et al., 2010). The GOM concentrations observed at both master sites show high variability and several episodes with high GOM values were probably due to local emission sources (such as domestic heating in small settlements) rather than to long-range transport from industrial and urbanized areas (Fu et al., 2015). GOM has a shorter atmospheric residence time that limits long-range transport (Lindberg and Stratton, 1998; Pirrone et al., 2008). However, with low RH and high winds, the possibility of regional transport of GOM cannot be ruled out. For example, the observations at MWA exhibit a number of high GOM events related to air plumes originating from industrial and urbanized centers that are about 90 km east of the sampling site (Fu et al., 2012a; Pirrone, 2016). MWA is a remote site situated at the edge of the northeastern part of the Qinghai-Xizang (Tibet) plateau. The monitoring station is relatively isolated from industrial point sources and there are no known local Hg sources around the site. Most of the Chinese industrial and populated regions associated with anthropogenic Hg emissions are situated to the east of MWA. Predominantly winds are from the west to southwest in cold seasons and the east in warm seasons (Pirrone, 2016). East Asia is, in fact, the largest Hg source region in the world, contributing to nearly 50 % of the global anthropogenic Hg emissions to the atmosphere (Streets et al., 2005, 2011; Pirrone et al., 2010; Lin et al., 2010).
4.2.3. Seasonal pattern analysis in the Southern Hemisphere
For the sites located in the Southern Hemisphere, the GEM concentrations highlight that the mean GEM values ranged between 0.84 and 1.09 ng m−3, in both 2013 and 2014, with a typical interquartile range of about 0.25 ng m−3 (see Figs. 3, 5 and 6). The mean GEM concentrations observed at the southern sites are lower than those reported in the Northern Hemisphere but in good agreement with the southern hemispherical background (1.1 ng m−3) (Lindberg et al., 2007; Sprovieri et al., 2010b; Lindberg et al., 2002; Dommergue et al., 2010b; Angot et al., 2014; Slemr et al., 2015; Soerensen et al., 2010a) and the expected range for remote sites in the Southern Hemisphere. As in the Northern Hemisphere, a seasonal variation of GEM concentrations was observed in the Southern Hemisphere. In particular, GEM concentrations from the coastal Global Atmosphere Watch station, Cape Point (CPT), South Africa, show seasonal variations with maxima during austral winter and minima in summer. The site is located in a nature reserve at the southernmost tip of the Cape Peninsula on a hill, 230 m a.s.l. It is characterized by dry summers with moderate temperatures and increased precipitation (cold fronts) during austral winter. During the summer months, biomass burning events sometimes occur within the southwestern Cape region, affecting GEM levels. The dominant wind direction at CPT is from the southeastern sector, advecting clean maritime air from the South Atlantic Ocean (Brunke et al., 2004, 2012) which occurs primarily during austral summer (December till February). Furthermore, the station is also at times subjected to air from the northern sector, mainly during austral winter. During such continental airflow events, anthropogenic emissions from the industrialized area in Gauteng, 1500 km to the northeast of CPT, can sometimes be observed (Brunke et al., 2012; Slemr et al., 2015). The GEM seasonal variability at CPT is hence in good agreement with the prevailing climatology at the site. Also GEM data at Amsterdam Island followed a similar trend, with slightly but significantly higher concentrations in winter (July-September) than in summer (December-February). Amsterdam Island is a remote and very small island of 55 km2 with a population of about 30 residents, located in the southern Indian Ocean at 3400 and 5000 km downwind from the nearest lands, Madagascar and South Africa, respectively (Angot et al., 2014). GEM concentrations at AMS were remarkably steady with an average hourly mean concentration of 1.03 ± 0.08 ng m−3 and a range of 0.72-1.55 ng m-3. A small seasonal cycle has been observed by Angot et al. (2014) and despite the remoteness of the island, wind sector analysis, air mass back trajectories and satellite observations suggest the presence of a long-range contribution from the southern African continent to the GEM regional/global budget from July to September during the biomass burning season extended from May to October (Angot et al., 2014). The higher GEM concentrations at AMS are comparable with those recorded at Calhau (Cabo Verde), Nieuw Nickerie (Paramaribo) and Sisal (Mexico) in the tropical zone, whereas the lower concentrations of GEM observed, less than 1 ng m−3, were associated with air masses coming from southern Indian Ocean and the Antarctic continent. Bariloche (BAR) master site in North Patagonia also shows higher concentrations during the austral winter (from end of May to September) and lower concentrations in other seasons (Diéguez et al., 2015). The Patagonian site has been established inside Nahuel Huapi National Park, a well-protected natural reserve, located east of the Patagonian Andes. The area is included in the Southern Volcanic Zone (SVZ) of the Andes, under the influence of at least three active volcanoes with high eruption frequency located at the west of the Andes cordillera (Daga et al., 2014). The climate of the region is influenced by the year-round strong westerly winds blowing from the Pacific which discharge the humidity in a markedly seasonal way (fall-winter) in the western area of the park. GEM records at BAR station show background concentrations comparable to those found in Antarctica and other remote locations of the South Hemisphere with concentrations ranging between 0.2 and 1.3 ng m−3, with an annual mean of 0.89± 0.15 ng m-3. Previous records of GEM concentrations from a short-term survey in 2007 along a longitudinal transect across the Andes with Bariloche as the eastern endpoint reported concentrations below 2 ng m−3 close to BAR (Higueras et al., 2014). In this survey, the highest GEM concentrations were recorded in the proximity and downwind from the volcanic area, reaching concentrations up to 10 ng m−3 (Higueras et al., 2014). Similarly to the seasonal trends at other GMOS sites in the Southern Hemisphere, GEM concentrations were at their lowest level in summer on the Antarctic Plateau at Concordia Station (DMC, altitude 3220 m) but at their highest level in fall (Angot et al., 2016b). GEM concentrations reached levels of 1.2 ng m−3 from mid-February to May (fall) likely due to a low boundary layer oxidative capacity under low solar radiation limiting GEM oxidation and/or a shallow boundary layer (~ 50 m in average) limiting the dilution. In summer (November to mid-February), the DMC GEM data showed a high variability with a concentration range varying from below the detection limit to levels comparable to those recorded at midlatitude background Southern Hemisphere stations due to an intense chemical exchange at the air/snow interface. Additionally, the mean summertime GEM concentration at DMC was ~ 25 % lower than at other Antarctic stations in the same period of the year, suggesting a continuous oxidation of GEM as a result of the high oxidative capacity of the Antarctic plateau boundary layer in summer. GEM depletion events occurred each year in summer (January-February 2012 and 2013) with GEM concentrations remaining low (~ 0.40 ng m−3) for several weeks. These depletion events did not resemble the ones observed in the Arctic. They were not associated with depletion of ozone and occurred as air masses stagnated over the Plateau, which could favor an accumulation of oxidants within the shallow boundary layer. These observations suggest that the inland atmospheric reservoir in Antarctica is depleted in GEM and enriched in GOM in summer. Measurements at DDU on the East Antarctic coast were dramatically influenced by air masses exported from the Antarctic Plateau by strong katabatic winds (Angot et al., 2016a). These results, along with observations from earlier studies, demonstrate that, in Antarctica, the inland atmospheric reservoir can influence the cycle of atmospheric Hg at a continental scale (Sprovieri et al., 2002; Temme et al., 2003; Pfaffhuber et al., 2012; Angot et al., 2016b, a). Observations at DDU also highlighted that the Austral Ocean is a net source of GEM in summer and a net sink in spring, likely due to enhanced oxidation by halogens over sea-ice-covered areas.
