

Abstract
Thioester reductase domains catalyze two- and four-electron reductions to release natural products following assembly on nonribosomal peptide synthetases, polyketide synthases, and their hybrid biosynthetic complexes. This reductive off-loading of a natural product yields an aldehyde or alcohol, can initiate the formation of a macrocyclic imine, and contributes to important intermediates in a variety of biosyntheses, including those for polyketide alkaloids and pyrrolobenzodiazepines. Compounds that arise from reductase-terminated biosynthetic gene clusters are often reactive and exhibit biological activity. Biomedically important examples include the cancer therapeutic Yondelis (ecteinascidin 743), peptide aldehydes that inspired the first therapeutic proteasome inhibitor bortezomib, and numerous synthetic derivatives and antibody drug conjugates of the pyrrolobenzodiazepines. Recent advances in microbial genomics, metabolomics, bioinformatics, and reactivity-based labeling have facilitated the detection of these compounds for targeted isolation. Herein, we summarize known natural products arising from this important category, highlighting their occurrence in Nature, biosyntheses, biological activities, and the technologies used for their detection and identification. Additionally, we review publicly available genomic data to highlight the remaining potential for novel reductively tailored compounds and drug leads from microorganisms. This thorough retrospecive highlights various molecularfamilies with especially privileged bioactivity while illuminating challenges and prospects toward accelerating the discovery of new, high value natural products.
For TOC Only
Thioester reductases arm natural products, like the peptide aldehydes and the anti-cancer drug Yondelis, with unique structures and bioactivity.
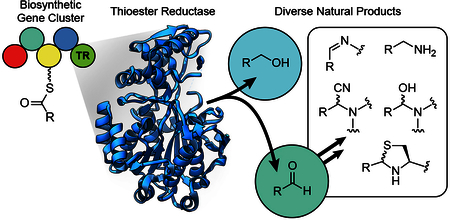
1. Introduction
Nonribosomal peptide synthetase (NRPS) and polyketide synthase (PKS) gene clusters encode for the production of nonribosomal peptides and polyketides, the most abundant classes of microbial secondary metabolites.1 Both classes and their hybrids represent immense structural diversity with a wide range of biological activities and utility as pesticides, antibiotics, immunosuppressives, toxins, siderophores, cytostatics, and antiproliferative agents.2 Notable examples such as penicillin G, daptomycin, rimocidin, vancomycin, and actinomycin are among the most clinically relevant antifungal, antibacterial, and anticancer medicines currently in use.3 Lovastatin, a polyketide isolated from the fungus Aspergillus terreus, serves as the basis for the statins, one of the most commercially successful and frequently-prescribed classes of drugs.4,5
1.1. NRPS and PKS Biosynthetic Pathways
NRPS and PKS enzymes are responsible for most microbial natural products (NPs) and are among the largest proteins in bacteria and fungi, with molecular weights in the megadaltons. This biosynthetic machinery has been described extensively elsewhere;2,6–8 what follows is a brief overview.
Biosynthetic processes for both classes follow a similar thiothemplate mechanism, with NRPSs most commonly catalyzing the polymerization of the 20 proteinogenic amino acids, as well as hundreds of less common residues such as D- amino acids, N-terminal fatty acyl chains, and non-canonical amino acids, all commonly modified by various dehydrations, methylations, hydroxylations, acetylations, phosphorylations, glycosylations, halogenations, cyclizations, and other enhancements. Monomer incorporation by PKSs involves the polymerization of acetyl, malonyl, or methylmalonyl units, with a suite of modifications analogous to that of NRPS systems (Fig. 1). These two classes can be hybridized with one another and with other biosynthetic systems (terpene, lanthipeptide, shikimate, etc.) to yield additional diversity. Together, the assembled monomers and their structural diversifications coalesce to yield molecules with the necessary biochemical properties for interacting with distinct biological targets.
Fig. 1.
