

Abstract
Fibro-Adipogenic Progenitors (FAPs) are typically activated in response to muscle injury, and establish functional interactions with inflammatory and muscle stem cells (MuSCs) to promote muscle repair. We found that denervation causes progressive accumulation of FAPs, without concomitant infiltration of macrophages and MuSC-mediated regeneration. Denervation-activated FAPs exhibited persistent STAT3 activation and secreted elevated levels of IL6, which promoted muscle atrophy and fibrosis. FAPs with aberrant activation of STAT3-IL6 signaling were also found in mouse models of spinal cord injury, Spinal Muscular Atrophy (SMA), Amyotrophic Lateral Sclerosis (ALS) and in muscles of ALS patients. Inactivation of STAT3-IL6 signaling in FAPs effectively countered muscle atrophy and fibrosis in mouse models of acute denervation and ALS (SODG93A mice). The activation of pathogenic FAPs upon loss of integrity of neuromuscular junctions further illustrates the functional versatility of FAPs in response to homeostatic perturbations and suggests their potential contribution to the pathogenesis of neuromuscular diseases.
Introduction
Skeletal muscle homeostasis is maintained by the equilibrium of physical and functional interactions between myofibers and different cell types that compose the muscle environment, including muscle (satellite) stem cells (MuSCs), motoneurons, and a variety of interstitial cells, such as vessel-associated cells, fibroblasts, PW1-expressing interstitial cells (PICs), and fibro-adipogenic progenitors (FAPs)1–10. Environmental perturbations trigger dynamic alterations in cell type composition and functional interactions between these cells, to activate a compensatory response toward restoring the original homeostasis. For instance, in response to acute injury, a timely coordinated appearance of the inflammatory infiltrate and expansion of interstitial cells instruct MuSCs to regenerate the damaged muscle5–7. However, in conditions of chronic injury loss of coordination between these cell types results in maladaptive fibrosis and ectopic fat deposition, as observed during progression of muscular dystrophies2,8.
Among the interstitial cells, FAPs are emerging as key effectors of compensatory or maladaptive repair of injured muscles9–11. In unperturbed skeletal muscles, FAPs are present in a steady state; however, they rapidly expand in response to muscle injury9,10. FAPs accumulation is preceded by the appearance of the inflammatory infiltrate and is followed by MuSC activation12–15. This temporal pattern suggests a key role for FAPs in converting inflammatory cues into pro-regenerative signals. FAP expansion and activation are also restricted within a precise window of time by signals derived from the inflammatory infiltrate14,15. Impaired FAP clearance leads to their abnormal accumulation and formation of fibrotic scars and fat deposition, which inhibit muscle regeneration16–18.
It is currently unknown whether environmental perturbations that do not activate an inflammatory response, such as muscle denervation, can stimulate FAP expansion and eventually promote functional properties different from those reported in injured muscles.
Results
Abnormal accumulation of FAPs during muscle atrophy by acute denervation
We compared the temporal kinetics of appearance of macrophages (MPs), FAPs and MuSCs in skeletal muscles subjected to either acute injury (by cardiotoxin) or denervation (by sciatic nerve transection), which trigger two opposite responses. A transient deposition of collagen (Fig. 1a-d; Suppl. Fig. 1a) and reduction of myofiber caliber (Fig. 1e) during the first 7 days post acute injury were typically ensued by a prompt resolution of fibrotic infiltration (Fig. 1a-d; Suppl. Fig. 1a) and recovery of the myofiber size (Fig. 1e). By contrast, muscle denervation induced progressive accumulation of fibrosis (Fig. 1a-d; Suppl. Fig. 1a) and reduction of myofiber cross-sectional area (CSA) (Fig. 1e). These opposite responses reflect the differential ability of injured muscles to regenerate, as compared to denervated muscles, with embryonal-Myosin Heavy Chain (eMyHC)-positive regenerating fibers observed in acutely injured muscles, but not in denervated muscles (Suppl. Fig. 1b). FACS (Fig. 1f; Suppl. Fig. 1c) and immunofluorescence (Suppl. Fig. 2a-f) analyses showed that while coordinated dynamics of MP, FAP and MuSC activation were observed in response to acute injury, a progressive accumulation of FAPs was detected in denervated muscles, in the absence of any significant increase in MP and MuSC number. Of note, we found higher EdU incorporation in CD90-positive interstitial cells (another FAP marker)10 from denervated muscles, as compared to unperturbed controls (Suppl. Fig. 1d). This evidence reveals that FAPs can be activated in response to different perturbations and within distinct cellular and functional contexts, and suggests the possibility that FAP’s activities could also differ depending on the type of perturbation.
Figure 1: Abnormal accumulation of FAPs during muscle atrophy by acute denervation.
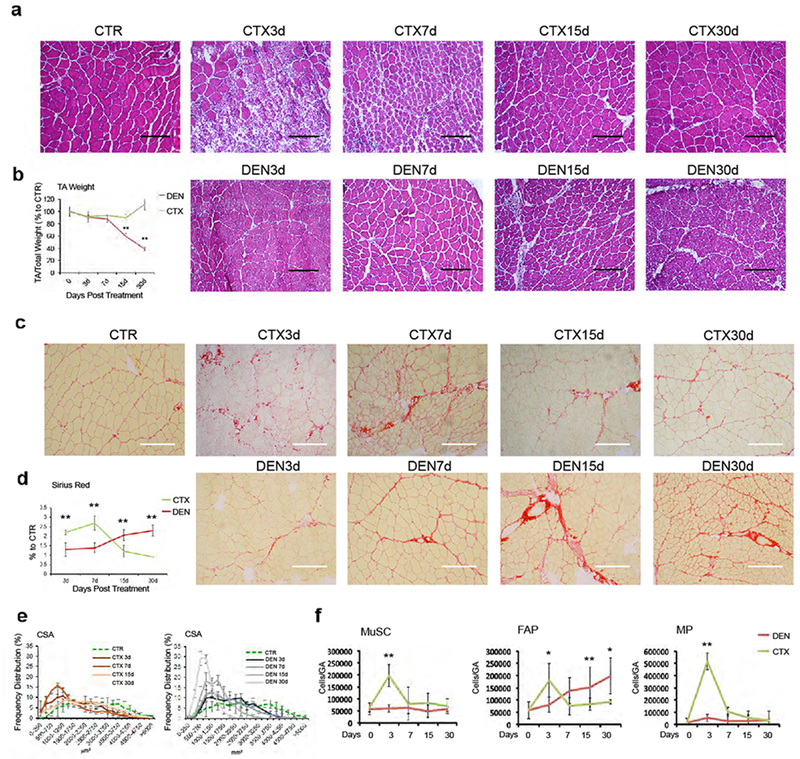
(a) Hematoxylin-Eosin staining of control (CTR), injured (CTX) or denervated (DEN) TA muscle cryosections at the indicated time points. Scale bar 200 μm. Data shown represent 3 independent experiments, (b) Quantification of normalized TA muscle weight. Values represent mean ± s.d. **P = 0.001; by one-way ANOVA (n=3 animals/group) (c) Representative images of Sirius red staining of control (CTR), injured (CTX) and denervated (DEN) TA muscle cryosection. Scale bar 200 μm. Data shown represent 3 independent experiments) (d) Quantification of collagen staining shown in c. Values represent mean ± s.d. **P < 0.01; by one-way ANOVA (n=3 animals/group), (e) Frequency distribution of fiber Cross-Sectional Area (CSA) in injured (CTX) (left panel) and denervated (DEN) (right panel) TA muscles. Values represent mean ± s.d. (n=3 animals/group), (f) Cyto-fluorimetric analysis of indicated cell types in injured (CTX) or denervated (DEN) muscles at the indicated time points. Values represent mean ± s.d. **P < 0.01, *P<0.05; by one-way ANOVA (n=12 CTR, n=20 DEN15d, n=6 DEN3d, CTX7d, n=4 CTX3d, CTX15d, n=3 DEN7d, DEN30d, CTX30d; “n” refers to animal numbers).
FAPs from denervated muscles exhibit distinct functional properties and gene expression profiles
We next investigated the functional differences between FAPs isolated from injured (FAP CTX) or denervated (FAP DEN) muscles. FAPs isolated from regenerating muscle post-injury typically enhance skeletal myoblast fusion into multinucleated myotubes9,10,16,18. We exploited the transwell co-culture system to determine whether FAPs from denervated muscles could exert similar or different effects on skeletal myoblasts. While FAPs isolated from regenerating muscles (at day 3 post-acute injury) could significantly increase the formation of multinucleated myotubes from co-cultured myoblasts, FAPs from denervated muscles (isolated at day 15 post denervation) showed much reduced ability to enhance myoblast fusion into myotubes (Fig. 2a and b). Since FAP activation upon denervation coincides with the progressive muscle atrophy caused by loss of integrity of neuromuscular junctions (NMJs), we evaluated the potential contribution of FAP DEN to muscle atrophy, by measuring the changes in caliber of pre-formed myotubes upon transwell co-culture with FAP DEN, as compared to FAP CTX or controls (i.e. myotubes co-cultured with FAPs from unperturbed muscles - FAP NT - or without any co-cultured cells). Only co-culture with FAP DEN caused a drastic reduction of myotubes caliber (Fig. 2c, d).
Figure 2: FAPs from denervated muscles exhibit distinct functional properties and gene expression profiles.