4.2.4. Seasonal pattern analysis in the tropical zone
Relatively few observations of atmospheric Hg had been carried out in the tropics, before the start of GMOS. Until recently atmospheric Hg data for the tropics were only available from short-term measurement campaigns. To date, therefore, there is no information in the tropical area that can be used to establish long-term trends. Observations in this region may provide a valuable input to our understanding of key exchange processes that take place in the Hg cycle considering that the Intertropical Convergence Zone (ITCZ) passes twice each year over this region and the northern and southern hemispheric air masses may well influence the evolution of Hg concentrations observed in this region. As can be seen in Fig. 3, five GMOS sites are located in the tropics: Sisal (SIS) in Mexico, Nieuw Nickerie (NIK) in Suriname, Manaus (MAN) in Brazil, Calhau (CAL) in Cabo Verde and southern Kodaikanal (KOD) in southern India. GEM concentrations observed in 2013 and 2014 at all sites are comparable with Hg levels recorded at remote sites in the Southern Hemisphere (1.1 to 1.3 ng m−3; Lindberg et al., 2007). Among these sites, the Kodaikanal site (KOD) shows the highest monthly mean GEM concentrations (see Figs. 5 and 6 as well as Tables S1 and S2) ranging between 1.25 ng m−3 (5th percentile) to 1.87 ng m−3 (95th percentile) during 2013 with an annually based statistic mean of 1.54 ± 0.20 ng m−3 and between 1.20 ng m−3 (5th percentile) to 2.03 ng m−3 (95th percentile) during 2014 with an annually average of 1.48 ±0.26 ng m-3. KOD is a Global Atmospheric Watch (GAW) regional site which is operated by the Indian Meteorological Department. It is worthwhile to point out that the other tropical GMOS sites are close to sea level and on the coast, whereas KOD is a high-altitude site (2333 m a.s.l.). Therefore different meteo-climatic conditions influence the long-range transport of air masses to this site. This site is also influenced by anthropogenic sources such as the well-known, but not close, Hg thermometer plant, 2150 m far away from the monitoring station at Kodaikanal (Karunasagar et al., 2006). Due to this anthropogenic influence, atmospheric Hg concentrations from 3 to 8 ng m−3 for the years 2000 and 2001 have been reported (Rajgopal and Mascarenhas, 2006). India is the third largest hard coal producer in the world after the People’s Republic of China and the USA (Pirrone et al., 2010; Mason, 2009; Penney and Cronshaw, 2015). For the past 3 decades, India has increased the production of metals, cement, fertilizers and electricity through burning of coal, natural gas and oil, becoming one of the most rapidly growing economies (Mukherjee et al., 2009; Karunasagar et al., 2006). Relatively little attention has been paid to potential Hg pollution problems due to mining operations, metal smelting, energy and fuel consumption, which could impact on ecosystem health (Mohan et al., 2012). Hg concentrations are in fact enhanced in India due to industrial emissions of Hg mostly from coal combustion (the major source category (48 %), followed by waste disposal (31 %), the iron and steel industry, chloralkali plants, the cement industry and other minor sources (i.e., clinical thermometers) (Mukherjee et al., 2008; UNEP, 2008). Unfortunately, details of Hg emissions from these facilities and atmospheric Hg data in general are scarce. Therefore it is necessary for India as well as for the other place in the world where Hg measurement are yet lacking to generate continuous data, which can then be used by scientists for modeling applications to improve emission inventories in order to prevent inaccurate assessments of Hg emission and deposition.
GEM levels observed at Sisal (SIS), Mexico, were below the expected global average concentration (~ 1.5 ng m−3). Monthly mean GEM concentrations ranged between 1.0 and 1.47 ng m−3 in 2013 with an annual average of 1.20±0.24 ng m−3 (5th and 95th percentiles: 0.8 and 1.58 ng m−3), whereas in 2014 the range varied from 0.82 to 1.45 ng m−3, with an annual average of 1.11 ±0.37 ng m−3 (5th and 95th percentiles: 0.82 and 1.45 ng m−3). GEM measurements at SIS showed, in addition, very little variability over the sampling period, indicating that this relatively remote site on the Yucatán Peninsula was not subject to any significant anthropogenic sources of Hg at all. During 2013 and 2014, the SIS site was typically influenced by the marine air originating from the Atlantic Ocean before entering the Gulf of Mexico (Sena et al., 2015). Average GEM concentrations reported at SIS are lower than those recorded in other rural places in Mexico, such as Puerto Angel (on the Pacific coast in Oaxaca state) and Huejutla (a rural area in the state of Hidalgo), where average values of 1.46 and 1.32 ng m−3 were determined, respectively (de la Rosa et al., 2004). Low GEM concentrations were recorded in 2013 during the later part of the wet season (July/October). Those values may indicate a slight decrease, probably due to deposition processes since the site is a coastal station and subject to frequent episodes with high humidity caused by rain (Sprovieri et al., 2016). These findings have also been confirmed through wind roses and backward trajectories that show the predominant wind direction from east-southeast most of the time and sometimes from east-northeast (Atlantic Ocean) (Sprovieri et al., 2016). In addition, the ITCZ moves north of the Equator passing over the Yucatán peninsula during the northern hemispheric summer, causing tropical rain events which could contribute to the slight decrease of Hg concentrations. Highest GEM levels were observed during the winter period (December-January) in 2013, whereas 2014 had the lowest GEM concentration in January and higher GEM levels during spring and summer. The background Hg concentrations measured at Sisal are closely comparable to those recorded at Nieuw Nickerie (NIK), Paramaribo, Suriname, located on the northeastern coast of the South American continent, the first long-term measurement site in the tropics which has been in operation since 2007 (Muller et al., 2012). Analysis of data shows that the annual mean GEM for 2013 and 2014 at NIK are a little lower than those at SIS: 1.13 ± 0.42 and 1.28 ± 0.46 ng m−3, respectively (see Tables S1 and S2). NIK is also a background site because most of the time the air masses arriving at the site come from the clean marine air of the Atlantic Ocean and the influence of possible local anthropogenic sources and continental air is minimal. As the ITCZ crosses Suriname twice each year, the NIK site samples both northern and southern hemispheric air masses. Occasionally higher values are seen: 1.57 ng m−3 in February/March 2013 and 1.51 in August/September 2014 (see Figs. S1 and S2). Manaus (MAN) in Amazonia (Brazil) is a GMOS master site located in the Amazon region, an area with a history of important land use change and significant artisanal and small-scale gold mining activities since the 1980s. Burning of natural vegetation to produce agriculture lands or pastures represents an important diffuse source of Hg to the atmosphere in Brazil (Lacerda et al., 2004; do Valle et al., 2005). The analysis of atmospheric Hg species at this site is thus important for the determination of the dynamics of atmospheric Hg. Annual mean Hg concentrations in 2013 and 2014 at MAN are slightly lower than those at both SIS and NIK, with little variability between the two years (see Tables S1 and S2). The measurements from MAN station may therefore suggest that although the Hg emissions from regional biomass burning and artisanal and small-scale gold mining represent the major emission sources in the Amazon basin as reported in a study performed by Artaxo et al. (2000), they may not have a significant impact locally but contribute to the global Hg background (concerning Hg from biomass burning see De Simone et al. (2015). Unfortunately the emissions from both these sources are associated with large uncertainties and vary over time. Quantifying their impact in South America is extremely important and there is a strong case for expanding the number of GMOS measurement sites in the region. MAN is in fact a very remote site, inside the campus of the Embrapa Amazonia Oriental and upwind from the three main gold mining areas in the Amazon basin, which are located in Rondonia, Mato Grosso and in the south of the Para states (Artaxo et al., 2000). Previous Hg measurements performed by Artaxo et al. (2000) during an aircraft experiment over different sites in the Amazon basin highlighted Hg concentrations between 0.5 and 2 ng m−3 at pristine sites (and among them also MAN) not impacted by air masses enriched with emissions from gold mining areas and/or biomass burning. Those data collected from August to September 1995 are comparable to ours observed in 2013 and 2014 at MAN during the same period, whereas other sites over areas with intense biomass burning and near areas with strong Hg emissions (Alta Floresta and Rondonia, for example) reported very high Hg levels (5-14 ng m−3)(Artaxo et al., 2000). These high Hg concentrations were never observed at MAN during the 2013 and 2014 period. Monthly mean GEM concentrations at MAN ranged between 1.01 and 1.18 ng m−3 in 2013 and between 0.94 and 1.10 ng m−3 in 2014. Also PBM and GOM recorded during 2013 show little variation and varied between 1.35 and 12.70 pgm−3 (5th and 95th percentile, respectively) with a median value of 3.17 pg m-3. In 2014, the range was from 0.53 to 5.24 pg m−3 (5th and 95th percentile, respectively) with a median value of 1.48 pg m-3. The MAN Hg concentrations therefore seem not to be influenced by regional emissions. However, a number of parameters, such as the intense air mass convection occurring in the Amazon basin and meteorological condition in general, clearly contribute to the observed Hg concentrations, and they do not necessarily reflect only regional emissions (Artaxo et al., 2000; do Valle et al., 2005). Most of the air masses that reach the site in 2013 and 2014 come from tropical Atlantic and travel for about 1500 km over pristine forest before reaching the site (Artaxo et al., 2015); the prevailing winds during the wet seasons (from January-March) were from north-northeast, northeast and east-northeast, whereas during the dry seasons (from August to October) they were from north and north-northeast as well as north-northwest (Artaxo et al., 2015).
