

Abstract
Mitoxantrone (MTO) is a topoisomerase II inhibitor which has been used to treat various forms of cancer either as solo chemotherapy regimen or as component in cocktail treatments. However, as with other anti-neoplastic agents, MTO has severe cardiac side effects. Therefore, a drug delivery approach holds promise to improve safety and applicability of this chemotherapy. Here, we report the application of a plant virus-based nanotechnology derived from tobacco mosaic virus (TMV) as a delivery vehicle for MTO towards cancer therapy. TMV is a high aspect-ratio, soft-matter nanotube with dimensions of 300×18 nm and a 4-nm wide channel. The surface chemistry of the interior and exterior surfaces is distinct and we established charge-driven drug loading mechanisms to accommodate therapeutics inside the channel for drug delivery. We demonstrate effective MTO loading into TMV yielding ~ 1,000 MTO per TMV carrier. Treatment efficacy of MTO-loaded TMV (MTOTMV) was assessed in in-vitro and in-vivo models. In-vitro testing confirmed that MTO maintained its efficacy when delivered by TMV in a panel of cancer cell lines. Drug delivery in-vivo using a mouse model of triple negative breast cancer demonstrated superior efficacy of TMV-delivered MTO vs. free MTO. This study demonstrates the potential of plant virus-based nanotechnology for cancer therapy and drug delivery.
Keywords: Tobacco mosaic virus, drug delivery, mitoxantrone, cancer therapy, nanotechnology
TOC
Introduction
Mitoxantrone (MTO) is a clinically approved chemotherapeutic used to treat metastatic breast cancer, advanced prostate cancer, as well as forms of leukemia and lymphoma.1 It is frequently used as cocktail with other chemotherapeutics (e.g. doxorubicin, vincristine, prednisone). MTO is a topoisomerase II inhibitor, and as such interferes will the cell cycle leading to apoptosis. While a potent anti-cancer therapeutic, MTO confers intense toxicity when delivered systemically, particularly to the heart. Dose-dependent cardiotoxicity of MTO results in the reduction of left ventricular ejection fraction along with congestive heart failure.2 These side effects limit its clinical utility. To bypass these severe cardiac side effects, we proposed the use of plant-virus based nanoparticles as delivery vehicles for MTO.
Plant-virus based nanotechnologies provide an exciting alternative to the more traditional and more frequently exploited synthetic nanoparticles.3 Plant viruses, or viruses in general, can be considered as nature’s delivery vehicles; viruses are designed to penetrate cells and deliver cargo. While mammalian viruses have been used to deliver genes for nucleic acid therapy,4 plant viruses offer a safer alternative due to their inability to infect or replicate in mammalian cells. Like other biologics, plant virus-based nanoparticles can be manufactured through a variety of homologous and heterologous expression systems at high yields and with high quality control and assurance.3, 5 Plant viruses are monodisperse and many of their structures known to near atomic resolution; therefore enabling structure-based design of high precision nanodrug delivery systems.6
In this work, we investigated the use of tobacco mosaic virus (TMV) as a drug delivery vehicle for mitoxantrone (MTO); efficacy of the plant viral drug delivery approach was evaluated in in-vitro and in-vivo tumor models. TMV is a soft matter, hollow nanotube measuring 300×18 nm with a 4-nm wide channel. This high aspect-ratio (AR=17) is advantageous when used in the context of drug delivery to tumors, because elongated materials have superior biodistribution and tumor homing compared to spherical particles.7, 8 Furthermore, our previous biodistribution studies have demonstrated that TMV does not accumulate in the heart when injected intravenously;9, 10 this is of importance because a major limitation of MTO is its cardiotoxicity. Another positive attribute of high aspect ratio nanoparticles is that they excel in immune system evasion,8 which reduces non-specific deposition in phagocytes and may help avoid or reduce potential immune responses against the carrier. Besides its shape-related attributes, the structure of TMV is known and its chemistry well understood.11–13 TMV offers chemically distinct interior and exterior surfaces, therefore allowing structure-based engineering of designer nanoparticles. Here, we developed a drug delivery protocol to encapsulate MTO into TMV; we report drug loading and release as well as its anti-tumor effects in various cell lines in-vitro. Finally we demonstrate superior efficacy of TMV-delivered MTO vs. free MTO in a murine model of triple negative breast cancer.