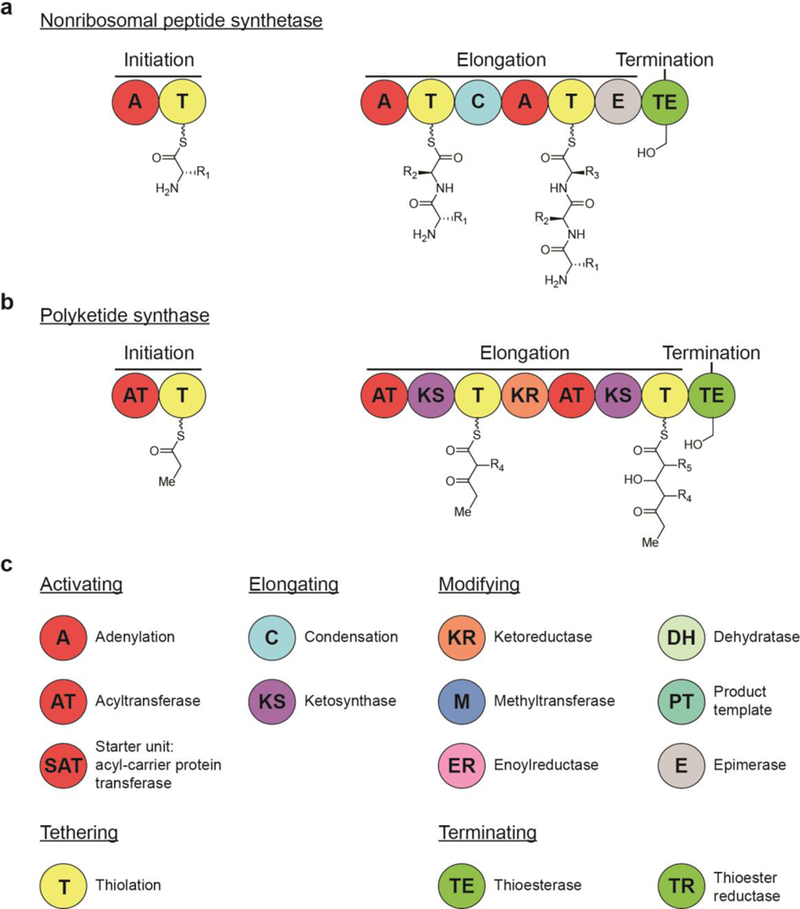
Logic of natural product thiothemplate biosynthesis: (a) Basic steps for NRPS biosynthesis of nonribosomal peptides. (b) Basic steps for PKS biosynthesis of polyketides. (c) A key to the most common domains in NRPS and PKS biosynthesis, organized based on function.
All NRPS and PKS megacomplexes consist of modules for substrate initiation, chain extension, and termination that are further divided into domains that perform each catalytic step on the growing substrate (Fig. 1). A typical NRPS module contains an adenylation domain (A) that recognizes and activates an amino acid monomer as an adenylate followed by acyl transfer to a peptidyl carrier protein (PCP; known more generally as the thiolation domain, T). This thiolation domain loads the activated amino acid on a 4’-phosphopantetheine (4’- Ppant) arm, tethering it for peptide bond formation with an amino acid on the subsequent module, a reaction that is catalyzed by a condensation domain. PKS modules are organized similarly, with an acyltransferase (AT) that activates an acetyl-CoA, malonyl-CoA, or methylmalonyl-CoA monomer for tethering to the 4’-Ppant arm of an acyl carrier protein (ACP; often referred to with the same name as its NRPS counterpart, the thiolation domain, T). A subsequent ketosynthase (KS) transiently binds the monomer to catalyze a Claisen condensation.2
1.2. Termination and release of substrate from NRPS and PKS pathways
Typically, a thioesterase (TE) domain terminates NRPS- and PKS- containing biosynthetic systems, catalyzing either hydrolysis that leads to the formation of a C-terminal carboxylic acid or cyclization to a macrolactone or macrolactam.9 Alternatively, a thioester reductase (TR) domain performs an alternative termination step by catalyzing an NAD(P)H-dependent two- or four-electron reductive release of a 4’-Ppant-tethered thioester peptide or polyketide chain to afford an aldehyde or an alcohol (Fig. 2).10
Fig. 2.
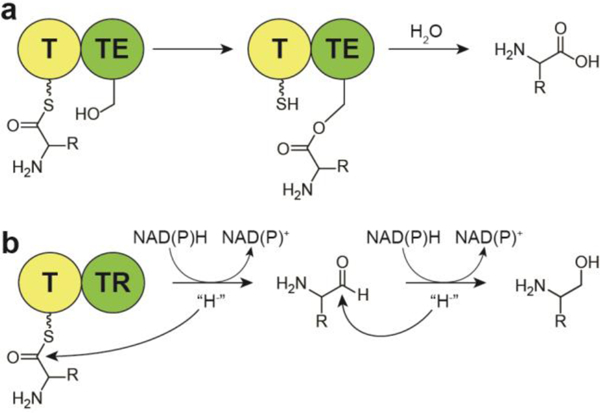
Mechanisms for the termination of multi-modular biosynthetic pathways, with NRPSs as the example, (a) Thioesterase mediated release commonly leads to carboxylic acids, while (b) reductive release can lead to two-electron reduction to an aldehyde or four-electron reduction to an alcohol. T, acyl carrier protein (ACP) or peptidyl carrier protein (PCP) thiolation domains; TE, thioesterase domain; Red, thioester reductase domain.