The Cape Verde Atmospheric Observatory’s Calhau Station (CAL) contributes data from the eastern tropical Atlantic Ocean, where GMOS provides the only existing dataset. CAL is an important GAW station located on Sao Vicente Island, approximately 50 m from the coastline. GEM measurements from 2012 to 2014 were broadly consistent with previously published oceanographic campaign measurements in the region, with typical Hg values between 1.1 and 1.4 ng m-3. The prevailing wind was from the northeast open ocean, bringing air masses from the tropical Atlantic and from the African continent (Mendes, 2014). Due to its relatively long residence time in the atmosphere, the ground-level background GEM concentration tends to be relatively constant over the year in tropical regions, unlike midlatitude and polar regions where a more noticeable seasonal variation has been observed. When compared with measurements from cruise campaigns from North to South Atlantic, we can see that the GEM data at CAL are similar to previously reported South Atlantic data, where Hg concentrations are lower than the northern part of the Atlantic. Monthly mean GEM concentrations in 2013 ranged from 1.12 to 1.38 ng m−3, with an annually based mean of 1.22±0.14 ng m−3 (5th and 95th percentile equal to 1.04 and to 1.46 ng m−3, respectively), whereas in 2014 the monthly mean observed varied from 1.12 to 1.33 ng m−3, with an annually based mean of 1.20±0.09 ng m−3 (5th and 95th percentile equal to 1.08 and to 1.36 ng m−3, respectively). The highest GEM concentrations in air originating from central Africa have been recorded at CAL when the relative humidity was lowest (occasionally during dust events) (Carpenter, 2011). All tropical GMOS sites show little atmospheric Hg variability through both the years (2013 and 2014) with small GEM fluctuations during the months, which agrees well with a relatively long atmospheric lifetime of Hg in the background troposphere and small variations in the source strength (Ebinghaus et al., 2002). However, clear diurnal cycles of Hg have been conversely observed.
5. Conclusions
The higher Hg concentrations and spatiotemporal variability observed in the Northern Hemisphere compared to the tropical area and Southern Hemisphere confirm that the majority of emissions and re-emissions are located in the Northern Hemisphere. The inter-hemispherical gradient with higher GEM concentrations in the Northern Hemisphere has remained nearly constant over the years, confirmed by the observations carried out in the Southern Hemisphere and other locations where previously GMOS Hg measurements were lacking or absent. Previous results on all cruises carried out over the oceans highlighted that in the Northern Hemisphere GEM mean values are almost generally higher than those obtained in the Southern Hemisphere, with a rather homogeneous distribution of GEM in the Southern Hemisphere. The stability of these background concentrations can be seen as evidence that the atmospheric lifetime of Hg is reasonably long to explain the extent of its dispersion but would not be in accord with the most recent theoretical and experimental studies of the reaction rates of Hg with atmospheric oxidants. The oxidation of atmospheric Hg can occur with extraordinary rapidity in the polar troposphere during the springtime Hg depletion events as well as within the MBL due to the reactions between Hg and bromine compounds, although there are other possible reactants that can enhance Hg oxidation depending upon environmental factors and setting. These uncertainties highlight several Hg issue which have to be improved to better understand the atmospheric transport and transformation mechanisms of Hg. One such issue concerns the chemical composition of the oxidized phase of atmospheric Hg, GOM and PBM, which are currently operationally defined but still not well understood. Field and laboratory studies highlighted analytical interferences within the methods currently adopted to measure oxidized Hg species which suggest the variation of the chemical compounds across space and time. This has significant implications for refining existing measurement methods and developing new techniques/methodologies capable of distinguishing between Hg compounds within different environmental compartments. Knowing the precise chemical composition of GOM would immediately provide impetus to those who study reaction kinetics to refine rate constants and reaction mechanisms as well as allow modeling studies chemical mechanisms to be verified, thus improving our understanding of the important processes characterizing the atmospheric transport and transformation of Hg. The variation of observed Hg concentration across GMOS network shows increased amplitude in areas strongly influenced by anthropogenic sources. There are, however, uncertainties in the emission estimates especially for the tropical region and the Southern Hemisphere and not enough long-term information in either areas to identify long-term trends. The lack of an advanced global emission inventory for regional- and global-scale models application represented another important objective of the GMOS network. In the last years several modeling studies have highlighted the discrepancy between modeled and observed concentrations of GEM at background sites primarily due to existing gaps in biomass burning, artisanal small-scale gold mining and open-coal bed fire contributions within the emission inventories for anthropogenic sources. Therefore, long-term atmospheric Hg measurements across the GMOS global network and additional new GMOS ground-based sites increasingly incorporated into strategic areas are crucial to continue in the next future in order to provide high-quality measurement datasets which can give new insights and information about the worldwide trends of atmospheric Hg. The overarching benefit of this coordinated Hg monitoring network would clearly be the advancement of knowledge about Hg processes on a global scale due to model/measurement comparisons, models development and validation on different spatial and temporal scales, and assessing trends with significant implications within the Task Force on Hemispheric Transport of Air Pollutants (HTAP-TF) in the context of a global model intercomparison aimed to study long-range transport pathways of pollutants and their precursors. The experience gained during GMOS, the development of SOPs for Hg monitoring and the establishment of the Spatial Data Infrastructure (SDI; http://www.gmos.eu/sdi/) (along GEOSS lines), which includes the G-DQM system, provide a template to aid countries complying with the requirements of Article 22 of the Minamata Convention.
6. Data availability
Mercury data discussed in this manuscript are reported within the GMOS central database and are available upon request at http://sdi.iia.cnr.it/geoint/publicpage/GMOS/gmos_historical.zul (GMOS Database, 2014). The GMOS database coordinated by the CNR-IIA is part of the GMOS CyberInfrastructure (CI), which supports advanced data acquisition, storage, management, integration, mining and visualization. All GMOS stations provide near-real-time raw data that are archived and managed for the QA/QC process by the GMOS-Data Quality Managment System (G-DQM), a web-based application which is part of the GMOS-CI, developed by the CNR-IIA in 2011, and is currently ongoing.
Supplementary Material
Acknowledgements.
The number of people and institutions to acknowledge for their great contribution during field studies is quite significant for a project like GMOS, but we greatly acknowledge the European Commission for funding GMOS as part of the FP7 (contract no. 26511). Special thanks to those research scientists and technicians that helped to set up aircraft-based platform for UTLS measurements, ground-based sites in remote locations including EV-K2 in Karakorum, Amsterdam Island and Dome C in Antarctica, and all those involved in GMOstral 1028 (IPEV) project for field logistics and technical support.
Footnotes
The Supplement related to this article is available online at doi:10.5194/acp-16-11915-2016-supplement.