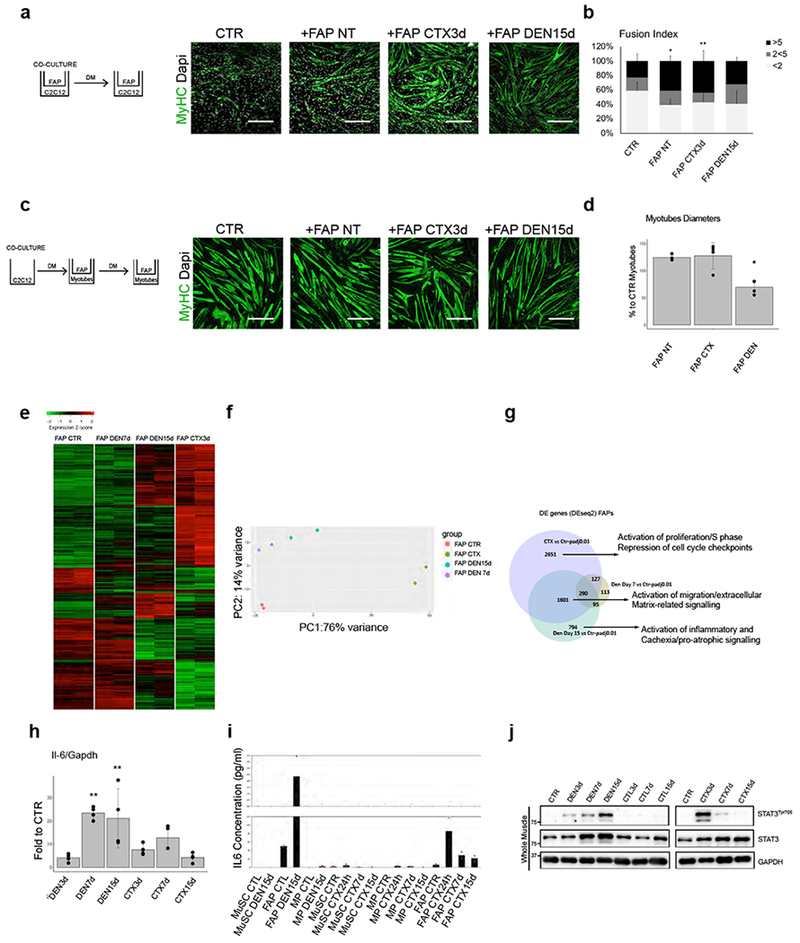
(a) Experimental setting and MyHC (Green) and DAPI (White) immunofluorescence of C2C12 myoblasts cultured alone (CTR) or in co-culture with FAP NT, FAP CTX3d or FAP DEN15d. Scale bar 200 μm. Data shown represent 3 biological independent experiments. (b) Fusion index of C2C12 myoblasts as described in a. “<2”: less than 2 nuclei in MyHC− or in MyHC+ mononucleated cells; “2<5”: MyHC+ myotubes with 2 to 5 nuclei; “> 5”: MyHC+ myotubes with than 5 nuclei. Values represent mean ±s.d. *P=0.021 **P=0.007; by one-way ANOVA (n=6 biological independent experiments). (c) MyHC (Green) and DAPI (White) immunofluorescence of myotubes cultured for 72h alone (CTR) or co-cultured with FAP NT, FAP CTX3d and FAP DEN15d. Scale bar 200 μm. Data shown represent 3 independent experiments. (d) Quantification of C2C12 myotube diameter. Values represent mean ±s.d., *P<0.05; by one-way ANOVA (n=4 independent experiments. CTR, FAP NT, FAP DEN; n=3 FAP CTX. (e) Heatmap of DE genes in FAPs DEN7d and DEN15d, or FAPs CTX3d, as compared to FAPs NT. Gene expression is represented as z-score calculated across the rows. Data shown are from two RNA-seq experiments. (f) PCA from RNA-seq in FAPs NT, FAP CTX3d and FAP DEN7 and DEN 15). Data from two independent RNA-seq experiments. (g) Venn diagram of genes common or uniquely in FAPs CTX3d and FAPs DEN7d and DEN15d, compared with FAPs NT. Statistical method was Deseq2. (h) qPCR analysis of IL-6 transcripts in FAPs CTX or FAP DEN. Values represent mean ±s.d. *P=0.021 **P=0.075; by one-way ANOVA (n=4 biologically independent samples). (i) ELISA quantification of secreted IL-6 by MuSCs, FAPs and MPs from the described conditions. (n=2 biologically independent samples (j) Representative Immunoblot of phosphoSTAT3 in the described conditions out of 3 independent experiments. See Supplementary Table 1 for source data shown in panels b, I and d.
We next performed RNA-seq analysis of FAPs isolated from denervated muscles at two time-points post-denervation −7 days (FAP DEN7d) and 15 days (FAP DEN15d). These transcriptional profiles were compared to those from RNA-seq analysis of FAP NT and FAP CTX (3 days post- injury). While FAP CTX showed extensive patterns of gene up- and down-regulation, as compared to FAP NT, FAP DEN exhibited a significantly smaller amount of differentially expressed (DE) genes, with a trend toward progressive increase in both up- and down-regulated genes from day 7 to day 15 post-denervation (Suppl. Fig. 3a). Heatmap representation of RNA-seq analysis revealed clear differences in the gene expression profiles of FAPs from these conditions (Fig. 2e). Principal Component Analysis (PCA) further demonstrated that FAP DEN (at both time points) clustered in the opposite direction from FAP CTX, with both PC1 and PC2 discriminating them (Fig. 2f). Venn diagram representation of common and unique gene expression profiles revealed the presence of a large amount of condition-specific as well as commonly expressed genes (Fig. 2g and Suppl. Fig. 3b). Functional annotation of biological differences predicted by the Ingenuity Pathway Analysis (IPA) revealed that commonly expressed genes included the cell identity surface markers, Seal, PDGF-alpha receptor and CD909,10, which allow isolation of FAPs in both experimental conditions, but also genes implicated in migration and extra-cellular signaling and fibrosis (Fig. 2g and Suppl. Fig. 3b). Fibrosis is indeed a common outcome for both conditions, although it was transiently induced by CTX, but persisted upon denervation (Suppl. Fig. 1A). Among condition-specific DE genes, FAP CTX-specific gene expression was enriched in genes associated with cell proliferation, DNA synthesis, mitosis and cell cycle checkpoints, while FAP DEN-specific gene expression patterns were enriched in functional categories implicated in the activation of inflammatory processes that included potential soluble mediators of pro-atrophic and catabolic signaling (Fig. 2g and Suppl. Fig. 3b). Among them, the concurrent activation of STAT3 and IL6, which have been reported to be functionally interconnected19–22, stood out as the main activated pathway from both IPA (Fig. 2g and Suppl. Fig. 3b) and upstream regulator analysis (Table 1). Progressive and persistent activation of IL6 gene expression was indeed detected by qPCR in FAP DEN at various time points following denervation, at variance with FAP CTX, which exhibited transient and less robust up-regulation of IL6 transcripts at day 7 post acute injury (Fig. 2h). ELISA analysis of IL6 concentrations in the supernatant from cultures with FAPs, MuSCs and MPs revealed that FAP DEN are the most abundant source of IL6 secretion, among these intra-muscular cell populations (Fig. 2i). Consistent with a potential paracrine activity of FAP-derived IL6 in denervated muscles, we detected a progressive activation STAT3 in total lysates from denervated muscles (7 days post-sciatic nerve section), while only a transient STAT3 activation was detected in regenerating muscles at 3 days post-injury (Fig. 2j).
Table 1: Denervated FAPs upstream regulators predictions.
Upstream regulators (by IPA analysis) of uniquely altered genes in FAP DEN compared to FAPs CTR. For each regulator, significance is reported as the overlap p-value between each regulator known targets and the dataset (one-sided Fisher’s exact test; Kramer et al., 2014). (n=2 biologically independent samples)
| ©2000-2018 QIAGEN. All rights reserved. | |||
|---|---|---|---|
| Upstream Regulator | Molecule Type | Activation z-score | p-value of overlap |
| RICTOR | other | 5.895 | 4.36E-15 |
| ESR1 | ligand-dependent nuclear receptor | 3.486 | 2.31E-08 |
| GATA2 | transcription regulator | 3.464 | 0.514 |
| IL2 | cytokine | 3.135 | 0.00000574 |
| RELA | transcription regulator | 3.098 | 0.0115 |
| ADCYAP1 | other | 3.064 | 0.00817 |
| KDM5A | transcription regulator | 3.004 | 0.00000267 |
| IL1B | cytokine | 2.873 | 0.0000314 |
| SMARCA4 | transcription regulator | 2.799 | 0.0129 |
| SP1 | transcription regulator | 2.798 | 0.0000112 |
| IL6 | cytokine | 2.768 | 0.0325 |
| Cg | complex | 2.668 | 0.00000914 |
| PTH | other | 2.635 | 0.0424 |
| IRF8 | transcription regulator | 2.6 | 0.0981 |
| GCG | other | 2.588 | 0.0123 |
| SMARCB1 | transcription regulator | 2.58 | 0.234 |
| IL6R | transmembrane receptor | 2.564 | 0.0174 |
| Vegf | group | 2.562 | 0.00969 |
| MITF | transcription regulator | 2.556 | 0.202 |
| IL1A | cytokine | 2.555 | 0.0493 |
| FOXO3 | transcription regulator | 2.555 | 0.00145 |
| HGF | growth factor | 2.427 | 0.0000532 |
| GDF2 | growth factor | 2.425 | 0.089 |
| EGR2 | transcription regulator | 2.402 | 0.00027 |
| INHA | growth factor | 2.395 | 0.0102 |
| STAT3 | transcription regulator | 2.393 | 0.0185 |
| KITLG | growth factor | 2.378 | 0.0585 |
| TWIST1 | transcription regulator | 2.354 | 0.0011 |
Persistent activation of IL6-STAT3 pathway in FAPs and myofibers from denervated muscles and prevention of myofiber atrophy and fibrosis by IL6 blockade
Since muscles are composed of different cell types, we sought to further determine the identity of the cells in which STAT3 was activated, by their anatomical localization. Immunofluorescence of muscle sections from denervated (DEN) or unperturbed (CTR) mice showed that phospho-STAT3 signal was predominantly localized in nuclei of cells within the muscle interstitial space as well as in the myonuclei of muscle fibers (Fig. 3a). Western blot with protein extracts from FACS-isolated FAPs showed that STAT3 signaling was robustly activated in FAP DEN, but not in MuSC DEN, consistent with a selective activation of STAT3 in FAPs by denervation. The phospho-STAT3 signal detected in the myonuclei (Fig. 3a) predicted an activation of STAT3 pathway also in denervated myofibers. Indeed, immunofluorescence analysis of single fibers isolated from unperturbed (CTR) or DEN muscles showed the presence of phospho-STAT3 signal localized in the typical myonuclear position only in DEN fibers (Fig. 3c). Accordingly, western blot analysis of the protein lysate from the same fibers confirmed the STAT3 activation in DEN fibers (Fig. 3d). RNA-seq analysis of single fibers from denervated muscles revealed extensive alterations in gene expression induced by denervation (Suppl. Fig. 4a-b), with an activation of IL6-STAT3 signaling, among other putative effectors of muscle atrophy and adaptation (Suppl. Fig. 4c-e)23–25.