2. Thioester reductase domains
In NRPS and PKS pathways, thioester reductase (TR) domains occur at the terminal position of a biosynthetic module and catalyze the NAD(P)H-dependent reductive release of acyl-S- polyketides or peptidyl-S-nonribosomal peptides. These domains possess cofactor binding and catalytic motif similarities to the short-chain dehydrogenase/reductase (SDR) superfamily, whose members’ most conserved feature is a characteristic N-terminal α/β Rossmann fold (six to seven parallel β-strands flanked by three to four α-helices).11 This structural motif is responsible for binding nucleotide-based enzyme cofactors (N AD (P)(H)) for a tyrosine-dependent oxidoreductase mechanism of catalysis. The SDR family of NAD(P)H-dependent oxidoreductases play central roles in metabolism of amino acids, lipids, carbohydrates, cofactors, hormones, and xenobiotics as well as in redox sensing.11
Biosynthetic gene clusters (BGCs) containing TR domains often adorn their substrates with electrophilic warheads responsible for bioactivity.2,9 The majority of NPs that arise from reductase- terminated assembly lines are bioactive in commonly used assays, with many showing antiproliferative and antitumor activity. Examples that will be discussed in later sections include the nostocyclopeptides,12,13 scytonemide A,14 anthramycin,15,16 peptide aldehyde NPs,17–20 and the important clinical oncology drug ET-743 (Yondelis).21
2.1. Structure and mechanism of thioester reductase domains
In 2012, Chhabra and coworkers solved the crystal structure of a mycobacterial NRPS TR domain (Fig. 3), specifically demonstrating that it performs two consecutive, two-electron reductions to release a thioester-bound lipopeptide as the corresponding alcohol, using a non-processive mechanism.22 The overall enzyme structure is composed of a 144 amino acid C-terminal domain intertwined with a 282 amino acid N- terminal domain that has an extended α/β Rossmann fold with seven parallel beta strands β2–6 and β8–10). The Rossmann fold is necessary for the binding of NAD(P)H which provides the hydride source for tyrosine-dependent reductive release of the substrate at the thioester. The crystal structure is consistent with a four-electron, non-processive mechanism. Displacement of a committed aldehyde intermediate generated in the first reduction step is necessary to accommodate a second equivalent of NADPH cofactor for the second reduction to the alcohol, as the NADPH binding site is physically obstructed by the bound intermediate. This second reduction to the alcohol is approximately 15-fold more efficient than the first reduction of the thioester-bound substrate to the initially formed aldehyde. An additional study by the same group illuminated a “concerted loop movement model” cofactor binding mechanism mediated by intermodular communication between the TR domain and the preceding T domain, which distinguishes TR domains from the canonical single domain SDR family of enzymes.23 In this model, the absence of a fully mature product at the T domain limits flexible gating and catalytic loop motifs of the reductase domain to sampling of conformations that prevent compatibility with NADPH binding. Only upon formation of a fully mature product on the 4’-Ppant arm of the T domain does a re-orientation of the covalent T-TR linker region occur that triggers a coordinated transition of the gating and catalytic loops from the apo to the NADPH-bound state.
Fig. 3.
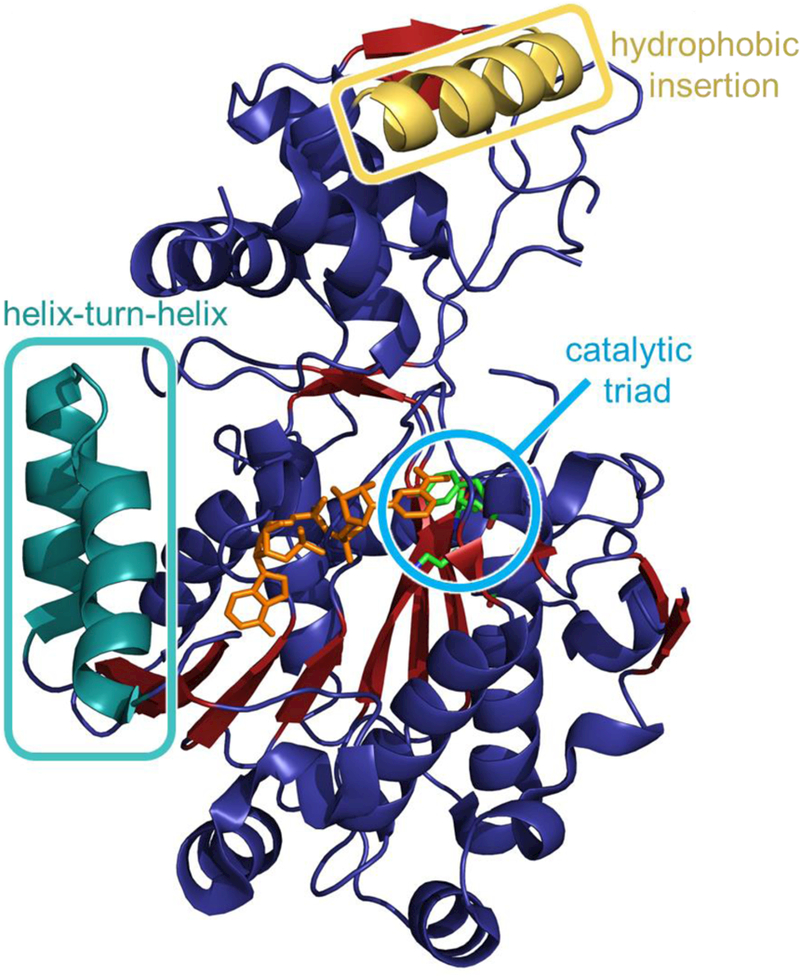
Crystal structure of a reductase domain isolated from a Mycobacterium tuberculosis NRPS pathway (PDB ID 1W6U). The catalytic triad of threonine, tyrosine, and lysine are shown in the cyan circle. NADPH is orange and present near the active site. The hydrophobic insertion region and helix-turn-helix motif common to reductase domains are shown. Adapted from Chhabra et a I., Proc. Natl. Acad. Sci., 2012.22
In 2015, high-resolution structures of the NRPS TR domain of the myxalamid biosynthetic pathway were established with and without bound NADPH (PDB ID 4U7W).24 The gross structural motifs were in alignment with those from the aforementioned mycobacterial reductase, including the canonical SDR tyrosine- dependent catalytic triad. Computational and biochemical characterization revealed a highly flexible C-terminal sub- domain that ceases movement upon selective binding of the myxalamid intermediate, while mutational analysis confirmed the distinctive helix-turn-helix motif as responsible for the substrate specificity and binding. Kinetics experiments were in agreement with the previous study when revealing that the first two-electron reduction in myxalamid biosynthesis was significantly slower and rate limiting.