References
- Abdollahnezhad K, Babanezhad M, and Jafari AA: Inference on Difference of Means of two Log-Normal Distributions; A Generalized Approach, Journal of Statistical and Econometric Methods, 1, 125–131, 2012. [Google Scholar]
- AMAP/UNEP: Technical Background Report for the Global Mercury Assessment 2013, Tech. rep., Arctic Monitoring and Assessment Programme, Oslo, Norway/UNEP Chemicals Branch, Geneva, Switzerland, 2013. [Google Scholar]
- An Z: The history and variability of the East Asian paleomonsoon climate, Quaternary Sci. Rev, 19, 171–187, doi: 10.1016/S0277-3791(99)00060-8, 2000. [DOI] [Google Scholar]
- Angot H, Barret M, Magand O, Ramonet M, and Dommergue A: A 2-year record of atmospheric mercury species at a background Southern Hemisphere station on Amsterdam Island, Atmos. Chem. Phys, 14, 11461–11473, doi: 10.5194/acp-14-11461-2014, 2014. [DOI] [Google Scholar]
- Angot H, Dion I, Vogel N, Legrand M, Magand O, and Dommergue A: Multi-year record of atmospheric mercury at Dumont d’Urville, East Antarctic coast: continental outflow and oceanic influences, Atmos. Chem. Phys, 16, 8265–8279, doi: 10.5194/acp-16-8265-2016, 2016a. [DOI] [Google Scholar]
- Angot H, Magand O, Helmig D, Ricaud P, Quennehen B, Gallée H, Del Guasta M, Sprovieri F, Pirrone N, Savarino J, and Dommergue A: New insights into the atmospheric mercury cycling in central Antarctica and implications on a continental scale, Atmos. Chem. Phys, 16, 8249–8264, doi: 10.5194/acp-16-8249-2016, 2016b. [DOI] [Google Scholar]
- Ariya P, Skov H, Grage M, and Goodsite M: Gaseous elemental mercury in the ambient atmosphere: review of the application of theoretical calculations and experimental studies for determination of reaction coefficients and mechanisms with halogens and other reactants, Adv. Quantum Chem, 55, 43–55, doi: 10.1016/S0065-3276(07)00204-3, 2008. [DOI] [Google Scholar]
- Artaxo P, de Campos R, Fernandes E, Martins J, Xiao Z, Lindqvist O, Fernàndez-Jimènez M, and Maenhaut W: Large scale mercury and trace element measurements in the Amazon basin, Atmos. Environ, 34, 4085–4096, 2000. [Google Scholar]
- Artaxo P, de Brito JF, Barbosa H, Morais F, Sprovieri F, and Bencardino M: Mercury concentrations in Central Amazon Basin, available at: http://www.gmos.eu/public/artaxo_poster_2015.pdf (last access: 4 September 2016), 2015.
- Brooks S, Saiz-Lopez A, Skov H, Lindberg S, Plane J, and Goodsite M: The mass balance of mercury in the springtime arctic environment, J. Environ. Monitor, 33, L13812, doi: 10.1029/2005GL025525, 2006. [DOI] [Google Scholar]
- Brown R, Pirrone N, van Hoek C, Horvat M, Kotnik J, Wängberg I, Corns W, Bieber E, and Sprovieri F: Standardization of a European measurement method for the determination of total gaseous mercury: results of the field trial campaign and determination of a measurement uncertainty and working range, Accredit. Qual. Assur, 15, 359–366, doi: 10.1007/s00769-010-0636-2, 2010a. [DOI] [PubMed] [Google Scholar]
- Brown R, Pirrone N, van Hoek C, Sprovieri F, Fernandez R, and Toté K: Standardisation of a European measurement method for the determination of mercury in deposition: results of the field trial campaign and determination of a measurement, J. Environ. Monitor, 12, 689–695, doi: 10.1039/b924955a, 2010b. [DOI] [PubMed] [Google Scholar]
- Brunke E-G, Labuschagne C, Parker B, Scheel H, and Whittlestone S: Baseline air mass selection at Cape Point, South Africa: application of 222Rn and other filter criteria to CO2, Atmos. Environ, 38, 5693–5702, doi: 10.1016/j.atmosenv.2004.04.024, 2004. [DOI] [Google Scholar]
- Brunke E-G, Ebinghaus R, Kock HH, Labuschagne C, and Slemr F: Emissions of mercury in southern Africa derived from long-term observations at Cape Point, South Africa, Atmos. Chem. Phys, 12, 7465–7474, doi: 10.5194/acp-12-7465-2012, 2012. [DOI] [Google Scholar]
- Carpenter LJ: Seasonal characteristics of tropical marine boundary layer air measured at the Cape Verde Atmospheric Observatory, J. Atmos. Chem, 67, 87–140, doi: 10.1007/s10874-011-9206-1, 2011. [DOI] [Google Scholar]
- Cinnirella S, D’Amore F, Bencardino M, Sprovieri F, and Pir-rone N: The GMOS cyber(e)-infrastructure: advanced services for supporting science and policy, Environ. Sci. Pollut. R, 21, 4193–4208, doi: 10.1007/s11356-013-2308-3, 2014. [DOI] [PubMed] [Google Scholar]
- Daga R, Guevara S, Poire D, and Arribére M: Characterization of tephras dispersed by the recent eruptions of volcanoes Calbuco (1961), Chaitén (2008) and Cordón Caulle Complex (1960 and 2011), in Northern Patagonia, J. S. Am. Earth Sci, 49, 1–14, doi: 10.1016/j.jsames.2013.10.006, 2014. [DOI] [Google Scholar]
- D’Amore F, Bencardino M, Cinnirella S, Sprovieri F, and Pirrone N: Data quality through a web-based QA/QC system: implementation for atmospheric mercury data from the Global Mercury Observation System, Environmental Sciences: Processes and Impacts, 17, 1482–1492, doi: 10.1039/C5EM00205B, 2015. [DOI] [PubMed] [Google Scholar]
- Dastoor A and Durnford D: Arctic Ocean: Is it a sink or a source of atmospheric mercury?, Environ. Sci. Technol, 48,1707-1717, 2014. [DOI] [PubMed] [Google Scholar]
- Dastoor A, Davignon D, Theys N, Roozendael MV, Steffen A, and Ariya P: Modeling dynamic exchange of gaseous elemental mercury at polar sunrise, Environ. Sci. Technol, 42, 1482–1492, doi: 10.1021/es800291w, 2008. [DOI] [PubMed] [Google Scholar]
- de la Rosa D, Volke-Sepulveda T, Solarzano G, Green C, Tordon R, and Beauchamp S: Survey of atmospheric total gaseous mercury in Mexico, Atmos. Environ, 38, 4839–4846, 2004. [Google Scholar]
- De Simone F, Cinnirella S, Gencarelli CN, Yang X, Hedgecock IM, and Pirrone N: Model Study of Global Mercury Deposition from Biomass Burning, Environ. Sci. Technol, 49, 6712–6721, 2015. [DOI] [PubMed] [Google Scholar]
- De Simone F, Cinnirella S, Gencarelli CN, Carbone F, Hedgecock IM, and Pirrone N: Particulate-Phase Mercury Emissions during Biomass Burning and Impact on Resulting Deposition: a Modelling Assessment, Atmos. Chem. Phys. Discuss, doi: 10.5194/acp-2016-685, in review, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diéguez M, García P, and Sprovieri F: Atmospheric mercury fluxes in North Patagonia: first continuous records of the EMMA station (Global Mercury Observation System, Bariloche, Argentina), in: proceedings ICMGP, 14-19 June 2015, Jeju, Republic of Korea, 2015. [Google Scholar]
- Dommergue A, Larose C, Fain X, Clarisse O, Foucher D, Hintelmann H, Schneider D, and Ferrari C: Deposition of mercury species in the Ny-Alesund area (79° N) and their transfer during snowmelt, Environ. Sci. Technol, 44, 901–907, doi: 10.1021/es902579m, 2010b, 2010a. [DOI] [PubMed] [Google Scholar]
- Dommergue A, Sprovieri F, Pirrone N, Ebinghaus R, Brooks S,Courteaud J, and Ferrari CP: Overview of mercury measurements in the Antarctic troposphere, Atmos. Chem. Phys, 10, 3309–3319, doi: 10.5194/acp-10-3309-2010, 2010b. [DOI] [Google Scholar]