Experimental
Synthesis of MTOTMV.
T158K mutant of TMV (in the following referred to as TMV) was produced by mechanical inoculation of Nicotiana benthamiana plants and established purification protocols.14 1 mg ml−1 of TMV was mixed in the dark with 10,000-fold molar excess of mitoxantrone (Sigma-Aldrich, MTO) in 10 mM potassium phosphate (KP) buffer at pH 7.4 for 18 hours. The reaction mix was purified by centrifugation at 112,000 g for 1 hour over a 40% (w/v) sucrose cushion to remove any excess of free MTO. The resulting MTOTMV pellet was resuspended in 10 mM KP buffer pH 7.4 and further purified by centrifugation at 16,000 g for 10 minutes to remove any particle aggregates. Finally, the MTOTMV were eluted through a GE Healthcare PD Minitrap G-25 column to remove any remaining free MTO. UV-visible spectroscopy (UV-vis) was used to determine the respective concentration of TMV and MTO. Samples were also analyzed by transmission electron microscopy (TEM) and size exclusion chromatography (SEC) to confirm particle monodispersity and integrity.
UV-Vis spectroscopy.
The UV-vis Spectra of TMV and MTOTMV were analyzed with a NanoDrop spectrophotometer (Thermo Scientific). The molar ratio of MTO loading was determined by comparing the ratio of MTO:TMV coat protein concentration, which was determined by analyzing their respective absorbance and Beer-Lambert law. The extinction coefficients of TMV and MTO are as follows: TMV ε(260 nm) = 3 mL.mg−1cm−1, MTO ε(622 nm) = 25,000 M−1cm-1. The molecular weight of TMV and MTO are 39.4 × 106 g mol−1 and 514.71 g mol−1 respectively. It should be noted that because MTO also absorbs at 260 nm, the MTO contribution at 260 nm is therefore subtracted when determining the TMV concentration.
Size exclusion chromatography (SEC) by fast protein liquid chromatography (FPLC).
Samples (200 µL at 1 mg ml−1 of protein) were eluted through a Superose6 column on the ÄKTA Explorer chromatography system (GE Healthcare) using a flow rate of 0.5 mL/min in 10 mM KP pH 7.4. The absorbance at 260 (TMV RNA), 280 (TMV protein), and 622 nm (MTO) was recorded.
Transmission electron microscopy (TEM).
A 20 µL drop of TMV or MTOTMV at 1 mg ml−1 protein concentration was added to Formvar carbon film coated copper TEM grids (FCF400-CU, Electron Microscopy Sciences) for 2 min at room temperature. After two washing steps with deionized water, the grids were stained twice with 2% (w/v) uranyl acetate in deionized water for 45 s. A Tecnai F30 transmission electron microscope was used to image the prepared samples at 300 kV.
MTO drug release from MTOTMV.
MTOTMV formulations (500 μL, 1 mg ml−1) were placed in Slide-A-Lyzer MINI dialysis units (69570, Fisher) and dialyzed against 2 l of 10 mM KP buffer pH 5.0 and pH 7.4 over 72 hours and at 4°C or room temperature (RT). 10 μL samples were removed at t = 12, 16, 24, 48, and 72 hours after the start of dialysis, and analyzed by UV-vis spectroscopy to quantify the percent of MTO released from MTOTMV. Drug release was calculated by comparing the remaining drug in the particle solution to the initial drug concentration.
MTOTMV vs. MTO cell uptake.