Figure 3: Persistent activation of IL6-STAT3 pathway in FAPs and myofibers from denervated muscles and prevention of myofiber atrophy and fibrosis by IL6 blockade.
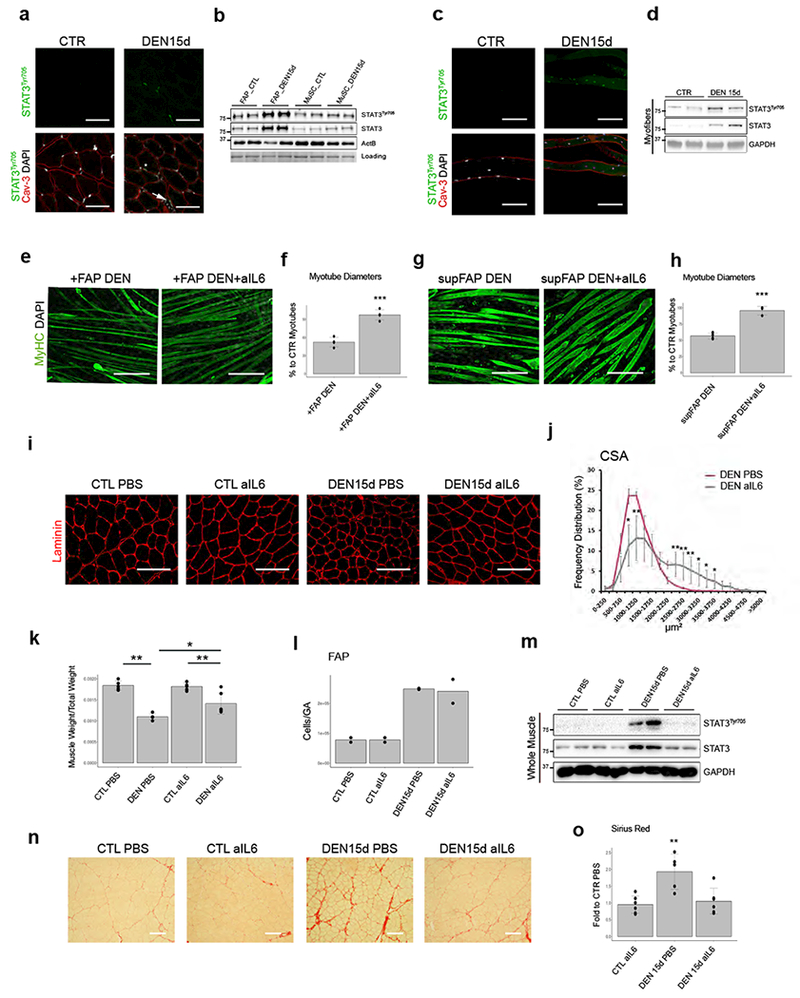
(a) Caveolin-3 (red), phospho-STAT3 (green) and DAPI (white) immunofluorescence in TA muscles from CTR or DEN muscle. Scale bar 200 μm. Data shown represent 3 independent experiments. (b) Immunoblot of phospho-STAT3 in FAPs and MuSCs from CTR or DEN muscles. Data shown represent 2 independent experiments. (c) Caveolin-3 (red), phospho-STAT3 (green) and DAPI (white) immunofluorescence in single myofibers from CTR or DEN muscles. Scale bar 200 μm. Data shown represent data from 3 animals/group. (d) Representative Immunoblot of phospho-STAT3 from single myofiber out of 2 independent experiments. (e) MyHC (Green) and DAPI (White) immunofluorescence of myotubes co-cultured with FAPs DEN, treated or not with alL6 antibodies (72h). Scale bar 200 μm. (f) Quantification of myotube diameter. Values represent mean ±s.d.***P = 0.0002; by two side student t-test (n=4 biologically independent samples). (g) MyHC (Green) and DAPI (White) immunofluorescence of myotubes cultured with FAP DEN-conditioned media, with or without alL6 antibodies (72h). Scale bar 200 μm. (h) Quantification of myotube diameter. Values represent mean ± s.d. **P = 0.003; by two side student t-test. (n=6 supFAP DEN, n=3 supFAP DENalL6 independent samples) (i) Laminin (red) immunofluorescence from DEN or contralateral (CTL) TA muscles, treated or not with alL6 antibody. Scale bar 100 μm. Data from 4 animals/group. (j) Frequency distribution of fiber CSA in denervated (DEN) TA muscle treated or not with alL6 antibodies. Line graph represents relative frequencies as percentage (n=4 animals). Values represent mean ± s.d. *P < 0.05, **P < 0.01; by two side student t-test. (k) Normalized TA muscle weight in the indicated conditions. Values represent mean ±s.d. *P<0.05, **P<0.01; by one-way ANOVA (n=3 animals/group).(I) FAP quantification by FACS. Values represent mean n=2 animals. (m) Immunoblot of phospho-STAT3 from whole TA muscles. Data shown represent data from 4 animals/group. (n) Sirius-red of TA muscle from contralateral (CTL) or DEN muscle treated or not with alL6 antibody. Scale bar 100 μm. (o) Quantification of collagen area. Values represent mean ±s.d. **P=0.002; by one-way ANOVA (n=5 animals/group.
As IL6-STAT3 signaling has been implicated in skeletal muscle atrophy19–22, we first determined whether incubation with neutralizing antibodies against IL6 (anti-IL6) could counter FAP DEN-induced atrophy of co-cultured myotubes. Figure 3e-f shows that exposure to anti-IL6 almost completely prevented the reduction of myotube caliber caused by co-culture with FAP DEN. Effects observed in co-culture systems typically entail the possibility that both or either co-cultured cell types are affected by the experimental conditions. For instance, anti-IL6 can interrupt the IL6-STAT3 feed-forward loop in FAPs, thereby decreasing FAP-derived IL6; however, IL6 has been also reported to directly promote muscle atrophy22. In order to determine whether anti-IL6 selectively affects FAP-mediated myotube atrophy, we incubated supernatants from FAPs (supFAP) DEN or CTR with anti-IL6, and observed an inhibition of the reduction in myotube caliber caused by DEN supFAP, as compared to CTR supFAPs (Fig. 3g and h). We then evaluated whether systemic delivery of anti-IL6 for 15 days could reduce myofiber atrophy following sciatic nerve transection in 2-month-old mice. Anti-IL6 treatment effectively prevented CSA reduction (Fig. 3i-j) and loss of weight (Fig. 3k) in tibialis anterior (TA) muscles of denervated mice. Of note, anti-IL6 did not alter the increase in number of DEN FAPs (Fig. 3I), although it could completely prevent STAT3 activation in whole muscle (Fig. 3m), indicating an effective neutralization of FAP-derived IL6 signaling, and suggesting that FAP expansion following denervation occurs independent on IL6. Importantly, IL6 blockade was sufficient also to prevent denervation-induced fibrosis (Fig. 3n and o), another effect potentially induced by DEN FAPs.
STAT3 inhibition in DEN FAPs, but not in myotubes, prevents denervation-mediated muscle atrophy
We next tested whether pharmacological inhibition of the IL6 downstream effector STAT3 could replicate the effects of anti-IL6 in denervated muscles. Systemic delivery of STAT3 inhibitor (STAT3i) countered CSA reduction (Fig. 4a and b) and loss of weight (Fig. 4c), as well as fibrosis (Fig. 4d and e) in denervated muscles of mice subjected to sciatic nerve severing. STAT3i could also effectively prevent the reduction of myotube caliber upon transwell co-culture with FAP DEN (Fig. 4f and g); however, when STAT3i was added to DEN supFAP prior to incubation with myotubes, it failed to prevent the myotube atrophy (Fig. 4h and i), indicating that the anti-atrophic effect of systemic STAT3i delivery is accounted for by the inhibition of IL6-STAT3 signaling in FAPs. Together with data shown in Fig. 3e-h, this evidence indicates that while IL6-STAT3 signaling drives the pathogenic phenotype of FAP DEN, FAP-derived IL6 promotes atrophy of cultured myotubes independent on STAT3 activation in the myonuclei. Accordingly, selective genetic ablation of STAT3 in skeletal myofibers or satellite cells, by crossing STAT3fl/fl with either MCKCRE (Suppl. Fig. 5a-e) or with Pax7CRE mice (Suppl. Fig. 5f-g), respectively, did not prevent denervation-induced myofiber atrophy, further indicating that STAT3 activation in myofibers or MuSCs does not contribute to denervation-induced muscle atrophy.