- Douglas T, Loseto L, MacDonald R, Outridge P, Dommergue A, and Poulain A: The fate of mercury in Arctic terrestrial and aquatic ecosystems, a review, Environ. Chem, 9, 321–355, doi: 10.1071/en11140, 2012. [DOI] [Google Scholar]
- do Valle C, Santana G, Augusti R, EgrejaFilho F, and Windmoller C: Speciation and quantification of mercury in Oxisol, Ultisol, and Spodosol from Amazon (Manaus, Brazil), Chemo-sphere, 58, 779–792, 2005. [DOI] [PubMed] [Google Scholar]
- Ebinghaus R, Kock HH, Coggins AM, Spain TG, Jennings SG, and Temme C: Long-term measurements of atmospheric mercury at Mace Head, Irish west coast, between 1995 and 2001, Atmos. Environ, 36, 5267–5276, 2002. [Google Scholar]
- Ebinghaus R, Jennings S, Kock H, Derwent R, Manning A, and Spain T: Decreasing trends in total gaseous mercury observations in baseline air at Mace Head, Ireland from 1996 to 2009, Atmos. Environ, 45, 3475–3480, 2011. [Google Scholar]
- Feng X, Shang L, Wang S, Tang S, and Zheng W: Temporal variation of total gaseous mercury in the air of Guiyang, China, J. Geophys. Res.-Atmos, 109, D03303, doi: 10.1029/2003JD004159, 2004. [DOI] [Google Scholar]
- Fisher JA, Jacob DJ, Soerensen AL, Amos HM, Steffen A, and Sunderland EM: Riverine source of Arctic Ocean mercury inferred from atmospheric observations, Nat. Geosci, 5, 499–504, doi: 10.1038/NGEO1478, 2012. [DOI] [Google Scholar]
- Fu X, Feng X, Zhu W, Wang S, and Lu J: Total gaseous mercury concentrations in ambient air in the eastern slope of Mt. Gongga, South-Eastern fringe of the Tibetan plateau, China, Atmos. Environ, 42, 970–979, 2008a. [Google Scholar]
- Fu X, Feng X, Zhu W, Zheng W, Wang S, and Lu JY: Total particulate and reactive gaseous mercury in ambient air on the eastern slope of the Mt. Gongga area, China, Appl. Geochem, 23, 408–418, 2008b. [Google Scholar]
- Fu XW, Feng X, Dong ZQ, Yin RS, Wang JX, Yang ZR, and Zhang H: Atmospheric gaseous elemental mercury (GEM) concentrations and mercury depositions at a high-altitude mountain peak in south China, Atmos. Chem. Phys, 10, 2425–2437, doi: 10.5194/acp-10-2425-2010, 2010. [DOI] [Google Scholar]
- Fu XW, Feng X, Liang P, Deliger, Zhang H, Ji J, and Liu P: Temporal trend and sources of speciated atmospheric mercury at Waliguan GAW station, Northwestern China, Atmos. Chem. Phys, 12, 1951–1964, doi: 10.5194/acp-12-1951-2012, 2012a. [DOI] [Google Scholar]
- Fu XW, Feng X, Shang LH, Wang SF, and Zhang H: Two years of measurements of atmospheric total gaseous mercury (TGM) at a remote site in Mt. Changbai area, Northeastern China, Atmos. Chem. Phys, 12, 4215–4226, doi: 10.5194/acp-12-4215-2012, 2012b. [DOI] [Google Scholar]
- Fu XW, Zhang H, Yu B, Wang X, Lin C-J, and Feng XB: Observations of atmospheric mercury in China: a critical review, Atmos. Chem. Phys, 15, 9455–9476, doi: 10.5194/acp-15-9455-2015, 2015. [DOI] [Google Scholar]
- Gay DA, Schmeltz D, Prestbo E, Olson M, Sharac T, and Tordon R: The Atmospheric Mercury Network: measurement and initial examination of an ongoing atmospheric mercury record across North America, Atmos. Chem. Phys, 13, 11339–11349, doi: 10.5194/acp-13-11339-2013, 2013. [DOI] [Google Scholar]
- Gencarelli C, Simone FD, Hedgecock I, Sprovieri F, Yang X, and Pirrone N: European and Mediterranean mercury modelling:Local and long-range contributions to the deposition flux, Atmos. Environ, 117, 162–168, doi: 10.1016/j.atmosenv.2015.07.015, 2015. [DOI] [Google Scholar]
- Gencarelli CN, Bieser J, Carbone F, De Simone F, Hedgecock IM, Matthias V, Travnikov O, Yang X, and Pirrone N: Sensitivity model study of regional mercury dispersion in the atmosphere, Atmos. Chem. Phys. Discuss, doi: 10.5194/acp-2016-663, in review, 2016. [DOI] [Google Scholar]
- GMOS Database: Land-based Monitoring Sites, available at: http:// sdi.iia.cnr.it/geoint/publicpage/GMOS/gmos_historical.zul, last access: 28 December 2014.
- Goodsite M, Plane J, and Skov H: A theoretical study of the oxidation of Hg0 to HgBr2 in the troposphere, Environ. Sci. Technol, 38, 1772–1776, doi: 10.1021/es034680s, 2004. [DOI] [PubMed] [Google Scholar]
- Goodsite M, Plane J, and Skov H: Correction to a theoretical study of the oxidation of Hg0 to HgBr2 in the troposphere, Environ. Sci. Technol, 46, 5262–5262, doi: 10.1021/es301201c, 2012. [DOI] [PubMed] [Google Scholar]
- Gustin MS, Amos HM, Huang J, Miller MB, and Heidecorn K: Measuring and modeling mercury in the atmosphere: a critical review, Atmos. Chem. Phys, 15, 5697–5713, doi: 10.5194/acp-15-5697-2015, 2015. [DOI] [Google Scholar]
- Hedgecock I and Pirrone N: Modelling chemical and physical processes of Hg compounds in the marine boundary layer, in: Dynamics of Mercury Pollution on Regional and Global Scales, edited by: Pirrone N and Mahaffey K, Springer Verlag Publishers, Norwell, USA, 2005. [Google Scholar]
- Hedgecock IM and Pirrone N: Chasing Quicksilver: Modeling the Atmospheric Lifetime of Hg0(g) in the Marine Boundary Layer at Various Latitudes, Environ. Sci. Technol, 38, 69–76, 2004. [DOI] [PubMed] [Google Scholar]
- Hedgecock IM, Pirrone N, Trunfio GA, and Sprovieri F: Integrated mercury cycling, transport, and air-water exchange (MECAWEx) model, J. Geophys. Res.-Atmos, 111, D20302, doi: 10.1029/2006JD007117, 2006. [DOI] [Google Scholar]
- Hedgecock IM, Pirrone N, and Sprovieri F: Chasing quicksilver northward: mercury chemistry in the Arctic troposphere, Environ. Chem, 5, 131–134, 2008. [Google Scholar]
- Higueras P, Oyarzun R, Kotnik J, Esbrí J, Martínez-Coronado A, Horvat M, López-Berdonces M, Llanos W, Vaselli O, Nisi B, Mashyanov N, Ryzov V, Spiric Z, Panichev N, McCrindle R, Feng X, Fu X, Lillo J, Loredo J, García M, Alfonso P, Villegas K, Palacios S, Oyarzún J, Maturana H, Contreras F, Adams M, Ribeiro-Guevara S, Niecenski L, Giammanco S, and Huremović J: A compilation of field surveys on gaseous elemental mercury (GEM) from contrasting environmental settings in Europe, South America, South Africa and China: separating fads from facts, Environ. Geochem. Health, 36, 713–734, doi: 10.1007/s10653-013-9591-2, 2014. [DOI] [PubMed] [Google Scholar]
- Holmes CD, Jacob DJ, Mason RP, and Jaffe DA: Sources and deposition of reactive gaseous mercury in the marine atmosphere, Atmos. Environ, 43, 2278–2285, 2009. [Google Scholar]
- Hynes AJ, Donohoue DL, Goodsite ME, and Hedgecock IM: Our current understanding of major chemical and physical processes affecting mercury dynamics in the atmosphere and at the air-water/terrestrial interfaces, in: Mercury Fate and Transport in the Global Atmosphere: Emissions, Measurements and Models, edited by: Pirrone N and Mason RP, chap. 14, 427–457, Springer, New York, USA, 2009. [Google Scholar]