Most recently, the Leys group from the University of Manchester obtained crystal structures of carboxylic acid reductases (CAR) from Nocardia iowensis (PDB ID 5MSC), Mycobacterium marinum (PDB ID 5MSO), and Segniliparus rugosus (PDB ID 5MSR), which are closely related to NRPSs and consist of adenylation, thiolation, and terminal reductase domains for catalyzing the reduction of aromatic and aliphatic carboxylic acids into the corresponding aldehydes.25 Interestingly, while CAR enzymes do not form peptide bonds, CAR terminal reductase domains maintain ~50% similarity to the aforementioned crystallized NRPS reductase enzymes.22–25 As opposed to other structural and mechanistic studies, this work illuminated the mechanism behind the ability for some reductases to catalyze only the two-electron ATP- and NADPH- dependent reduction of carboxylic acids to an aldehyde, stopping short of further reduction to the alcohol. Analysis of crystal structures revealed that 4’-Ppant docking in the reductase active site induced reorientation of a bound, non- catalyticaIly oriented nicotinamide moiety to the necessary catalytic position, a change proposed to limit reduction to the aldehyde product. Mutagenesis of an Asp residue involved in the nicotinamide reorientation supported this observation when it permitted four-electron reduction to the related alcohol product.
Mechanistically, thioester reductions catalyze the transfer of hydride from NAD(P)H to form a thiohemiacetal intermediate, which is stabilized by tyrosine in the catalytic triad and remains covalently bound to the enzyme (Fig. 4a).22,26 Cleavage of the thiohemiacetal then releases the aldehyde from the active site. If an alcohol is to be formed, reintroduction of the aldehyde is required for a second reduction. Investigations of thiohemiacetal formation thermodynamics have revealed that the bound intermediate is in equilibrium with the free thiol and aldehyde. The equilibrium constant K for this process has some dependency on acyl group identity. For example, thiohemiacetal formation is favored in the presence of electron- withdrawing groups as they stabilize the electron-rich thiohemiacetal, reducing the value of K (so that K is between 1 and 0.1). In contrast, electron-donating groups favor free thiol and aldehyde so that K > 1, facilitating product release.26
Fig. 4.
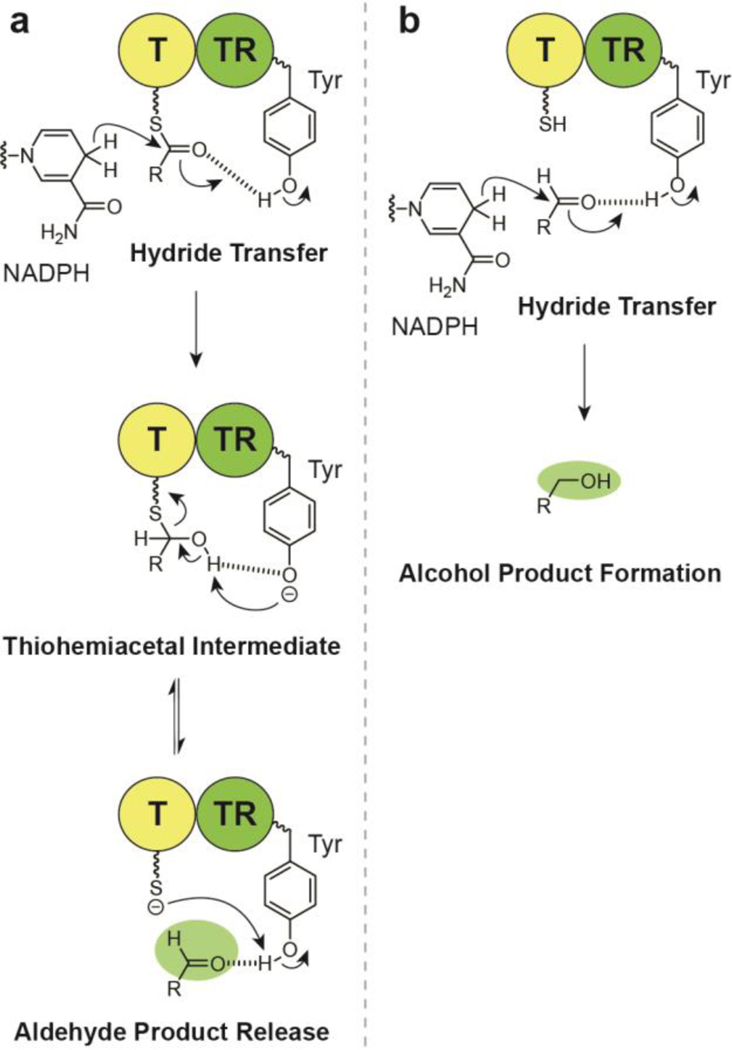
(a) Mechanism of thioester reductase NADPH-dependent aldehyde substrate release, (b) Mechanism of alcohol product formation.