Figure 4: STAT3 inhibition prevents the atrophic effect of denervation-derived FAPs:
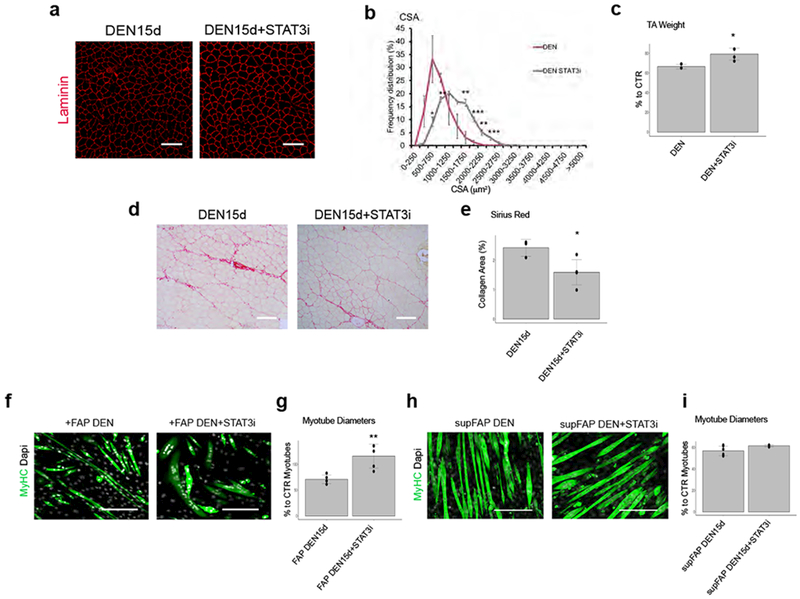
(a) Immunofluorescence of Laminin (red) of TA muscle derived from DEN muscle with or without STAT3i treatment. Scale bar 100 μm. Data shown represent 4 independent experiments. (b) Frequency distribution of fiber Cross-Sectional Area (CSA) TA muscle. Values represent mean ±s.d. *P<0.05 **P<0.01 ***P<0.001; by one-way ANOVA (n=4 animals/group). (c) Normalized TA muscle weight for the indicated conditions. Values represent mean ± s.d. *P = 0.018; by two side student t-test (n=6 CTR, n=3 DEN, n=4 DEN STAT3i animals). (d) Representative Sirius-red stained DEN muscles with or without STAT3i treatment. Scale bar 100 μm. Data shown represent 3 independent experiments. (e) Quantification of collagen staining. Values represent mean ±s.d. *P=0.017; by student two side t-test (n=3 DEN15d n=5 DEN15d+STAT3i animals). (f) Immunofluorescence of MyHC (Green) and DAPI (White) of C2C12 myotubes co-cultured for 72h with FAP DEN15d pretreated or not with STAT3i for 24h. Scale bar 200 μm. (g) Quantification of myotubes diameter from experiments shown in f. Values represent mean ±s.d. **P=0.003; by student two side t-test (n=5 biologically independent samples). (h) Immunofluorescence of MyHC (Green) and DAPI (White) of C2C12 myotubes cultured alone (control) or with FAP-conditioned media with or without STAT3 inhibitor for 72h. Scale bar 200 μm. (i) Quantification of myotubes diameter from experiments shown in h. Values represent mean ± s.d.(n=6 supFAP DEN; n=3 supFAP DEN STAT3i biological independent samples).
To explore the specific contribution of FAP DEN IL6-STAT3 signaling to myofiber atrophy in vivo, we tested whether FAP DEN could be sufficient to induce myofiber atrophy, upon their transplantation into skeletal muscles of WT mice. In one experimental approach, FAPs isolated from denervated muscles of GFP mice (GFP-FAP DEN) were incubated ex vivo for 3 hours with STAT3i or vehicle (DMSO) and then injected into muscles of WT mice (Fig. 5a). Transplantation of GFP-FAP DEN led to a significant reduction of CSA in myofibers of recipient muscle (Fig. 5a-c); however, when GFP-FAP DEN were exposed to STAT3i prior to the transplantation, their pro-atrophic effect was prevented (Fig. 5a-d). In another approach, freshly sorted FAP DEN from R26RTdTStat3fl/fl transgenic mice (or R26RTdTStat3+/+, as control), were infected with Cre-expressing adenovirus prior to transplantation into muscles of WT mice. Transplanted FAP DEN were visualized by TdT expression, and led to CSA reduction of fibers from recipient mice (Fig. 5e-g); however, this effect was not observed upon genetic deletion of STAT3 (Fig. 5e-h). Of note, we observed that transplanted muscles did not show features of interstitial fibrosis in these experimental conditions. Collectively, these data indicate that DEN FAPs acquire a cell-autonomous pro-atrophic potential that relies on activation of STAT3 signaling and IL6 secretion, which are initially triggered by denervation, but persists even in the absence of denervation. By contrasts denervation-induced fibrosis appears to require the concomitant presence of muscle denervation, in addition to DEN FAPs.
Figure 5: Selective STAT3 inhibition in FAPs is sufficient to counter myofiber atrophy:
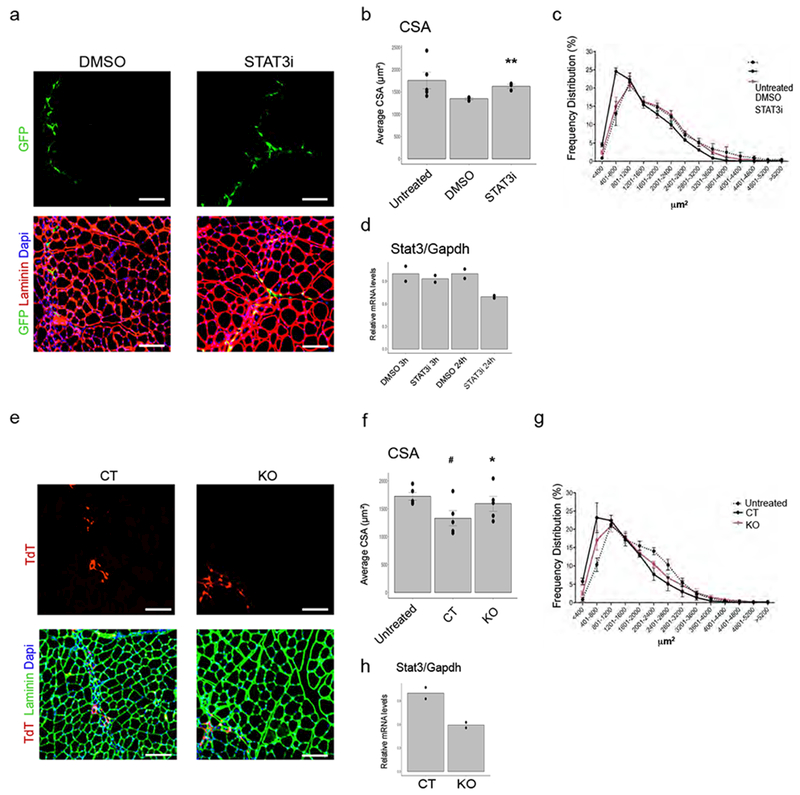
FAPs were isolated from denervated muscles 15 days after transection of the sciatic nerve. Donor EGFP mice were used. (a) Representative Images of GFP (green), Laminin (red) immunofluorescence staining of muscle derived from mice transplanted with FAP DEN incubated for 3 hours with DMSO or STAT3 inhibitor ex vivo. Scale bar 100 μm. Data shown represent results from n=5 untreated, n=3 DMSO and STAT3i animals. (b-c) Mean CSA and frequency distribution of myofiber CSA from untreated mice or mice transplanted with FAP DEN incubated for 3 hours with DMSO or STAT3 inhibitor. Values represent mean ± S.E.M. **P<0.01 (p=0.008) vs DMSO; Two-tail student’s t-test (Data shown represent results from n=5 untreated, n=3 DMSO and STAT3i animals). (d) qPCR analysis of STAT3 transcripts in FAP DEN treated with the STAT3 inhibitor for 3 or 24 hours in vitro. Values represent mean. (n=2 technical replicates). (e-h) Donor R26RTdTSTAT3flox/floxTdTf/f mice and control R26RTdTSTAT3+/+animals were used. (e) Representative Images of TdT (red), Laminin (green) immunofluorescence staining of muscle derived from mice transplanted with FAP DEN infected O/N with adenoviruses expressing Cre Recombinase. Scale bar 100 μm (f-g) Mean CSA (f) and frequency distribution (g) of myofiber CSA from untreated mice or mice transplanted with FAP DEN infected 0/N with adenoviruses expressing Cre Recombinase (KO) or Adeno control (CTR). Values represent mean ± S.E.M. **P<0.01 (p=0.003) vs CT; #P<0.05 (p=0.033) vs Untreated; Two-tail student’s t-test (n=5 animals/group). (h) qPCR analysis of STAT3 transcripts in FAP DEN 24 hours after infection. Values represent mean ±s.d. *P<0.05 (p=0.036) vs CT; Two-tail student’s t-test (n=2 technical replicates).
Thus, selective inhibition of IL6-STAT3 signaling in DEN FAPs is sufficient and necessary to counter muscle atrophy and fibrosis, presumably by interrupting a denervation-activated feed-forward loop that leads to chronic secretion of abnormal levels of IL6. Indeed, our evidence that STAT3 activation and IL6 secretion are consensually inhibited in FAPs isolated from denervated muscles of mice treated with either anti-IL6 or STAT3i (Suppl. Fig. 5h) supports the existence of a reciprocal positive feedback between IL6 and STAT3 cascades in DEN FAPs. The upregulation of IL6 receptor observed in FAP DEN by RNAseq (Table 1) likely propels this feedback.
FAPs with aberrant IL6-STAT3 activation accumulate upon loss of integrity of neuromuscular junctions (NMJs)
The accumulation of FAPs in skeletal muscles upon acute denervation suggests a previously unrecognized functional relationship between the integrity of NMJs and FAP activation. We therefore hypothesized that FAPs with an aberrant activation of IL6-STAT3 could accumulate in conditions of acute traumatic damage of NMJs (e.g. spinal cord injury — SCI) and/or progressive denervation, within physiological (aging) or pathological (e.g. Spinal Muscle Atrophy — SMA; Amyotrophic Lateral Sclerosis - ALS) conditions.
The dramatic reduction of the CSA (Fig. 6a) and induction of fibrosis (Fig. 6b,c) in muscles of 4 month-old mice subjected to SCI, coincided with an increased number of FAPs (Fig. 6d-f) that exhibited deregulated activation of STAT3-IL6 signaling and patterns of gene expression similar to those observed upon sciatic nerve severing (Fig. 6g,h). By contrast, the slowly progressive and milder denervation occurring in aged (2 year-old) mice correlated with a proportionally milder reduction of CSA (Suppl. Fig. 6a) and fibrotic deposition (Suppl. Fig. 6b,c), without increase in FAP’s number (Suppl. Fig. 6d) or expression of IL6 and other genes typically induced by denervation (Suppl. Fig. 6e).