Cell uptake of MTOTMV vs. MTO was assessed using MDA-MB-231 cells (triple negative breast cancer), HT1080 cells (fibrosarcoma), and PC-3 cells (prostate cancer). Cell lines were obtained from ATCC. MDA-MB-231 and HT1080 cells were cultured in high glucose Dulbecco’s modified Eagle medium (DMEM) with 4 mM L-glutamine (Fisher). PC-3 cells were cultured in Rosewell Park Memorial Institute (RPMI) 1640 medium. All media were supplemented with 10% (v/v) FBS and 1% (v/v) penicillin-streptomycin. Cells were grown to confluency at 37°C and 5% CO2. Cells were seeded into an untreated V-bottomed 96-well plate at 250,000 cells per well in 200 µL of media. Triplicates of MTO or MTOTMV were added at a concentration of 100,000 particles/cell and incubated for 16 hours at 37°C, 5% CO2. Cells were then washed with 7.4 pH PBS containing 5% (v/v) FBS and 0.1% (w/v) sodium azide, and then fixed with 2% (v/v) paraformaldehyde 7.4 pH PBS for 15 minutes. Cells were sorted by fluorescence using a R660 filter (405 nm Em / 660 nm Ex) and a Accuri C6 Flow Cytometer. All experiments were carried out at least twice and triplicate samples were analyzed using FlowJo software.
MTOTMV vs. MTO cytotoxicity.
Cell toxicity was evaluated using the MTT assay (ATCC) and MDA-MB-231, HT1080, and PC-3 cell lines. Cells were exposed to free MTO, MTOTMV, TMV, and PBS for 24 hours in culture medium at 37°C and 5% CO2; MTO concentrations ranged from 10 µM to 100 pM with increments of factor 10; TMV concentration were matched to MTOTMV. The assay was performed as per manufacturer’s recommendation; a BioTek Synergy HT multidetection microplate reader was used for read-out.
MTOTMV vs. MTO therapy using the MDA-MB-231 mouse model of triple negative breast cancer.
All animal studies were performed according to Case Western Reserve University’s Institutional Animal Care and Use Committee-approved procedures. Female NCR nu/nu mice were injected subcutaneously into the right flank using 2 × 106 MDA-MB-231 cells suspended in 100 µL of media and Matrigel (Corning) at a 1:1 ratio. Once established, tumors were monitored every other day, with total tumor volume calculated using the formula , where l is the length and w the width of the tumor. Treatment injections were started when tumors reached a volume of 100 mm3. Groups of n=5 animals were treated with MTO, MTOTMV, TMV, and PBS. Treatment was delivered via IV injection at 1 mg kg−1 normalized to MTO on days 1, 5, and 10. Injection volumes did not exceed 300 µL.
Results and Discussion
MTOTMV synthesis and characterization
TMV was propagated in and purified from Nicotiana benthamiana plants at yields of up to 500 mg pure TMV per 100 gram of infected leaf tissue. TMV consists of 2,130 identical coat proteins (made of 158 amino acids) arranged in a helical structure around the single-stranded viral RNA. TMV virions form a cylindrical structure, its inner surface is lined with 4,260 solvent-exposed glutamic acid residues (Glu 97 and 106).11–13 These exposed carboxylic groups provide a negatively charged environment, which we have previously exploited for the encapsulation of positively charged therapeutics, such as chemotherapies, photosensitizers, and pestidcides.10, 13, 15–17 MTO in its native state contains a +2 charge (Figure 1) and measures approximately 1.4 nm across its longest axis; therefore MTO has attributes making it an attractive candidate to be encapsulated into TMV via charge-driven interactions. To achieve this, MTO and TMV were mixed at a 10,000:1 MTO:TMV ratio overnight in 10 mM potassium phosphate (KP) buffer at pH 7.4. Following purification to remove any excess and MTO, the resulting MTO-loaded TMV particles, denoted MTOTMV, were analyzed to confirm their structural integrity and assess drug loading and release (Figure 2).
Figure 1:

MTO loading into TMV. TMV forms a 300 × 18 nm nanorod (left), with a 4 mm-wide channel lined with glutamic acids. The negative charge of the glutamic acids allows for electrostatic interactions with the positively charged MTO, allowing for pH dependent drug -loading and -release.