2.2. Grouping of thioester reductase domains
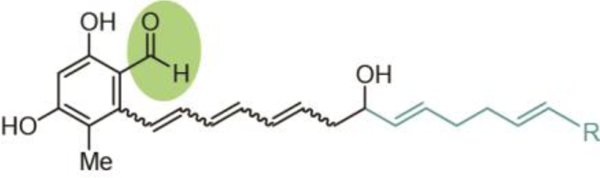
The differing extent of reduction that results in the formation of either an alcohol (four-electron reduction) or an aldehyde (two- electron reduction) gives rise to the two corresponding general categories. In the case of four-electron reductase domains, the most common products contain alcohols, which represent a diversity of biological activities.27 For two-electron reductase domains, the resultant aldehyde functionality is either maintained in the final product (e.g., flavopeptin17) or serves as an intermediate that undergoes a subsequent reaction such as intramolecular cyclization to form a cyclic imine (e.g., nostocyclopeptide12,13,28,29), transamination and cyclization to form a polyketide alkaloid (e.g., coelimycin P130, 31), transamination to form an amine (e.g., myxochelin B32), formation of a hemiaminal (e.g., anthramycin15), intramolecular Pictet-Spengler cyclization (e.g., ecteinascidin 74333), or Dieckmann condensation to form a heterocycle (e.g., tenellin34–37). Compounds with aldehyde warheads are often identified as protease and proteasome inhibitors due largely to their electrophilic nature, and as such have received extensive investigation for antiproliferative activity.38
3. Primary alcohol-containing natural products
The four-electron reductive release of a specialized metabolite substrate from a multi-modular NRPS or PKS megacomplex results in a primary alcohol, which is commonly maintained in the final product. As stated previously, the four-electron reduction of a thioester to an alcohol is completed as two two- electron reductions. The first step leads to the formation of an aldehyde (Fig. 4a), which coordinates with an active site tyrosine. The intermediate aldehyde is then reduced again by NAD(P)H, leading to the alcohol product (Fig. 4b).39 This offloaded alcohol is not electrophilic, but rather a weak nucleophile, and therefore the wide range of biological activities observed by these compounds are often-times dependent on the other sub-structural features of this group of compounds.
3.1. Myxochelin A
Myxochelin A was first discovered in the myxobacteria Angiococcus disciformis An d30 and is composed of a lysine modified with two flanking 2,3-dihydroxybenzoic acid residues, which are responsible for the compound’s iron chelating activity.27, 40 It is a catecholate-type siderophore that exhibits antioxidant properties, inhibits tumor cell invasion without cytotoxicity, and is a weak inhibitor of Gram-positive bacteria. The biosynthetic pathway (Fig. 5a) was characterized from the myxobacterium Stigmatella aurantiaca Sg a15 and represents an unusual instance of a TR domain with the ability to alternate between two- or four-electron reduction of an T domain-bound thioester rather than the more common case of reduction by one mode or the other.32, 41 The condensation domain of MxcG iteratively condenses the two amino groups of a MxcG PCP- bound lysine with MxcF T-bound 2,3-dihydroxybenzoic acids, which are synthesized upstream by proteins MxcC-MxcF. The resulting thioester is then reductively released by the MxcG TR domain to yield an aldehyde intermediate, which either undergoes an additional reduction (four-electron total) to give rise to the alcohol in myxochelin A or is acted upon by an aminotransferase, MxcL, to form myxochelin B (Section 6.2).32 Myxochelin B also exhibits iron chelating activity, but has not shown to inhibit tumor cell invasion. This suggests the 4- electron reduction to the alcohol may be necessary for the observed activity of myxochelin A.
Fig. 5.