Figure 6: FAPs with aberrant IL6-STAT3 activation accumulate in skeletal muscles from SCI.
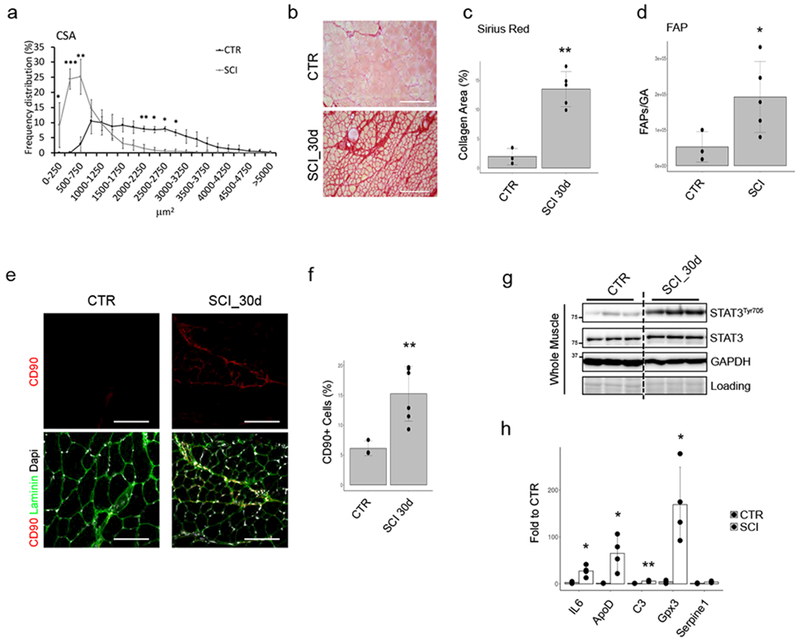
(a) Frequency distribution of fiber CSA in Quadriceps of control (CTR) or Spinal Cord Injury (SCI) animal model. Values represent mean ±s.d. *P<0.05, **P<0.01, ***P<0.001; by one-way ANOVA (n=3 CTR and n=5 SCI animals; two independent experiments). (b) Sirius-red staining of CTR or SCI muscles. Scale bar 200 μm. (n=3 CTR and n=5 SCI animals; Data shown represent 2 independent experiments). (c) Quantification of data in b. Values represent mean ±s.d., **P<0.01; by student t-test. (n=3 CTR and n=5 SCI animals)(d) Cyto-fluorimetric count of FAPs from CTR or 15days SCI mice. Values represent mean ±s.d., *P<0.05; by t-test. (n=3 CTR and n=5 SCI animals). (e) Immunofluorescence of Laminin (Green), DAPI (White) and CD90 (Red) in CTR and SCI muscle sections (n=3 CTR and n=5 SCI animals). (f) CD90-positive cells quantification. Values represent mean ±s.d., **P=0.003; by two side student t-test (n=3 CTR and n=5 SCI animals). (g) Immunoblot of phospho- and total-STAT3 from whole muscles (n=3 animals/group) of CTR or SCI mice. Samples are loaded on the same gel; intermediate time points were removed. Representative blot out of 2 independent experiments. (h) qPCR analysis of indicated genes in freshly isolated FAPs from WT and SCI. Values represent mean ±s.d. *P<0.05, **P<0.01; by t-test (n=3 CTR and n=4 SCI animals).
We next investigated the activation of FAPs by denervation within mouse models of two neuromuscular disorders, such as SMA and ALS. SMAΔ7 is a mouse model of SMA, in which mice are born smaller (Suppl. Fig. 6f) and with reduced muscle mass (Suppl. Fig. 6g), and typically die by the third week of age26. While the neonatal lethality and reduced muscle mass clearly limit the analysis of these animals and precludes a direct comparison with other models of denervation in adult mice, we could still observe a notable increase of FAPs relative to the reduced muscle mass of 9 day old SMAΔ7 mice (Suppl. Fig. 6h-i). Moreover, FAPs isolated from 9-day old SMAΔ7 mice showed increased expression of IL6 as well as other genes typically induced by denervation (Suppl. Fig. 6j). Finally, we investigated the mouse model of ALS — the SOD G93A mice. In these mice, a progressive reduction of CSA (Fig. 7a,c and d) and loss of muscle weight (Fig. 7b), concomitant with development of muscle fibrosis (Fig. 7e and f), are typically observed within the first 5 months of life, with symptoms and signs manifesting between 2 and 3 months of life27,28. We detected an increased number of FAPs in ALS muscles already at 3 months of life, leading to an accumulation that persisted throughout the disease progression (Fig. 7g). However, only FAPs isolated from muscles of 140 day-old SOD G93A mice exhibited increased levels of IL6 transcripts (Fig. 7h) and activated STAT3 signaling (Fig. 7i-j), indicating a correlation between IL6-STAT3 activation in FAPs and muscle atrophy and fibrosis at symptomatic stages of ALS, as also observed by Gonzales et al.11. We finally investigated whether the presence of FAPs in muscle biopsies of ALS patients could also discriminate pre-symptomatic from symptomatic stages of human disease progression. As Seal is not expressed in humans, and because of the inefficient detection of FAPs by anti-PDGFalpha receptor antibodies in human muscle sections, we used an alternative cell surface marker (CD90)10, which permits isolation of prospectively equivalent FAPs (Suppl. Fig. 7a and b), to detect FAPs in muscles of ALS patients. Human ALS muscles showed reduction of CSA and fibrosis (Fig. 7k and I) and CD90-positive interstitial cells (Fig. 7m) at symptomatic stages of disease..We also detected interstitial phospho-STAT3 signal (Fig. 7n) that coincided with CD90 positive cells (Suppl. Fig. 7c). We therefore explored the potential ability of STAT3i to alleviate the consequence of FAP-derived pro-atrophic and fibrotic signaling in SOD G39A mice. The STAT3 Inhibitor VI S3I-20129 was administered systemically to 8-week old SOD G39A mice until they reached 12 weeks of age. This treatment was sufficient to prevent muscle atrophy (Fig. 7o) and to reduce the formation of intramuscular fibrosis (Fig. 7p and q).
Figure 7: FAPs with aberrant IL6-STAT3 activation accumulate in ALS skeletal muscles:
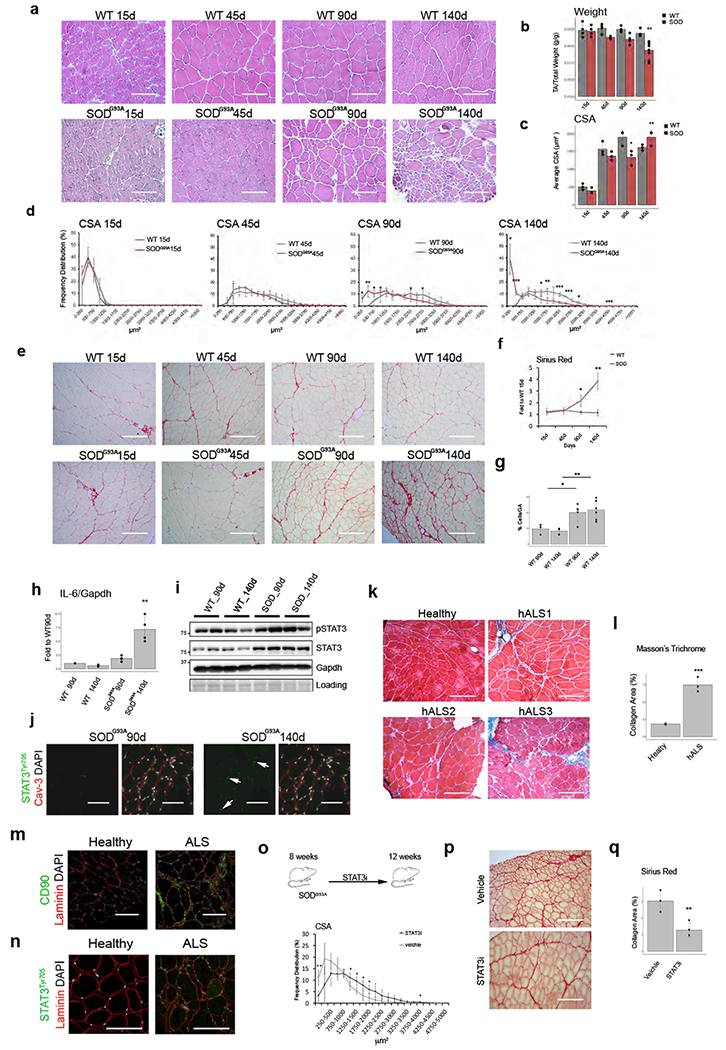
a) Hematoxylin-Eosin staining of TA muscles from WT and SODG93A mice. Scale bar 200 μm. Data represent 3 independent experiments. (b) Normalized TA muscle weight of WT and SODG93Amice. Values represent mean ±s.d. **P<0.01; by one-way ANOVA (n=4 animals for all experiments, but SOD140 n=10). (c) Mean CSA of fibers from WT and SODG93A mice. Values represent mean ±s.d. *P<0.05, **P<0.01; by one-way ANOVA (n=3 animals/group)(d) Frequency distribution of fiber CSA in WT and SODG93A mice. Values represent mean ±s.d. *P<0.05, **P<0.01, ***P<0.001; by one-way ANOVA (n=3 animals/group). (e) Sirius-red staining of TA muscles from WT and SODG93A mice. Scale bar 200 μm. Data represent results from 3 independent experiment. (f) Quantification of data in e. Values represent mean ±s.d. *P<0.05, **P<0.01; by one-way ANOVA. (n=3 animals/group). (g) FACS quantification of FAPs from WT and SODg93a mice. Values represent mean ±s.d. *P<0.05, **P <0.01; by one-way ANOVA. (n=4 WT90d, n=5 SOD90d, n=6 WT140d and SOD140d animals). (h) qPCR analysis of IL6 transcripts in FAPs from WT and SODG93A muscle. Values represent mean ±s.d. **P<0.01; by one-way ANOVA (n=3 WT n=4 SOD animals). (i) Immunoblot of phospho-STAT3 from TA muscles. Data shown represent 2 independent experiments. (j) Immunofluorescence of Laminin (Red), DAPI (White) and phospho-STAT3 (Green) in TA sections from SODG93A mice. Scale bar 200 μm. (k) Masson-Trichrome staining in control or 3 ALS patient muscles. (3 section/condition analyzed). Scale bar200 μm(I) Quantification of data in k. Values represent mean ±s.d. ***P<0.0007; by two side student t-test. (n=3 independent samples/groups)(m) CD90 (Green) immunofluorescence of control or ALS patient muscles. Scale bar 200 μm (n) phospho-STAT3 (Green) immunofluorescence of control and ALS patient muscles. Scale bar 200 μm. Representative images out of 3 independet experiment. (o) Top: Scheme of treatment. Bottom: Frequency distribution of fiber CSA of TA muscle. Values represent mean ±s.d. *P<0.05, **P<0.01; by two-side t-test. n=4 animals/group.. (p) Sirius-red staining of TA muscles from Control and STAT3i-treated mice. (q). Quantification of data in p. Values represent mean ±s.d. *P<−0.05; by t-test. n=4 animals/group.