Figure 2:

MTOTMV particle characterization. FPLC using a Superose6 column and ÄKTA purifier (A, detectors: 260 nm for TMV’s RNA, 280 nm for TMV’s protein, and 622 nm for MTO) and TEM of negative-stained samples (C) were used to confirm MTOTMV particle integrity. Drug loading was determined using UV-Vis spectroscopy (B), with absorbance peaks at 260 and 280 nm corresponding to TMV, and 622 nm corresponding to MTO. Drug release (D) was performed via dialysis in varying buffer conditions, with samples removed at designated time-points for analysis and quantification of MTO content per TMV by UV-Vis.
Size exclusion chromatography (SEC) by fast liquid protein chromatography (FPLC) and transmission electron microscopy (TEM) confirmed that MTOTMV particles maintained their structural integrity after MTO loading (Figure 2A+C). TEM imaging shows high aspect ratio nanorods. It should be noted that while native TMV measures 300 nm, TEM imaging typically shows a distribution of particle lengths which likely is an artifact from sample preparation leading to broken or fragmented particles; however, no differences were noted between TMV and MTOTMV indicating stability of the MTOTMV formulation (Figure 2C). The FPLC elution profile also shows the typical TMV profile with elution from the Superose6 column at ~ 8 mLs; the ratio of 260:280 of 1.2 is indicative of intact TMV and overlap of the 622 nm peak with the 260/280 nm peaks indicates co-elution of MTO with TMV; free MTO was not detected in the preparation (Figure 2A).
MTO drug loading into TMV was then quantified by UV-Vis spectroscopy (Figure 2B) using Beer Lambert Law and the TMV and MTO-specific extinction coefficients; we determined the loading with approximately ~1,000 MTO molecules per TMV particle. The degree of drug loading is comparable to other TMV-drug delivery systems that we previously described. For example, we reported similar degree of drug loading using a porphyrin-based photosensitizer.17 It should be noted that with platinum drugs, using either cisplatin or its monofunctional derivatives phenanthriplatin, loading with up to 2,000 drugs per TMV could be achieved.10, 16 A recent structure-function study using a distinct set of phenanthriplatin analogs indicates that the net charge of phenanthriplatin analogs and their ionic mobilities have no effect on loading – however an increased number of heteroaromatic rings of the platinum ligand appears to enhance loading efficiency, possibly by stabilizing the hydrophobic interactions and stacking inside the TMV channel.13 MTO is known to interact and bind to proteins via hydrophobic interactions;18, 19 therefore we propose that MTO-TMV drug loading is driven by a combination of charge interactions and hydrophobic stacking. In fact, the possibility exists that MTO is not exclusively bound to the interior channel, but could also be bound to the exterior surface of the particle through non-specific drug-protein interactions.
Lastly, we assessed the release rate of MTO from TMV using dialysis against KP buffer pH 5.0 vs. 7.4 corresponding to the acidic tumor microenvironment and physiological conditions. 20 We also considered testing at 4°C and 22°C to assess stability under storage conditions in the fridge or room temperature. While temperature only had a modest effect on drug release; faster drug release rates were observed under acidic conditions (t1/2 ~ 7–8 hours) vs. physiologic pH 7.4 (t1/2 ~ 13–25 hours). Only testing at pH 5.0 achieved complete drug release post 24 hours; at pH 7.4 ~ 40% of the drug remained associated with TMV post 72 hours; a plateau is established after ~2 days thus indicating that longer incubation periods would not achieve further drug release at pH 7.4. The drug release profile of MTOTMV is similar to other nanoparticle-MTO formulations, e.g. mesoporous silica nanorods.23 Increased stability under physiologic conditions, e.g. during circulation (pH 7.4), and increased drug release at lower pH (pH 5.0) is expected and desired: under acidic conditions, the carboxylic acid will be protonated thus weakening the charge interactions with MTO triggering its release. This pH dependent release provides favorable conditions for delivery of MTO to the tumor microenvironment and its cells. Tumors generally exhibit acid microenvironments;21 in particular, more aggressive forms of breast cancer such as MDA-MB-231 are known to acidify their surroundings more actively compared to both healthy cells.20 Furthermore, we previously demonstrated that TMV is taken up by cancer cells including MDA-MB-231 cells. Upon cell uptake, TMV traffics to the endolysosomal compartment where the drug cargo is released and the protein carrier degraded by hydrolysis and proteolysis.22
Compared to covalent drug loading strategies, we find the non-covalent drug loading to be more efficient: for example, we have previously demonstrated that doxorubicin can be covalently attached to the interior glutamic acids of TMV using carbodiimide chemistries; however, the maximum drug loading capacity was found to be 270 drug molecules per particle,24 which is roughly 4x lower than our strategy described here. Furthermore, the drug release mechanism of non-covalent drug delivery systems is favorable as it is markedly faster than drug release in covalently-bound drugs, where the rate-limiting factor for release is the degradation of the particle via hydrolase and protease activity.22 Given the increased stability of the MTOTMV complex at pH 7.4, mimicking conditions the particles experience during circulation, with t1/2 between 13–25 hours, and the rather short circulation half-life of TMV (on the order of minutes9), we hypothesize that the non-specific drug release during systemic administration would be minimal.