- Karunasagar D, Krishna MB, Anjaneyalu Y, and Arunachalam J: Studies of mercury pollution in a lake due to a thermometer factory situated in a tourist resort; Kodaikanal, India, Environ. Pollut, 143, 153–158, 2006. [DOI] [PubMed] [Google Scholar]
- Kock H, Bieber E, Ebinghaus R, Spain T, and Thees B: Comparison of long-term trends and seasonal variations of atmospheric mercury concentrations at the two European coastal monitoring stations Mace Head, Ireland, and Zingst, Germany, Atmos. Environ, 39, 7549–7556, 2005. [Google Scholar]
- Krishnamoorthya K and Mathewb T: Inferences on the means of lognormal distributions using generalized p-values and generalized confidence intervals, J. Stat. Plan. Infer, 115, 103–121, doi: 10.1016/S0378-3758(02)00153-2, 2003. [DOI] [Google Scholar]
- Kurokawa J, Ohara T, Morikawa T, Hanayama S, Janssens-Maenhout G, Fukui T, Kawashima K, and Akimoto H: Emissions of air pollutants and greenhouse gases over Asian regions during 2000-2008: Regional Emission inventory in Asia (REAS) version 2, Atmos. Chem. Phys, 13, 11019–11058, doi: 10.5194/acp-13-11019-2013, 2013. [DOI] [Google Scholar]
- Lacerda L, de Souzab M, and Ribeiro M: The effects of land use change on mercury distribution in soils of Alta Floresta, Southern Amazon, Environ. Pollut, 129, 247–255, 2004. [DOI] [PubMed] [Google Scholar]
- Landis M, Stevens R, Schaedlich F, and Prestbo E: Development and characterization of an annular denuder methodology for the measurement of divalent inorganic reactive gaseous mercury in ambient air, Environ. Sci. Technol, 36, 3000–3009, doi: 10.1021/es015887t, 2002. [DOI] [PubMed] [Google Scholar]
- Lin C-J, Pan L, Streets DG, Shetty SK, Jang C, Feng X, Chu H-W, and Ho TC: Estimating mercury emission outflow from East Asia using CMAQ-Hg, Atmos. Chem. Phys, 10, 1853–1864, doi: 10.5194/acp-10-1853-2010, 2010. [DOI] [Google Scholar]
- Lindberg S and Stratton W: Atmospheric mercury speciation: concentrations and behavior of reactive gaseous mercury in ambient air, Environ. Sci. Technol, 32, 49–57, doi: 10.1021/es970546u, 1998. [DOI] [PubMed] [Google Scholar]
- Lindberg S, Brooks S, Lin C, Scott K, Landis M, Stevens R, and Goodsite M: Dynamic oxidation of gaseous mercury in the arctic troposphere at polar sunrise, Environ. Sci. Technol, 36, 1245–1256, 2002. [DOI] [PubMed] [Google Scholar]
- Lindberg S, Bullock R, Ebinghaus R, Engstrom D, Feng X, Fitzgerald W, Pirrone N, Prestbo E, and Seigneur C: A synthesis of progress and uncertainties in attributing the sources of mercury in deposition, Environ. Sci. Technol, 36, 19–33, doi: 10.1579/0044-7447(2007)36[19:ASOPAU]2.0.CO;2, 2007. [DOI] [PubMed] [Google Scholar]
- Mason RP: Mercury emissions from natural processes and their importance in the global mercury cycle, in: Mercury Fate and Transport in the Global Atmosphere: Emissions, Measurements and Models, edited by: Pirrone N and Mason RP, chap. 7, 173–191, Springer, New York, USA, 2009. [Google Scholar]
- Mendes NLS: Total Gaseous Mercury (TGM) measurements at the Cape Verde Atmospheric Observatory (CVAO), Tech. rep., University of York, Chemistry (York), available at: http://etheses.whiterose.ac.uk/6616/ (last access: 15 June 2016), 2014.
- Mohan M, Deepa M, Ramasamy E, and Thomas A: Accumulation of mercury and other heavy metals in edible fishes of Cochin backwaters, Southwest India, Environ. Monit. Assess, 184, 4233–4245, doi: 10.1007/s10661-011-2258-5, 2012. [DOI] [PubMed] [Google Scholar]
- Mukherjee A, Bhattacharya P, Sarkar A, and Zevenhoven R: Mercury emissions from industrial sources in India, in: Mercury Fate and Transport in the Global Atmosphere: Measurements, models and policy implications, edited by: Pirrone N and Mason R, UNEP, available at: http://www.unep.org/chemicalsandwaste/Portals/9/Mercury/Documents/Full_Report.pdf (last access: 10 April 2015), 2008. [Google Scholar]
- Mukherjee AB, Bhattacharya P, Sarkar A, and Zevenhoven R: Mercury Emissions from Industrial Sources in India and its Effects in the Environment, in: Mercury Fate and Transport in the Global Atmosphere: Emissions, Measurements, and Models, edited by: Pirrone N and Mason R, 3–49, Springer, New York, USA, 2009. [Google Scholar]
- Müller D, Wip D, Warneke T, Holmes CD, Dastoor A, and Notholt J: Sources of atmospheric mercury in the tropics: continuous observations at a coastal site in Suriname, Atmos. Chem. Phys, 12, 7391–7397, doi: 10.5194/acp-12-7391-2012, 2012. [DOI] [Google Scholar]
- Munthe J, Wängberg I, Pirrone N, Iverfeldt A, Ferrara R, Ebinghaus R, Feng X, Gardfeldt K, Keeler G, Lanzillotta E, Lindberg SE, Lu J, Mamane Y, Prestbo E, Schmolke S,Schroeder WH, Sommar J, Sprovieri F, Stevens RK, Stratton W, Tuncel G, and Urba A: Intercomparison of methods for sampling and analysis of atmospheric mercury species, Atmos. Environ, 35, 3007–3017, 2001. [Google Scholar]
- Munthe J, Wängberg I, Iverfeldt A, Lindqvist O, Stomberg D, Sommar J, Gårdfeldt K, Petersen G, Ebinghaus R, Prestbo E, Larjava K, and Siemens V: Distribution of atmospheric mercury species in Northern Europe: final results from the MOE project, Atmos. Environ, 37, 9–20, 2003. [Google Scholar]
- Munthe J, Sprovieri E, Horvat M, and Ebinghaus R: SOPs and QA/QC protocols regarding measurements of TGM, GEM, RGM, TPM and mercury in precipitation in cooperation with WP3, WP4 and WP5, GMOS deliverable 6.1, CNR-IIA, IVL, available at: http://www.gmos.eu (last access: 10 October 2015), 2011.
- Nguyen QT, Skov H, Sørensen LL, Jensen BJ, Grube AG, Massling A, Glasius M, and Nøjgaard JK: Source apportionment of particles at Station Nord, North East Greenland during 2008-2010 using COPREM and PMF analysis, Atmos. Chem. Phys, 13, 35–49, doi: 10.5194/acp-13-35-2013, 2013. [DOI] [Google Scholar]
- Pacyna E, Pacyna J, Sundseth K, Munthe J, Kindbom K, Wilson S, Steenhuisen F, and Maxson P: Global emission of mercury to the atmosphere from anthropogenic sources in 2005 and projections to 2020, Atmos. Environ, 44, 2487–2499, doi: 10.1016/j.atmosenv.2009.06.009, 2010. [DOI] [Google Scholar]
- Penney K and Cronshaw I: Coal in India 2015, Tech. rep., Australian Government - Department of Industry and Science, availalbe at: http://www.industry.gov.au/Office-of-the-Chief-Economist/Publications/Documents/Coal-in-India.pdf (last access: 10 May 2016), 2015.
- Pfaffhuber KA, Berg T, Hirdman D, and Stohl A: Atmospheric mercury observations from Antarctica: seasonal variation and source and sink region calculations, Atmos. Chem. Phys, 12, 3241–3251, doi: 10.5194/acp-12-3241-2012, 2012. [DOI] [Google Scholar]
- Pirrone N: GMOS DATA POLICY, available at: http://www.gmos.eu/public/GMOS-Governance_Data_Policy_rev160705.pdf (last access: 22 May 2015), 2012.
- Pirrone N: Atmospheric mercury at High Altitude sites: transport patterns and emission source regions, Special Issue: Global Mercury Observation System - Atmosphere (GMOS-A), in preparation, 2016.