Discussion
Our results revealed a previously unappreciated function of FAPs as source of pro-atrophic and pro-fibrotic signals in denervated muscles. While it remains unknown the mechanism of FAP activation in denervated muscles, we hypothesize that loss of NMJ could be a key trigger event. We initially reasoned that myofiber atrophy could promote FAP activation, by perturbing the spatial equilibrium between myofibers and the interstitial space. However, we could not observe any comparable accumulation of FAPs in other pro-atrophic conditions that spared the functional integrity of NMJ, such as forced immobilization by casting or hind-limb suspension. Consistently, the slowly progressive deterioration of NMJs that accompanies milder reduction in CSA of aged muscles did not coincide with a significant increase in FAP’s number and IL6 expression. By contrast, activation of FAPs with aberrantly activated IL6-STAT3 signaling was invariably observed in all conditions of severe functional impairment of NMJs, irrespective of whether they were caused by acute trauma (SCI) or rapidly progressive neuromuscular disorders (SMA and ALS). Collectively, this evidence indicates that the degree of severity of denervation is a key determinant for FAPs to adopt a pro-atrophic and fibrotic phenotype. As chronic activation of IL6-STAT3 is a distinctive feature of DEN FAPs and mediates these pathological outcomes, we argue that persistent rise of IL6 from an intramuscular cellular source activated by denervation (e.g. DEN FAPs) accounts for the severity of atrophy and fibrosis that was observed in these conditions. In this regard, it is likely that reciprocal positive feedback between IL6 and STAT3 pathways sustain a feed-forward circuit that propels the persistent activation of IL6-STAT3 signaling in DEN FAPs, whereby FAP-secreted IL6 activates STAT3 (and viceversa) in a autocrine manner. It is interesting to note that the potential paracrine effect of FAP-derived IL6 on STAT3 activation in other neighboring cells within denervated muscles appears restricted to myonuclei, at variance with the activation of STAT3 signaling that is typically observed in MuSCs upon acute or chronic injury29. We argue that in denervated muscles the lack of infiltration by inflammatory cells (e.g. macrophages) together with the integrity of the key anatomical component of MuSC niche (e.g. the basal lamina) insulate MuSCs from environmental signals, such as FAP-derived IL6, thereby preventing an aberrant activation of STAT3 signaling in quiescent MuSCs.
While moderate increase of circulating IL6 has been observed during aging30, abnormally elevated concentration of circulating IL6 are found during cancer cachexia, which correlates with a rapid and dramatic loss of muscle mass31. Moreover, IL6-STAT3 signaling can be activated by inflammation-free perturbations of skeletal muscle, such as contraction and hypertrophy30,32, which lead to anabolic responses —the opposite biological outcome of denervation-induced muscle atrophy. Thus, differences in cellular sources and relative amounts of local or systemic IL6, duration of activation of downstream (e.g. STAT3) or parallel cascades, and co-stimulatory signals from the local milieu appear to determine the final biological effect of this IL6 on muscles.
Finally, our work indicates that targeting IL6-STAT3 signaling in DEN FAPs with FDA-approved drugs33–35 could be used to counter muscle atrophy and fibrosis upon acute denervation or during ALS progression.
Materials and Methods
Animals
All experiments in this study were performed in accordance with protocols approved by Italian Ministry of Health, National Institute of Health (IIS), and Santa Lucia Foundation (Rome) and by the Sanford Burnham Prebys Medical Discovery Institute Animal Care and Use Committee. The study is compliant with all relevant ethical regulations regarding animal research.
C57BL/6J and CD1 mice were provided by the Core Structure of the EMMA (European Mouse Mutant Archive) and by the SBP Animal Facility (La Jolla). Two to three month-old male mice were used for ex-vivo experiments. The hemizygous transgenic mice carrying the mutant human SOD1G93A(B6.Cg-Tg(SOD1*G93A)1Gur/J) gene were originally obtained from Jackson Laboratories (Bar Harbor, USA).
R26RTdT mice were purchased from Jackson Laboratories. Stat3flox/flox were a kind gift from S. Akira (described in 36). R26RTdT and Stat3flox/flox mice were used to generate the R26RTdTStat3flox/flox mice and control R26RTdTSTAT3+/+ animals. Pax7CreERStat3flox/flox mice were generated as described29. MCKCreStat3flox/flox were a kind gift from S. Schenk (described 37).
The SMA mouse model used was FVB.Cg-Tg(SMN2*delta7) 4299Ahmb Tg(SMN2)89Ahmb Smn1tm1Msd/J (The Jackson Laboratory), hereafter referred to as SMAΔ7 mice. Genomic DNA was isolated from the tail, amplified by PCR using the primers listed below, and genotyped as described previously26. SMAΔ7 mice were scarified at 8 days of postnatal life. Control littermate were used as controls (WT).
Denervation and muscle injury
Denervation was performed as previously described38. Briefly, unilateral hindlimb denervation was performed by transecting the sciatic nerve under anesthesia by intraperitoneal injection of 40 mg/kg ketamine (Zoletil®) and 10 mg/kg xylazine (Rampum®). Upon exposure of the sciatic nerve, a piece of 0.5cm was transected near to the head of the femur. The lesion was sutured after the operation.
Muscle injury was performed by intramuscular injection of 10μg/ml Cardiotoxin (CTX, Latoxan). Tissues were harvested after 3, 7, 15 and 30d for FACS or histological analyses.
STAT3 inhibitor and anti-IL-6 treatments
Neutralization with IL-6 (1,0 mg/ml) (LEAF™ purified anti-mouse IL-6, Cat. No. 504506, BioLegend) in vivo for 15 days was performed on denervated mice of two months old, by subcutaneous application of osmotic pump drug delivery system (Alzet®).
The STAT3 inhibition (STAT3 Inhibitor VI, S3I-201, CAS 501919–59-1 Calbiochem®, 5 mg/kg) was performed on denervated and SODG93A mice. Denervated mice were treated for 15 days by intraperitoneal injection with Stat3i. Injection was performed every 3 days starting from day zero — that is, the day in which denervation was performed. Likewise, SODG93A mice was treated every 3 days starting from 8 weeks for 4 weeks.
Tissue samples for cryosectioning were snap frozen in liquid nitrogen cooled-isopentane and stored at −80°C until processing. Tissue samples for RNA/DNA were also cooled in liquid nitrogen.
Spinal Cord Injury (SCI)
Three month-old CD1 female mice (Charles River Laboratories, Como, Italy or EMMA InfraFrontiers, Monterotondo, Italy) were used in SCI. To perform SCI, mice were deeply anesthetized with a mixture 1:1 of Rompun (Bayer 20mg/ml; 0.5ml/kg) and Zoletil (100mg/ml; 0.5ml/kg), the back hairs were shaved, skin disinfected with betadine and an incision to expose spinal cord was made. Animals were mounted on a stereotaxic apparatus with spinal adaptors connected to a cortical PinPoint precision impactor device (Stoelting) and maintained at 37°C throughout surgery. To induce a severe trauma the following parameters were set up: - middle, round and flat tip (#4); - velocity 3 m/sec; - depth 5 mm; - dwell time 800 ms. The impact was applied on thoracic level (vertebrae T10-T11). Analysis of the graphical impact parameters, operated by the PinPoint software, was used to identify potential outliers. Behavioral analyses were also used to corroborate differences in injury severity within groups. Slight lesions were excluded from the study based on these criteria.
Cell preparation and FACS isolation
Hind limb muscles were digested in PBS with 2 μg/mL Collagenase A (Roche), 2,4 U/mL Dispase I (Roche), 10 ng/mL DNase I (Roche), 0,4 mM CaCl2 and 5mM MgCl2 for 60 min at 37° C. Muscle Macrophages (MPs) cells were isolated as TER119−/CD45−/CD31− /F480+/CD11B+ cells; FAPs were isolated as TER119−/CD45−/CD31−/a7INTEGRIN−/SCA− 1+ cells; MuSCs were isolated as TER119−/CD45−/CD31−/a7INTEGRIN+/SCA-1− cells; as described in Saccone et al. 2014. FACS sorting gating strategy is shown in Supplemental Material (Suppl. Figure 1c).
FAP Transplants
R26RTdTSTAT3flox/flox mice and control R26RTdTSTAT3+/+ animals were denervated. 15 days after denervation, FAPs were isolated by FACS as CD31-, CD45-, Ter-, α7 integrin-, Sca1+ cells. After isolation, cells were plated overnight and infected with an adenovirus coding for the Cre Recombinase (50000 MOIS). After infection, FAPs were resuspended in PBS and transplanted into the tibialis anterior of C57BL6/J host mice. 50,000 FAPs were transplanted in each muscle.