MTOTMV cell uptake and cytotoxicity
In vitro efficacy of MTOTMV vs. MTO was assessed in a panel of cancer cell lines, including MDA-MB-231 (triple negative breast cancer), HT1080 cells (fibrosarcoma), and PC3 (prostate cancer). The results are consistent in all three cells lines and indicate that MTOTMV retains comparable efficacy compared to its free drug counterpart; IC50 values of the free MTO treatment for MDA-MB-231, HT1080, and PC3 cells were 575±45, 169±13, and 713±80 nM respectively. For the MTOTMV treatment, IC50 values were 641±81, 450±42, and 472±42 nM respectively (Figure 3B). At the maximum MTO concentrations tested (10 µM), cell viability was suppressed to roughly 20%. Cell uptake of MTOTMV vs. MTO was assessed using flow cytometry taking advantage of the drug’s natural fluorescence (Ex/Em 607/684 nm).25 In each cell line, MTOTMV showed improved cellular uptake compared to MTO. This is reflected by an increase in mean fluorescence intensity (Figure 3C+D). The increase in cell uptake however does not reflect an increase in cell killing efficiency of the MTOTMV formulation compared to free MTO – this may be explained by slower metabolism of the drug by the tumor cells, due to the added barrier of MTO encapsulation.26
Figure 3:

(A+B) Cell viability as a function of MTO concentration comparing free MTO and MTOTMV using the MTT assay. IC50 values of free MTO and MTOTMV against MDA-MB-231, HT1080, and PC3 cells. (C+D) Cellular uptake of free MTO and MTOTMV was quantified by flow cytometry (FACS). Histograms are shown in C and corresponding mean fluorescence intensities in D. Stats: Experiments were done in triplicates and repeated at least twice; mean values and standard deviations are shown.
Our data are consistent with other reports; drug efficacy is cell line dependent and MTO appears to be more effective in triple negative breast cancer models compared to HER2+ breast cancer subtypes: for example, liposomal MTO and free MTO exhibited IC50 values of 1.25 and 2.13 μM for MTO and encapsulated MTO respectively when delivered to HER2+ MCF-7 breast tumor cells.27 On the other hand, IC50 values for mesoporous silica nanorods loaded with MTO vs. free MTO against triple negative breast cancer cells MDA-MB-231 lied at 548 and 966 nM, respectively.23 The latter is in good agreement with our studies. We therefore chose the triple negative model for investigation of drug efficacy using the TMV delivery approach in-vivo.
In vivo drug delivery using MTOTMV in a mouse model of triple negative breast cancer
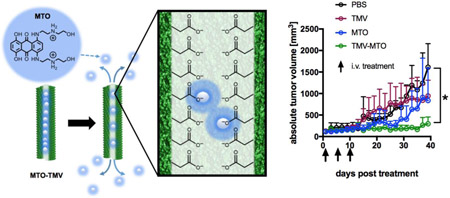
The efficacy of MTOTMV vs. free MTO was assessed in a mouse model of triple negative breast cancer, where MDA-MB-231 xenografts were induced into the subcutaneous space of the right flank of NCR nu/nu mice. Treatment was started when tumors reached 100 mm3; the treatment schedule comprised three treatments every 5 days of PBS (control), TMV (control), free MTO and MTOTMV at a dose of 1 mg kg−1 normalized to MTO (groups were assigned randomly with n=5). Each treatment of MTOTMV was prepared fresh the day of treatment to ensure maximum drug loading and avoiding premature release during extended periods of storage. Tumor burden was measured every other day as a function of tumor volume.