- Pirrone N, Ferrara R, Hedgecock I, Kallos G, Mamane Y, Munthe J, Pacyna J, Pytharoulis I, Sprovieri F, Voudouri A, and Wängberg I: Dynamic Processes of Mercury Over the Mediterranean Region: results from the Mediterranean Atmospheric Mercury Cycle System (MAMCS) project, Atmos. Environ, 37, 21–39, doi: 10.1016/S1352-2310(03)00251-6, 2003. [DOI] [Google Scholar]
- Pirrone N, Hedgecock I, and Sprovieri F: Atmospheric mercury, easy to spot and hard to pin down: impasse?, Atmos. Environ, 42, 8549–8551, doi: 10.1016/j.atmosenv.2008.09.004, 2008. [DOI] [Google Scholar]
- Pirrone N, Cinnirella S, Feng X, Finkelman R, Friedli H, Leaner J, Mason R, Mukherjee A, Stracher G, Streets D, and Telmer K: Global mercury emissions to the atmosphere from natural and anthropogenic sources, in: Mercury Fate and Transport in the Global Atmosphere: Emissions, Measurements, and Models, edited by: Pirrone N and Mason R, 3–49, Springer, New York, USA, 2009. [Google Scholar]
- Pirrone N, Cinnirella S, Feng X, Finkelman RB, Friedli HR, Leaner J, Mason R, Mukherjee AB, Stracher GB, Streets DG, and Telmer K: Global mercury emissions to the atmosphere from anthropogenic and natural sources, Atmos. Chem. Phys, 10, 5951–5964, doi: 10.5194/acp-10-5951-2010, 2010. [DOI] [Google Scholar]
- Pirrone N, Aas W, Cinnirella S, Ebinghaus R, Hedgecock I, Pacyna J, Sprovieri F, and Sunderland E: Toward the next generation of air quality monitoring: Mercury, Atmos. Environ, 80, 599–611, doi: 10.1016/j.atmosenv.2013.06.053, 2013. [DOI] [Google Scholar]
- Poulain A, Lalonde J, Amyot M, Shead J, Raofie F, and Ariya P: Redox transformations of mercury in an Arctic snowpack at springtime, Atmos. Environ, 38, 6763–6774, doi: 10.1016/j.atmosenv.2004.09.013, 2004. [DOI] [Google Scholar]
- Rajgopal T and Mascarenhas HRP: Epidemiologicalsurveillance of employees in a mercury thermometer plant: An occupational health study, Indian J. Occup. Environ. Med, 10, 11–18, 2006. [Google Scholar]
- Selin NE, Jacob DJ, Yantosca RM, Strode S, Jaeglé L, and Sunderland EM: Global 3-D land-ocean-atmosphere model for mercury: Present-day versus preindustrial cycles and anthropogenic enrichment factors for deposition, Global Biogeochem. Cy., 22, GB2011, doi: 10.1029/2007GB003040, 2008. [DOI] [Google Scholar]
- Sena F, Umlauf G, Ruiz AA, Islas MR, Trejo JAV, Cabrera FA, and Vargas IO: Wet deposition and atmospheric mercury monitoring in Sisal, Yucatán, México, as part of the Global Mercury Observation System (GMOS), Tech. rep., JRC - EUROPEAN COMMISION, doi: 10.2788/823558, 2015. [DOI] [Google Scholar]
- Sheu G, Lin N, Lee C, Wang J, Chuang M, Wang S,Chi K, and Ou-Yang C: Distribution of atmospheric mercury in northern Southeast Asia and South China Sea during Dongsha Experiment, Atmos. Environ, 78, 174–183, doi: 10.1016/j.atmosenv.2012.07.002, 2013. [DOI] [Google Scholar]
- Sholupov S and Ganeyev A: Zeeman atomic absorption spectrometry using high frequency modulated light polarization, Spectrochim. Acta B, 50, 1227–1236, doi: 10.1016/0584-8547(95)01316-7, 1995. [DOI] [Google Scholar]
- Sholupov S, Pogarev S, Ryzhov V, Mashyanov N, and Stroganov A: Zeeman atomic absorption spectrometer RA915+ for direct determination of mercury in air and complex matrix samples, Fuel Process. Technol, 85, 473–485, doi: 10.1016/j.fuproc.2003.11.003, 2004. [DOI] [Google Scholar]
- Skov H, Christensen JH, Goodsite ME, Heidam NZ, Jensen B, Wåhlin P, and Geernaert G: Fate of Elemental Mercury in the Arctic during Atmospheric Mercury Depletion Episodes and the Load of Atmospheric Mercury to the Arctic, Environ. Sci. Technol, 38, 2373–2382, 2004. [DOI] [PubMed] [Google Scholar]
- Skov H, Goodsite M, Lindberg S, Meyers T, Landis M, Larsen M, and McConville G: The fluxes of Reactive Gaseous mercury measured with a newly developed method using relaxed eddy accumulation, Atmos. Environ, 40, 5452–5463, doi: 10.1016/j.atmosenv.2006.04.061, 2006. [DOI] [Google Scholar]
- Slemr F, Brunke E-G, Ebinghaus R, and Kuss J: Worldwide trend of atmospheric mercury since 1995, Atmos. Chem. Phys, 11, 4779–4787, doi: 10.5194/acp-11-4779-2011, 2011. [DOI] [Google Scholar]
- Slemr F, Angot H, Dommergue A, Magand O, Barret M, Weigelt A, Ebinghaus R, Brunke E-G, Pfaffhuber KA, Edwards G, Howard D, Powell J, Keywood M, and Wang F: Comparison of mercury concentrations measured at several sites in the Southern Hemisphere, Atmos. Chem. Phys, 15, 3125–3133, doi: 10.5194/acp-15-3125-2015, 2015. [DOI] [Google Scholar]
- Soerensen A, Skov H, Soerensen DJB, and Johnson M: Global concentrations of gaseous elemental mercury and reactive gaseous mercury in the marine boundary layer, Environ. Sci. Technol, 44, 7425–7430, doi: 10.1021/es903839n, 2010a. [DOI] [PubMed] [Google Scholar]
- Soerensen A, Sunderland E, Holmes C, Jacob D, Yantosca R, Skov H, Christensen J, Strode S, and Mason R: An improved global model for air-sea exchange of mercury: high concentrations over the North Atlantic, Environ. Sci. Technol, 44, 8574–8580, doi: 10.1021/es102032g, 2010b. [DOI] [PubMed] [Google Scholar]
- Sommar J, Andersson ME, and Jacobi H-W: Circumpolar measurements of speciated mercury, ozone and carbon monoxide in the boundary layer of the Arctic Ocean, Atmos. Chem. Phys, 10, 5031–5045, doi: 10.5194/acp-10-5031-2010, 2010. [DOI] [Google Scholar]
- Sprovieri F, Pirrone N, Hedgecock I, Landis M, and Stevens R: Intensive atmospheric mercury measurements at Terra Nova Bay in antarctica during November and December 2000, J. Geophys. Res, 107, ACH 20-1-ACH 20-8, doi: 10.1029/2002JD002057, 2002. [DOI] [Google Scholar]
- Sprovieri F, Pirrone N, Landis M, and Stevens R: Oxidation of gaseous elemental mercury to gaseous divalent mercury during 2003 polar sunrise at Ny-Alesund, Environ. Sci. Technol, 39, 9156–9165, doi: 10.1021/es050965o, 2005a. [DOI] [PubMed] [Google Scholar]
- Sprovieri F, Pirrone N, Landis M, and Stevens R: Atmospheric mercury behaviour at different altitudes at NyAlesund during Spring 2003, Atmos. Environ, 39, 7646–7656, doi: 10.1016/j.atmosenv.2005.08.001, 2005b. [DOI] [Google Scholar]
- Sprovieri F, Hedgecock IM, and Pirrone N: An investigation of the origins of reactive gaseous mercury in the Mediterranean marine boundary layer, Atmos. Chem. Phys, 10, 3985–3997, doi: 10.5194/acp-10-3985-2010, 2010a. [DOI] [Google Scholar]
- Sprovieri F, Pirrone N, Ebinghaus R, Kock H, and Dommergue A: A review of worldwide atmospheric mercury measurements, Atmos. Chem. Phys, 10, 8245–8265, doi: 10.5194/acp-10-8245-2010, 2010b. [DOI] [Google Scholar]
- Sprovieri F, Gratz L, and Pirrone N: Development of a ground based atmospheric monitoring network for the Global Mercury Observation System (GMOS), in: E3S web Conference, doi: 10.1051/e3sconf/20130117007, 2013. [DOI] [Google Scholar]