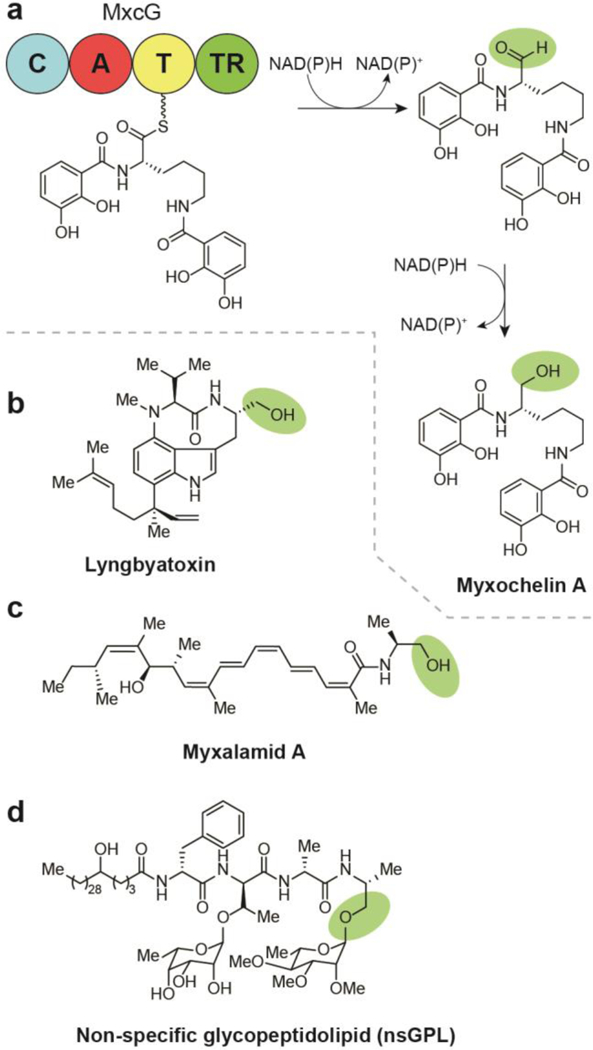
Biosynthesis of alcohol-containing natural products by four-electron reductive release, (a) Termination steps in the biosynthesis of myxochelin A. (b) Structure of lyngbyatoxin. (c) Structure of myxalamid A. (d) Structure of non-specific glycopeptidolipid (nsGPL).
3.2. (−)-lndolactam V natural products
The skin irritant, lyngbyatoxin A (Fig. 5b), was first discovered from the organic extract of the Hawaiian shallow-water cyanobacteria Moorea producens (formerly classified as Lyngbya majuscula Gomont).42,43, 44Lyngbyatoxin A was later found to be identical to teleocidin A-1, a toxic compound from Streptomyces mediocidicus, the broader class of which, along with O-methylated derivatives, the olivoretins, is produced by several strains of actinobacteria.45,46 All members of this class contain the (−)-indolactam V core—a cyclic nonribosomal dipeptide indole alkaloid—and exert potent tumor promotion activity via activation of protein kinase C and induction of ornithine decarboxylase.44, 47, 48 Efforts since the first lyngbyatoxin A isolation have resulted in discovery of various stereoisomers and analogs with hydroxylation on the geranyl chain.43, 44 The lyngbyatoxin BGC encodes a two-module NRPS (LtxA) specific for N-methylated L-Val followed by L-Trp. Subsequent terminal reduction off-loads the linear peptide intermediate, yielding an alcohol at the C-terminus of the tryptophan.10 The final steps of the biosynthesis include a proposed oxidation (LtxB) of the indole ring leading to cyclization with the N-methylvaline and formation of the nine- membered heterocycle. Additional tailoring enzymes (LtxC and LtxD) are responsible for the modifications seen in lyngbyatoxins B and C.49
3.3. Myxalamids
While screening a Myxococcus xanthus strain for activity against fungi and Gram-positive bacteria, Gerth and coworkers uncovered the myxalamids, a family of polyene antibiotics (Fig. 5c).50, 51 Myxalamid B exhibited inhibition of various Grampositive bacteria and fungi when tested using the agar diffusion method with paper disks. The myxalamids were also determined to be acutely toxic to mice with an LD1oo (the lowest dose to cause death in 100% of animals tested) of 3 mg/kg when administered subcutaneously and 10 mg/kg when administered orally. The mechanism of toxicity was attributed to inhibition of electron transport at the site of cytochrome b reduction by complex I (NADH:ubiquinone oxidoreductase) in bovine heart mitochondria. The same inhibition of electron transport mechanism was not observed in the mitochondria of S. cerevisiae, which lacked sensitivity to myxalamid B.51.