The in-vivo drug delivery study demonstrated that tumor growth rates were significantly suppressed when animals were treated using the MTOTMV formulation: at the endpoint, which was defined as the timepoint when all PBS-control animals had to be sacrificed based on tumor burden (40 days post first treatment), animals treated with MTOTMV exhibited tumors 5.4x smaller than control tumors (297 mm3 vs. 1,610 mm3 for MTOTMV vs. PBS, p < 0.0005). There was no statistical significance comparing the PBS vs. TMV vs. MTO groups (Figure 4).
Figure 4:

MTOTMV vs. free MTO treatment using a mouse model of triple negative breast cancer (MDA-MB-231 s.c. xenografts in NRCnu/nu mice, n=5). Treatments (PBS, TMV, MTO, MTOTMV) were given on days 1, 5, 10 at a dosage of 1 mg kg−1 MTO; TMV was normalized to the equivalent amount of TMV in MTOTMV.
The enhanced tumor efficacy of MTOTMV vs. free MTO may be explained by the favorable biodistribution of TMV vs. free MTO. In our previous study using phenanthriplatin-loaded TMV and the same MDA-MB-231 mouse model, we found that the amount of drug within the tumors tissue when delivered by TMV was increased by ~10-fold compared to drug administered systemically.10 Furthermore data from our previous biodistribution studies indicate that besides tumor accumulation, TMV is cleared through the liver and spleen with no detectable accumulation in the heart.8–10 This biodistribution profile matches other nanotechnologies and is one of the attributes that makes nanocarriers potentially powerful platforms for cancer therapy enabling safer administration of chemotherapy regimens that otherwise would lead to cardiotoxicity.28
Conclusion
In this work we report the encapsulation and delivery of the chemotherapy MTO using the plant virus-based nanotechnology derived from TMV. We demonstrate efficient drug loading achieving a load of 1,000 MTO per TMV particle. MTOTMV particles maintained their efficacy with comparable IC50 values to free MTO determined using a panel of cancer cell lines in-vitro. Most importantly we demonstrate increased efficacy of TMV-delivered MTO vs. free MTO in a mouse model of triple negative breast cancer. Nanotechnology holds great promise in cancer drug delivery with a myriad of nanotechnologies under development, both of synthetic and biological nature. While there is likely no magic bullet to cure cancer, it is critical to fuel the field of cancer nanotechnology with novel concepts, discussion and possible drug delivery platforms. Our results highlight the potential for plant viral nanoparticles in cancer therapy.
Acknowledgements
This work was funded by a Research Scholar Award from the American Cancer Society (RSG-15–144-01-CDD) to N.F.S. and a NIH Training grant (T32EB007509) to R.D.L. We thank Paul L. Chariou (CWRU) for assistance with TEM.