For the studies with the STAT3 inhibitor (STAT3 Inhibitor VI, S3I-201, CAS 501919–59-1 Calbiochem®), we isolated FAPs from denervated EGFP mice (15 days after denervation). After isolation, FAPs were incubated for 3 hours with the STAT3 inhibitor (100 μM) (or with the vehicle, DMSO) at 4°C with rotation. After treatment, 30,000 FAPs were transplanted into the tibialis anterior of C57BL6/J host mice.
Transplanted muscles from both experimental designs were harvested 7 days after transplantation to perform histological analysis.
Histology Immunofluorescence
For the histological analysis 8-10 μm muscle cryosection were analysed. Both cryosections and cultured cells were fixed in 4% PFA for 10 min and permeabilized with 100% acetone for 1 min at RT or with 0.25% Triton for 15 min at RT. Muscle sections and cultured cells were then blocked for 1h with a solution containing 4% BSA in PBS. The PAX7 staining was performed by an antigen retrieval protocol. The primary antibodies immunostaining was performed ON at 4°C and then the antibody binding specificity was revealed using secondary antibodies coupled to Alexa Fluor 488, 594, or 647 (Invitrogen). Sections were incubated with DAPI in PBS for 5 minutes for nuclear staining, washed in PBS, and mounted with mounting medium or glycerol (3:1 in PBS).
The primary antibodies used for immunofluorescences are: rabbit anti-Laminin (#L9393, Sigma, 1:400); mouse anti-Caveolin3 (#610420, BD Transduction Laboratories; 1:1000); rat anti-SCA1 (Ly-6A/E, eBioscence, 1:150); mouse anti-eMyHC (Developmental Studies Hybridoma Bank, DSHB, 1:20); rabbit Anti-STAT3 Tyr705 (#9131S, Cell Signaling, 1:150); mouse anti-MF20 (Developmental Studies Hybridoma Bank, DSHB, 1:20) mouse anti-PAX7 (Developmental Studies Hybridoma Bank, DSHB, 1:20), rat anti-CD90 (#14-0903-82, eBioscence, 1:150), anti-Collagen-1 (ab6308, Abeam, 1:100),mouse anti-αSMA (#C2817, Santa Cruz,1:100), ratanti-F480 (#MCA497G, Bio-Rad, 1:150).
For Hematoxylin and Eosin staining, sections were fixed in 4% PFA for 10 min, washed in PBS and then stained in haematoxylin for 8 min and eosin for 1 min. The muscle sections were further dried in gradual increasing concentration of ethanol/water solutions and, after fixation in 100% xylene, were mounted with EUKITT® mounting medium (Sigma-Aldrich). Sirius red staining was performed to analyse total collagen I and III content. Muscle cryosections were fixed for an 1h at 56°C in Bouin’s Solution and then stained in Picro-Sirius red (0.1%) solution for 1h protected from light. After a brief washing in acidified water 0.5% v/v, sections were fixed in 100% ethanol and the final dehydration was performed in xylene 100%. Sections were mounted with EUKITT® and visualized using a Nikon Eclipse 90i.
Masson’s trichrome staining was performed as described in Mozzetta et al.16.
The transverse sections were visualized on a Zeiss confocal microscope or a Nikon Eclipse 90i fluorescent microscope and then edited using the ImageJ® software. The figures reported are representative of all the examined fields.
Human Samples
Muscle biopsies from ALS patients were obtained for diagnostic purposes in the Hospital de la Santa Creu i Sant Pau, Barcelona, Spain. All patients signed informed consent to use the samples for research. After muscle was obtained from surgeon, the sample was frozen in isopentane cooled with liquid nitrogen. Five micrometers muscle sections were obtained using a cryostat CM 1950 from Leica.
Morphometric Analysis
Whole Tibialis anterior myofibers cross-sectional was performed with a staining for Laminin and quantified by ImageJ® software. The values shown as average percentage of frequency distribution.
Fibrotic areas were calculated from sections evaluating image analysis algorithms for color deconvolution. ImageJ was used for image processing, the original image was segmented with three clusters and the plugin assumes images generated by colour subtraction.
Proteins extraction and Western Blot
Total protein extract was obtained by homogenising TA muscles with TissueRuptor (Qiagen) proteins Lysis Buffer (50 mM Tris-HCI pH 7,5, Sigma; 1M NaCI, Sigma; 625Mm Saccarosio, Sigma; 10% Glicerolo, Sigma; 1% Triton x-100, Sigma) with protease and phosphatase inhibitors: 1mM PMSF, Sigma; 2 g/ml Aprotinin, Sigma;10 g/ml Leupeptin, Sigma; 10 g/ml Pepstatin, Sigma; 1 mM NaF, Sigma; 0,1 mM Na3V04, Sigma). After separation with 8% SDS—PAGE gels, proteins were transferred to nitrocellulose membranes (Trans-Blot® Turbo™, Bio-Rad, 170-4158) and then blocked in a solution of 5% skim milk (Sigma) in TTBS, and incubated ON at 4 °C with primary antibodies. Western blot was performed using antibodies against the following proteins: STAT3Tyr705 (#9131S, Cell Signaling, 1:1000); STAT3 (#4904S, Cell Signaling, 1:1000); GAPDH (#97166S, Cell Signaling, 1:1000). The membranes were then incubated 1h at RT with the corresponding secondary antibodies: goat polyclonal anti-rabbit (1:2000 in T-TBS) or anti-mouse (1:1000 in T-TBS) IgG antibody conjugated to peroxidase (HRP) (Santa Cruz). The signal was detected with a ChemiDoc MP Imaging System (Bio-Rad, Hercules, CA, USA) and the protein expression levels were then quantified by ImageJ® software and normalized on GAPDH.
RNA analysis by quantitative PCR
Total RNA from Tibialis anterior muscles was extracted using TRI® Reagent (Sigma) by following manufacturer’s protocol. Cells’ RNA was isolated with Qiagen RNeasy mini-kit following the manufacturer’s protocol. After quantification with Nanodrop 8000 spectrophotometer, the isolated RNA was retrotranscribed using the TaqMan® Reverse Transcription Reagents kit (Applied Biosystems). The cDNA was used as a template in Real-time qPCRs reactions, performed with SYBR green Master Mix reagents (Applied Biosystems) with an ABI PRISM 7000 sequence detection system, using primers provided in the Supplementary Materials. Values were further normalized on glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
ELISA assay
ELISA essay was performed using 72h conditioned media (BIOAMF-2 or GM) from each population with Mouse lnterleukin-6 (Mouse IL-6) ELISA Kit (#KMC0061, Invitrogen).
Coculture conditions of FAPs and C2C12
C2C12 and FAPs cells were cocultured by using inserts with 1μm porous membrane to avoid direct contact between populations. For fusion index analysis, C2c12 were grown independently from FAPs in GM (DMEM w/o pyruvate with 2% FBS (Gibco)) for 72h in 24-well culture plates (BD bioscience). For myotubes diameter analysis, C2c12 were grown independently from FAPs in GM for 72h and for 72h in DM (DMEM w/o pyruvate with 2% Horse Serum (Gibco) in 24-well culture plates. After C2C12 differentiation of after 72h of GM, freshly sorted FAPs cells were plated on the upper insert and transwell co-cultures were maintained for additional 72h.
In the co-cultures experiment, the treatment with anti-IL-6 was performed using alL6 neutralizing antibody (0,02 mg/ml. LEAF™ purified anti-mouse IL-6, Cat. No. 504506, BioLegend) for 72h.
For Red oil staining, FAPs cells were inducted to differentiate in adipocites as described in Mozzetta et al.16.
Myofiber isolation
For myofiber isolation, muscle digestion was performed for 1h at 37°C with digestion buffer (DMEM +Piruvate, 0.4% collagenase form Clostridium hystoliticum (C6885)). The content of the digestion buffer is then poured in horse serum-coated dishes for the direct isolation of myofibers at RT. The isolation of the single myofibers was realized by using a medium size pipette (the re-equilibration of the medium was performed by incubation at 37 °C, 5% C02 for 5 min minimun). This passage was repeated at least 3 times to remove debris. The single myofibers were incubated at 37°C, 5% C02. Immunostaining on myofibers was performed on floating following as described in Pasut et al., 2013 38.
RNA-sequencing
FAPs were isolated from mice GA muscle as described. RNA from FAPs was extracted using RNeasy Mini kit (Qiagen) following the manufacturer’s protocol. RNA was shipped to the sequencing IGA of Udine. The libraries for sequencing were prepared using NuGEN Ovation System V2 RNA-Seq. For each biological sample two independent experiments were carried out for the isolation of RNA. All duplicates are from different mice, sorted at different times.
RNA-sequencing data processing
For sequencing alignment, we used the human reference genome assembly GRCm38/mm10 (http://ftp.ensembl.org/pub/release-76/fasta/mus_musculus/dna/), while for transcriptome annotation we used the version85 of the GRCm38 (http://ftp.ensembl.org/pub/release85/qtf/mus_musculus/Mus_musculus.GRCm38.85.gtf.gz). We used the FASTQC package (v0.11.3) to assess the quality of sequenced libraries. All passed quality control. Reads were mapped to the reference genome using TopHat2 v.2.1.1 (Kim et al., 2013). We used the following non-default TopHat2 parameters: -p 48 -g 1 --library-type fr-firststrand. The number of mapped reads ranged between 23 and 44*106 and the percentage of mapping was between 79 and 92%. The quality control of the reads distribution along transcripts was performed using infer_experiment.py from RSeQC package v2.6.3. All samples had a uniform distribution of reads along transcripts. The sequenced read counts per annotated gene were derived with the use of htseq-count script distributed with HTSeq v0.5.4p5. We used the R library package DESeq2 v.1.12.4 for measuring differential gene expression between two different cell conditions, considering the two RNA-Seq experiments as biological replicates. We picked genes with adjusted p-value<0.01. Gene ontology analysis was performed using Ingenuity pathway analysis (IPA; http://www.1ingenuity.com).