- Sprovieri F, Pirrone N, Bencardino M, D’Amore F, Angot H, Barbante C, Brunke E-G, Arcega-Cabrera F, Cairns W, Comero S, Diéguez MDC, Dommergue A, Ebinghaus R, Feng XB, Fu X, Garcia PE, Gawlik BM, Hageström U, Hansson K, Horvat M, Kotnik J, Labuschagne C, Magand O, Martin L, Mashyanov N, Mkololo T, Munthe J, Obolkin V, Islas MR, Sena F, Somerset V, Spandow P, Vardè M, Walters C, Wängberg I, Weigelt A, Yang X, and Zhang H: Five-year records of Total Mercury Deposition flux at GMOS sites in the Northern and Southern Hemispheres, Atmos. Chem. Phys. Discuss, doi: 10.5194/acp-2016-517, in review, 2016. [DOI] [Google Scholar]
- Steffen A, Douglas T, Amyot M, Ariya P, Aspmo K, Berg T, Bottenheim J, Brooks S, Cobbett F, Dastoor A, Dommergue A, Ebinghaus R, Ferrari C, Gardfeldt K, Goodsite ME, Lean D, Poulain AJ, Scherz C, Skov H, Sommar J, and Temme C: A synthesis of atmospheric mercury depletion event chemistry in the atmosphere and snow, Atmos. Chem. Phys, 8, 1445–1482, doi: 10.5194/acp-8-1445-2008, 2008. [DOI] [Google Scholar]
- Steffen A, Scherz T, Olson M, Gay D, and Blanchard P: A comparison of data quality control protocols for atmospheric mercury speciation measurements, J. Environ. Monitor, 14, 752–765, 2012. [DOI] [PubMed] [Google Scholar]
- Streets DG, Hao J, Wu Y, Jiang J, Chan M, Tian H, and Feng X: Anthropogenic mercury emissions in China, Atmos. Environ, 39, 7789–7806, 2005. [Google Scholar]
- Streets DG, Devane MK, Lu Z, Bond TC, Sunderland EM, and Jacob DJ: All-Time Releases of Mercury to the Atmosphere from Human Activities, Environ. Sci. Technol, 45, 10485–10491, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subir M, Ariya PA, and Dastoor AP: A review of uncertainties in atmospheric modeling of mercury chemistry I. Uncertainties in existing kinetic parameters - Fundamental limitations and the importance of heterogeneous chemistry, Atmos. Environ, 45, 5664–5676, 2011. [Google Scholar]
- Subir M, Ariya PA, and Dastoor AP: A review of the sources of uncertainties in atmospheric mercury modeling II. Mercury surface and heterogeneous chemistry - A missing link, Atmos. Environ, 46, 1–10, 2012. [Google Scholar]
- Temme C, Einax JW, Ebinghaus R, and Schroeder WH: Measurements of Atmospheric Mercury Species at a Coastal Site in the Antarctic and over the South Atlantic Ocean during Polar Summer, Environ. Sci. Technol, 37, 22–31, 2003. [DOI] [PubMed] [Google Scholar]
- Temme C, Blanchard P, Steffen A, Banic C, Beauchamp S, Poissant L, Tordon R, and Wiens B: Trend, seasonal and multivariate analysis study of total gaseous mercury data from the Canadian atmospheric mercury measurement network (CAMNet), Atmos. Environ, 41, 5423–5441, 2007. [Google Scholar]
- UNEP: UNEP: Mercury fate and transport in the global atmosphere: Measurements, models and policy implications, Tech. rep., UNEP Global Mercury Partnership Mercury Air Transport and Fate Research partnership area, http://www.unep.org/gc/gc22/Document/UNEP-GC22-INF3.pdf (last access: 10 June 2016), 2008.
- van Donkelaar A, Martin R, Brauer M, Kahn R, Levy R, and Verduzco C: Global estimates of ambient fine particulate matter concentrations from satellite-based aerosol optical depth: development and application, Environ. Health Persp, 118, 847–855, doi: 10.1289/ehp.0901623., 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhang X, Chen Z, and Zhang Y: Mercury concentrations in size-fractionated airborne particles at urban and suburban sites in Beijing, Atmos. Environ, 40, 2194–2201, doi: 10.1016/j.atmosenv.2005.12.003, 2006. [DOI] [Google Scholar]
- Wang Z, Chen Z, Duan N, and Zhang X: Gaseous elemental mercury concentration in atmosphere at urban and remote sites in China, J. Environ. Sci, 19,176–180, doi: 10.1016/S1001-0742(07)60028-X, 2007. [DOI] [PubMed] [Google Scholar]
- Wängberg I, Munthe J, Pirrone N, Iverfeldt A, Bahlman E, Costa P, Ebinghaus R, Feng X, Ferrara R, Gardfeldt K, Kock H, Lanzillotta E, Mamane Y, Mas F, Melamed E, Osnat Y, Prestbo E, Sommar J, Schmolke S, Spain G, Sprovieri F, and Tuncel G: Atmospheric mercury distribution in Northern Europe and in the Mediterranean region, Atmos. Environ, 35, 3019–3025, 2001. [Google Scholar]
- Wängberg I, Munthe J, Amouroux D, Andersson M, Fajon V, Ferrara R, Gårdfeldt K, Horvat M, Mamane Y, Melamed E, Monperrus M, Ogrinc N, Yossef O, Pirrone N, Sommar J, and Sprovieri F: Atmospheric mercury at mediterranean coastal stations, Environ. Fluid Mech, 8, 101–116, 2008. [Google Scholar]
- Weigelt A, Ebinghaus R, Manning A, Derwent R, Simmonds P, Spain T, Jennings S, and Slemr F: Analysis and interpretation of 18 years of mercury observations since 1996 at Mace Head, Ireland, Atmos. Environ, 100, 85–93, doi: 10.1016/j.atmosenv.2014.10.050, 2015. [DOI] [Google Scholar]
- Weigelt A, Ebinghaus R, Pirrone N, Bieser J, Bödewadt J, Esposito G, Slemr F, van Velthoven PFJ, Zahn A, and Ziereis H: Tropospheric mercury vertical profiles between 500 and 10000min central Europe, Atmos. Chem. Phys, 16, 4135–4146, doi: 10.5194/acp-16-4135-2016, 2016. [DOI] [Google Scholar]
- Wu Y, Wang S, Streets DG, Hao J, Chan M, and Jiang J: Trends in Anthropogenic Mercury Emissions in China from 1995 to 2003, Environ. Sci. Technol, 40, 5312–5318, 2006. [DOI] [PubMed] [Google Scholar]
- Xiu G, Cai J, Zhang W, Zhang D, Bueler A, Lee S, Shen Y, Xu L, Huang X, and Zhang P: Speciated mercury in size-fractionated particles in Shanghai ambient air, Atmos. Environ, 43, 3145–3154, doi: 10.1016/j.atmosenv.2008.07.044, 2009. [DOI] [Google Scholar]
- Xu L, Chen J, Yang L, Niu Z, Tong L, Yin L, and Chen Y: Characteristics and sources of atmospheric mercury speciation in a coastal city, Xiamen, China, Chemosphere, 119, 530–539, doi: 10.1016/j.chemosphere.2014.07.024, 2015. [DOI] [PubMed] [Google Scholar]
- Zhang H, Fu X, Lin C-J, Shang L, Zhang Y, Feng X, and Lin C: Monsoon-facilitated characteristics and transport of atmospheric mercury at a high-altitude background site in southwestern China, Atmos. Chem. Phys. Discuss, doi: 10.5194/acp-2016-506, in review, 2016. [DOI] [Google Scholar]
- Zhang L, Wang SX, Wang L, and Hao JM: Atmospheric mercury concentration and chemical speciation at a rural site in Beijing, China: implications of mercury emission sources, Atmos. Chem. Phys, 13, 10505–10516, doi: 10.5194/acp-13-10505-2013, 2013. [DOI] [Google Scholar]
- Zhu J, Wang T, Talbot R, Mao H, Yang X, Fu C, Sun J, Zhuang B, Li S, Han Y, and Xie M: Characteristics of atmospheric mercury deposition and size-fractionated particulate mercury in urban Nanjing, China, Atmos. Chem. Phys, 14, 2233–2244, doi: 10.5194/acp-14-2233-2014, 2014. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mercury data discussed in this manuscript are reported within the GMOS central database and are available upon request at http://sdi.iia.cnr.it/geoint/publicpage/GMOS/gmos_historical.zul (GMOS Database, 2014). The GMOS database coordinated by the CNR-IIA is part of the GMOS CyberInfrastructure (CI), which supports advanced data acquisition, storage, management, integration, mining and visualization. All GMOS stations provide near-real-time raw data that are archived and managed for the QA/QC process by the GMOS-Data Quality Managment System (G-DQM), a web-based application which is part of the GMOS-CI, developed by the CNR-IIA in 2011, and is currently ongoing.