The myxalamids are nonribosomal peptide/polyketide hybrids encoded by an NRPS/PKS BGC comprised of six PKS genes with extensive dehydration and reduction tailoring domains, MxaBl, MxaB2, and MxaC-F, and a single NRPS gene, MxaA.52, 53 This terminal NRPS module installs a C-terminal alanine before a four-electron reductase domain releases the substrate as an alcohol.24
3.4. Non-tuberculosis mycobacterial glycopeptidolipids
Glycopeptidolipids (GPLs) are a class of species-specific lipids in non-tuberculosis mycobacteria (NTM) that comprise more than 70% of the outer layer of the mycobacterial cell wall (Fig. 5d).54 These lipids are important for motility, bacterial aggregation, biofilm formation, and cell wall integrity, while also contributing to pathogenicity.54–56 The glycopeptidolipids and other glycolipids were discovered during a search for antigens on the surface of NTM.57
In addition to modules for the incorporation of an N-terminal fatty acyl group, the BGC of the GLPs contains four NRPS modules. The terminal NRPS module incorporates L-alanine and contains a reductase domain for the four-electron thioester reductase-catalyzed release of the tripeptide-amino alcohol core (D-Phe-D-allo-Thr-D-Ala-L-alaninol). Subsequent glycosyltransferases then modify the C-terminal alcohol with an O-methylated rhamnose and the allo-threonine with a 6-deoxy- talose.22, 58 This basic structure comprises the non-specific glycopeptidolipid that is found across NTM. Further elaboration with oligosaccharides and incorporation of various O- methylations and O-acetylations result in biologically important cell surface antigens that are sub-species, serotype-specific. This GPL thioester reductase domain has received considerable structural and mechanistic characterization as summarized in Section 2.2 and maintains an overall homology of 45–50% with reductase domains that reduce the thioester to an aldehyde or an alcohol or catalyze a Dieckmann cyclization (Section 11).22, 23
4. Aldehyde-containing natural products
Aldehyde functional groups are relatively rare in NPs due to their high electrophilicity and generally low stability. As shown in Fig. 4 and Fig. 6a, this unique substructure is most commonly installed by two-electron TR domains. The first, naturally occurring peptide aldehyde was discovered in 1969 (leupeptin) and the first BGC for a peptide aldehyde was characterized in 2013 (flavopeptin).17 The highly electrophilic nature of the C- terminal aldehyde commonly equips this class with potent protease inhibition (see Section 12 for a full analysis of biological activities). Recognition of the privileged aldehyde warhead in recent years has inspired considerable interest by multiple research groups to identify novel members of this compound class. While aldehyde functional groups can be installed by additional mechanisms as in the biosynthesis of tylosin and rosamycin via alcohol oxidation by the P450 enzyme TylH1, or via reduction of carboxylic acids,25, 59, 60 this review will solely include those that are installed by reductase domains that terminate thiotemplate assembly lines.
Fig. 6.
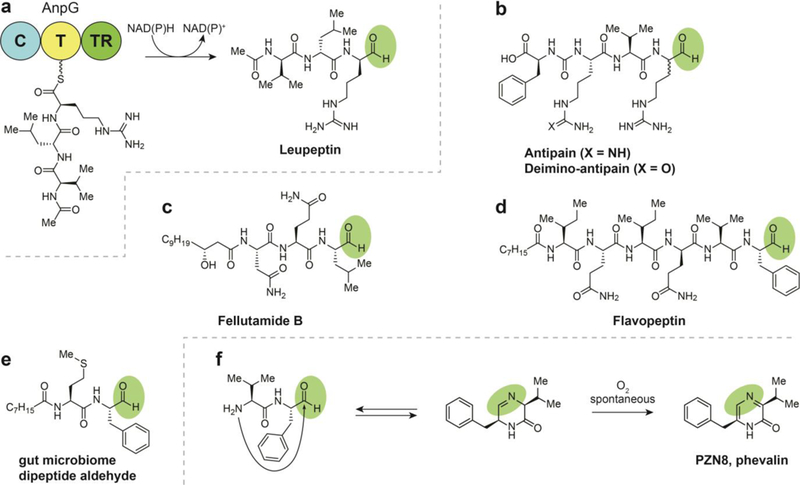
Biosynthesis of aldehyde-containing natural products by two electron reductive release, (a) Termination steps in the biosynthesis of fellutamide. (b) Structure of a flavopeptin. (c) Structure of leupeptin. (d) Structures of antipain and deimino-antipain. (e) Structure of dipeptide aldehyde from the gut microbiome. (f) Commensal microbe-derived dipeptide aldehydes are in equilibrium with their corresponding imine, but spontaneously convert to pyrazinones upon exposure to oxygen.
4.1. Leupeptin and ureido tetrapeptide aldehydes
Leupeptin is a cell-permeable protease inhibitor that was initially discovered in the soil isolate Streptomyces roseus MA839-A1 in the late 1960’s (Fig. 6a).61 This peptide trimer NRPS product is elaborated with an N-terminal acetyl group and a C-terminal aldehyde. The acetyl group may prevent macrocyclization of the N-terminus with the C-terminal aldehyde, though it has yet to be tested. The N-acyl “protecting group” is present in some form in many compounds of the peptide aldehyde class. The reduction step that results in the aldehyde was identified in the biosynthesis of leupeptin in the 1980s.62–64 Leupeptin is structurally and biosynthetically similar to the ureido tetrapeptide aldehydes, such as antipain.65, 66
Inspired to identify additional metabolites that exhibited similar biological activity of leupeptin and antipain, a reactivity-based screening of bacterial extracts discovered the peptide aldehyde deimino-antipain (Fig. 6b) and identified the TR-containing BGC responsible for its production.64 This approach takes advantage of the electrophilicity of aldehydes that enables their protease inhibition and other bioactivities as the means for labeling and detecting the responsible compound directly within an extract or fraction.64 A phylogenetic analysis of the deimino-antipain BGC led to the proposal that leupeptin and the ureido tetrapeptide aldehydes (e.g., antipain, elastatinal, chymostatin, and microbial alkaline protease inhibitor) likely share a common biosynthetic origin.