Primers sequences
| Genes | Sequences |
| GAPDH | Fwd: 5’-CACCATCTTCCAGGAGCGAG-3’ Rev: 5’-CCTTCTCCATGGTGGTGAAGAC-3’ |
| IL6 | Fwd: 5’-TCCTCTCTGCAAGAGACTTCC-3’ Rev: 5’-TTGTGAAGTAGGGAAGGCCG-3’ |
| C3 | Fwd: 5’-CCCCTGAGACAGCCAAGAAG-3’ Rev: 5’-ATCTACTCCAGAGGCCAGCA-3’ |
| Gpx3 | Fwd: 5’-ACGTAGCCAGCTACTGAGGT-3’ Rev: 5’-CGAACATACTTGAGACTGG-3’ |
| ApoD | Fwd: 5’-CATCTGATCTGAGAGTTAGAAACTG-3’ Rev: 5’-TGTCCTTTGGCTGTGGTGAA-3’ |
| SMAΔ7 | Fwd: 5’-TTTTCTCCCTCTTCAGAGTGAT-3’ Rev1: 5’-GCTGTGCCTTTTGGCTTATCTGG-3’ Rev2: 5’-GAGTAACAACCCGTCGGATTC-3’ |
| ActB | Fwd: 5’-CACTGTCGAGTCGCGTCC-3’ Rev: 5’-TCATCCATGGCGAACTGGTG-3’ |
| Rplp0 | Fwd: 5’ GCAGGTGTTTGACAACGGCAG-3’ Rev: 5’ GATGATGGAGTGTGGCACCGA-3- |
Statistics and Reproducibility
Data are presented as mean δ SD or SEM. Comparisons were made using the Student’s t-test assuming a two-tailed distribution or by On way Anova, with significance being defined as P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***). Where appropriate and possible the exact p value is reported. The number of biological replicates for each experiment is indicated in the figure legends. RNAseq data was performed in 2 independent samples derived from different animals. Statistical method was Deseq2. Right-tailed Fisher’s exact test and one-sided Fisher’s exact test was used for IPA analyses. Histological and Immunofluorescence images are representative of at least 3 different experiment/animals. Western Blot analysis was performed in independent animal samples as indicated in the figure legends. Unprocessed blots was shown in Supplementary Figures 8-9.
Data Availability
RNA-seq data of this study have been deposited in the Sequence Read Archive (SRA) under the accession code SRP14829. Source data of Figures 2b-2d, 3f-3h, 4g-4i are provided in Supplementary Table 1 .All other data supporting the findings of this study are available from the corresponding author on reasonable request.
Supplementary Material
Acknowledgments
This work was supported by Italian Ministry of Health (Grant n° GR-2013-02356592) to L.M.; NIH grants R01AR056712, R01AR052779, P30 AR061303, MDA grant and EPIGEN grant to P.L.P; NIH grants R01 AR064873 and P30 AR061303 and MDA grant to A.S.; California Institute for Regenerative Medicine (CIRM) training grant TG2-01162 and AFM-Telethon Postdoctoral fellowship (N° 21084) to D.S. We thank Emanuela Aleo at the Institute of Applied Genomics in Udine, Italy, for the RNA-seq library preparation and sequencing; Dr. Luca Battistini and Giovanna Borsellino for flow cytometry related discussions and advice; Dr. Libera Berghella for preliminary analysis. Dr. Roberto Rizzi for the support in the in-vivo treatments. We are grateful to Drs Guttridge, Akira and Schenk for sharing the mice generated in their labs. Dr. Constantine Heil for the help in graphic formatting.
References
- 1.Pannérec A, Marazzi G, Sassoon D. Stem cells in the hood: the skeletal muscle niche. Trends Mol Med. 10:599–606. (2012) [DOI] [PubMed] [Google Scholar]
- 2.Farup J, Madaro L, Puri PL & Mikkelsen UR Interactions between muscle stem cells, mesenchymal-derived cells and immune cells in muscle homeostasis, regeneration and disease. Cell Death Dis. 6, e1830 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bentzinger CF, Wang YX, Dumont NA & Rudnicki MA Cellular dynamics in the muscle satellite cell niche. EMBO Rep. 14, 1062–1072 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitchell KJ et al. Identification and characterization of a non-satellite cell muscle resident progenitor during postnatal development. Nat. Cell Biol 12, 257–266 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Collins CA et al. Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell 122, 289–301 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Kang J-S & Krauss RS Muscle stem cells in developmental and regenerative myogenesis. Curr. Opin. Clin. Nutr. Metab. Care 13, 243–248 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tidball JG Mechanisms of muscle injury, repair, and regeneration. Compr. Physiol 1, 2029–2062 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Serrano AL, Muñoz-Cánoves P. Fibrosis development in early-onset muscular dystrophies: Mechanisms and translational implications. Semin Cell Dev Biol. 64:181–190 (2017) [DOI] [PubMed] [Google Scholar]
- 9.Joe AWB et al. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 12, 153–163 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uezumi A, Fukada S, Yamamoto N, Takeda S & Tsuchida K Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol.12, 143–152 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Gonzalez D et al. ALS skeletal muscle shows enhanced TGF-β signaling, fibrosis and induction of fibro/adipogenic progenitor markers. PloS One 12, e0177649 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kharraz Y, Guerra J, Mann CJ, Serrano AL & Muñoz-Cánoves P Macrophage plasticity and the role of inflammation in skeletal muscle repair. Mediators Inflamm. 2013, 491497 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tidball JG Regulation of muscle growth and regeneration by the immune system. Nat. Rev. Immunol 17, 165–178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heredia JE et al. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 153, 376–388 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lemos DR et al. Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF- mediated apoptosis of fibro/adipogenic progenitors. Nat. Med. 21, 786–794 (2015) [DOI] [PubMed] [Google Scholar]
- 16.Mozzetta C et al. Fibroadipogenic progenitors mediate the ability of HDAC inhibitors to promote regeneration in dystrophic muscles of young, but not old Mdx mice. EMBO Mol. Med 5, 626–639 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uezumi A et al. Identification and characterization of PDGFRα+ mesenchymal progenitors in human skeletal muscle. Cell Death Dis. 5, e1186 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saccone V et al. HDAC-regulated myomiRs control BAF60 variant exchange and direct the functional phenotype of fibro-adipogenic progenitors in dystrophic muscles. Genes Dev. 28, 841–857 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sala D, Sacco A. Signal transducer and activator of transcription 3 signaling as a potential target to treat muscle wasting diseases. Curr Opin Clin Nutr Metab Care. 9(3):171–6 (2016. ) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonetto A et al. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PloS One 6, e22538 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonetto A et al. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am. J. Physiol. Endocrinol. Metab. 303, E410–421 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haddad F, Zaldivar F, Cooper DM & Adams GR IL-6-induced skeletal muscle atrophy. J. Appl. Physiol. Bethesda Md 1985 98, 911–917 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Tang H, et al. mTORC1 promotes denervation-induced muscle atrophy through a mechanism involving the activation of FoxO and E3 ubiquitin ligases. Sci Signal. 7(314):ra18 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Moresi V, et al. Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell. 1:35–45 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joassard OR, Bélanger G, Karmouch J, Lunde JA, Shukla AH, Chopard A, Legay C, Jasmin BJ. HuR Mediates Changes in the Stability of AChR β-Subunit mRNAs after Skeletal Muscle Denervation. J Neurosci. 35(31):10949–62. (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le TT, et al. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 14(6):845–57 (2005) [DOI] [PubMed] [Google Scholar]
- 27.Mead RJ et al. Optimised and Rapid Pre-clinical Screening in the SOD1G93A Transgenic Mouse Model of Amyotrophic Lateral Sclerosis (ALS). PLoS ONE 6, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olivan S et al. Comparative study of behavioural tests in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Exp. Anim. 64, 147–153 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tierney MT, Aydogdu T, Sala D, Malecova B, Gatto S, Puri PL, Latella L, Sacco A. STAT3 signaling controls satellite cell expansion and skeletal muscle repair. Nat Med. 20(10):1182–6 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muñoz-Cánoves P, Scheele C, Pedersen BK, Serrano AL. Interleukin-6 myokine signaling in skeletal muscle: a double-edged sword? FEBS J. 280,:4131–48. (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zimmers TA, Fishel ML, Bonetto A. STAT3 in the systemic inflammation of cancer cachexia. Semin Cell Dev Biol. 54:28–41 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Serrano AL, Baeza-Raja B, Perdiguero E, Jardí M, Muñoz-Cánoves P. Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 1:33–44. (2008) [DOI] [PubMed] [Google Scholar]
- 33.Rossi J-F et al. A phase I/II study of siltuximab (CNTO 328), an anti-interleukin-6 monoclonal antibody, in metastatic renal cell cancer. Br. J. Cancer 103, 1154–1162 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Rhee F et al. Siltuximab, a f anti-interleukin-6 monoclonal antibody, for Castleman’s disease. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 28, 3701–3708 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. February 6. doi: 10.1038/nrclinonc.2018.8 (2018) [DOI] [PMC free article] [PubMed] [Google Scholar]
References Associated to Methods Only
- 36.Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S. Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol. 161(9): 4652–60 (1998) [PubMed] [Google Scholar]
- 37.White AT, LaBarge SA, McCurdy CE, Schenk S. Knockout of STAT3 in skeletal muscle does not prevent high-fat diet-induced insulin resistance. Mol Metab. >13;4(8):569–75 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sala D, Ivanova S, Plana N, Ribas V, Duran J, Bach D, Turkseven S, Laville M, Vidal H, Karczewska-Kupczewska M, Kowalska I, Straczkowski M, Testar X, Palacin M, Sandri M, Serrano AL, Zorzano A. Autophagy-regulating TP53INP2 mediates muscle wasting and is repressed in diabetes. J Clin Invest. 124(5):1914–27 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pasut A, Jones AE & Rudnicki MA Isolation and culture of individual myofibers and their satellite cells from adult skeletal muscle. J. Vis. Exp. JoVE e50074 (2013). doi: 10.3791/50074 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data of this study have been deposited in the Sequence Read Archive (SRA) under the accession code SRP14829. Source data of Figures 2b-2d, 3f-3h, 4g-4i are provided in Supplementary Table 1 .All other data supporting the findings of this study are available from the corresponding author on reasonable request.