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.Evison BJ, Sleebs BE, Watson KG, Phillips DR and Cutts SM, Med Res Rev, 2016, 36, 248–299. [DOI] [PubMed] [Google Scholar]
- 2.Paul F, Dörr J, Würfel J, Vogel HP and Zipp F, Journal of Neurology, Neurosurgery, and Psychiatry, 2007, 78, 198–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wen AM and Steinmetz NF, Chem Soc Rev, 2016, 45, 4074–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vannucci L, Lai M, Chiuppesi F, Ceccherini-Nelli L and Pistello M, New Microbiol, 2013, 36, 1–22. [PubMed] [Google Scholar]
- 5.Hefferon K, Biomedicines, 2017, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steinmetz NF, Lin T, Lomonossoff GP and Johnson JE, Curr Top Microbiol Immunol, 2009, 327, 23–58. [DOI] [PubMed] [Google Scholar]
- 7.Shukla S, Ablack AL, Wen AM, Lee KL, Lewis JD and Steinmetz NF, Mol Pharm, 2013, 10, 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shukla S, Eber FJ, Nagarajan AS, DiFranco NA, Schmidt N, Wen AM, Eiben S, Twyman RM, Wege C and Steinmetz NF, Advanced healthcare materials, 2015, 4, 874–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruckman MA, Randolph LN, Vanmeter A, Hern S, Shoffstall AJ, Taurog RE and Steinmetz NF, Virology, 2014, 449, 163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Czapar AE, Zheng YR, Riddell IA, Shukla S, Awuah SG, Lippard SJ and Steinmetz NF, ACS Nano, 2016, 10, 4119–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Namba K and Stubbs G, Science (New York, N.Y, 1986, 231, 1401–1406. [DOI] [PubMed] [Google Scholar]
- 12.Schlick TL, Ding Z, Kovacs EW and Francis MB, J Am Chem Soc, 2005, 127, 3718–3723. [DOI] [PubMed] [Google Scholar]
- 13.Vernekar AA, Berger G, Czapar AE, Veliz FA, Wang DI, Steinmetz NF and Lippard SJ, J Am Chem Soc, 2018, 140, 4279–4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bruckman MA and Steinmetz NF, Methods in molecular biology, 2014, 1108, 173–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chariou PL and Steinmetz NF, ACS Nano, 2017, 11, 4719–4730. [DOI] [PubMed] [Google Scholar]
- 16.Franke CE, Czapar AE, Patel RB and Steinmetz NF, Mol Pharm, 2017, DOI: 10.1021/acs.molpharmaceut.7b00466. [DOI] [PubMed] [Google Scholar]
- 17.Lee KL, Carpenter BL, Wen AM, Ghiladi RA and Steinmetz NF, ACS Biomater Sci Eng, 2016, 2, 838–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chassany O, Urien S, Claudepierre P, Bastian G and Tillement JP, Cancer Chemother Pharmacol, 1996, 38, 571–573. [DOI] [PubMed] [Google Scholar]
- 19.Khan SN, Islam B, Yennamalli R, Sultan A, Subbarao N and Khan AU, Eur J Pharm Sci, 2008, 35, 371–382. [DOI] [PubMed] [Google Scholar]
- 20.Montcourrier ISP, Farnoud R, Bird I, Rochefort H, Clin Exp Metastasis, 1997, 15, 382–392. [DOI] [PubMed] [Google Scholar]
- 21.Liu J, Huang Y, Kumar A, Tan A, Jin S, Mozhi A and Liang XJ, Biotechnol Adv, 2014, 32, 693–710. [DOI] [PubMed] [Google Scholar]
- 22.Wen AM, Infusino M, De Luca A, Kernan DL, Czapar AE, Strangi G and Steinmetz NF, Bioconjugate chemistry, 2015, 26, 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wani A, Savithra GHL, Abyad A, Kanvinde S, Li J, Brock S and Oupický D, Scientific Reports, 2017, 7, 2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bruckman MA, Czapar AE, VanMeter A, Randolph LN and Steinmetz NF, J Control Release, 2016, 231, 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Consoli U, Van NT, Neamati N, Mahadevia R, Beran M, Zhao S and Andreeff M, Leukemia, 1997, 11, 2066–2074. [DOI] [PubMed] [Google Scholar]
- 26.Chourasiya V, Bohrey S and Pandey A, Materials Discovery, 2016, 5, 1–13. [Google Scholar]
- 27.Zhuang Y-G, Xu B, Huang F, Wu J-J and Chen S, Solid lipid nanoparticles of anticancer drugs against MCF-7 cell line and a murine breast cancer model, 2012. [PubMed] [Google Scholar]
- 28.Heath JR and Davis ME, Annu Rev Med, 2008, 59, 251–265. [DOI] [PMC free article] [PubMed] [Google Scholar]