Interestingly, the BGC of the novel ureido tetrapeptide deimino- antipain and its known counterpart, antipain, breaks collinearity with the amino acid sequence as it involves only three NRPS adenylation domains. These genes anpD-F contain A domains predicted to install Arg, Phe, and Val, respectively. The absence of a fourth Arg/Cit-specific A domain suggests that either the Arg-specific AnpD module loads an extra monomer non- consecutively or that AnpE also installs Arg/Cit, an organization similar to syringolin biosynthesis, especially if this single domain also included installation of the ureido group.67 Neither case is common, with the ability for a single A domain to load two dissimilar amino acids being completely unprecedented. The terminal module is comprised of a peptide carrier protein (T), C domains, and a thioester reductase domain for release of the product with a C-terminal aldehyde.
4.2. Fellutamides
The fellutamides (fellutamide B shown in Fig. 6c) were first discovered from Penicillium fellutanum Biourge, a fungus isolated from a marine fish.68 Since this initial discovery, the fellutamides have also been observed in Aspergillus nidulans.69 Fellutamide A and B were both reported to exhibit cytotoxic activities by inhibition of the proteasome. As with established proteasome inhibitors, the aldehyde serves as the pharmacophore of the fellutamides and forms a reversible covalent bond with nucleophilic N-terminal threonine side chains at one of three protease-like sites in the proteasome.18 In addition, the fellutamides have been found to induce nerve growth factor synthesis.
Despite their structural and biological characterization in the early 1990’s, the BGC and biosynthetic pathway of the fellutamides was not elucidated until 201 6.69, 70 A resistance gene-guided genome mining approach identified an NP BGC, inp, containing a proteasome-encoding resistance gene (inpE). Replacement of the native BGC promoter with an inducible promoter allowed for expression and isolation of a fellutamide B stereoisomer as well as detection of additional fellutamide analogs with varying aliphatic chain lengths. The BGC encodes an acyl-AMP ligase (InpC), and two NRPS domains (InpA, InpB). Biosynthesis is initiated by hydroxy-fatty acid tail incorporation by the InpC module, preventing cyclization via condensation with the N-terminus. Interestingly, the growing peptide chain was found to be passed directly from InpB to InpA at very low levels during knockout experiments. A putative esterase/lipase, InpF, is proposed to catalyze release and transfer of intermediate from InpB to InpA. Following InpB incorporation of two amino acids and InpA incorporation of one to form a polypeptide, a terminal reductase domain on InpA performs the two-electron release of the peptide chain that yields the C- terminal aldehyde.
4.3. Flavopeptins
The flavopeptins (Fig. 6d) were the first compounds with a C- terminal aldehyde that were definitively assigned to arise from an NRPS pathway containing a terminal reductase domain when they were discovered from Streptomyces flavogriseus.17 This class was discovered using a combination of proteomic, genomic, and metabolomics analysis. Notably, the proteomic natural product discovery platform developed in the Kelleher lab known as Proteomic Investigation of Secondary Metabolism (PrISM) was used for flavopeptin discovery. This unique approach involves the size-selective mass spectrometry-based screening for expressed natural product biosynthesis enzyme complexes, which are much larger than other members of the microbial proteome. This protein-first approach facilitates deorphanization of BGCs by simultaneous discovery of gene clusters and associated secondary metabolites. The flavopeptins were determined to exhibit cysteine protease and 20S proteasome inhibition as well as antiproliferative activity against myeloma and lymphoma cell lines.
Like the fellutamides and leupeptin, members of the flavopeptin family contain an N-terminal fatty acid. In the flavopeptin biosynthetic pathway, there are no fatty acid synthase ORFs. Instead, analysis of condensation domains suggest that the N-terminal fatty acid is incorporated into the flavopeptin core by a “starter” condensation domain, the first domain encoded in the FlavA NRPS machinery.71 This flavopeptin fatty tail was speculated to serve as preventive for polymerization or macrocyclization; however, a synthesized free N-terminal analogue exhibited less than 10% macrocyclization when analyzed by MS, suggesting a role other than cyclization inhibition.17 The FlavA NRPS is also responsible for incorporation of a pair of alternating isoleucine and glutamine residues, while the subsequent FlavB module first incorporates valine or isoleucine followed by a terminal phenylalanine. Flavopeptin analogs are differentiated based on their acyl chain length and penultimate incorporation of valine or isoleucine. Finally, the mature acylated hexapeptide is cleaved from the assembly complex by a terminal TR domain, revealing the C-terminal aldehyde.
4.4. Pyrazinones with dipeptide aldehyde intermediates
In 2017, a team led by the Fischbach group combined metagenomics of the human gut microbiome with synthetic biology and heterologous expression to isolate a family of novel pyrazinone natural products attributed to the anaerobic Grampositive Clostridia class as well as Gram-negative genera Bacteroides and Desulfovibrio.72 Curiously, while not containing aldehyde groups, these compounds’ BGCs contained terminal reductase domains. Extensive investigation indicated that these heterocycles were spontaneously formed from a corresponding set of protease-active dipeptide aldehydes, the true products of the BGCs. Under the physiological conditions simulated in the experiment, the dipeptide aldehydes have half-lives of hours that could be prolonged in the anaerobic environment of the gut - time enough for systemic distribution and biological activity in a human host. These aldehydes exist in equilibrium with the corresponding cyclic imine until exposed to oxygen, when the dihydropyrazinone oxidizes irreversibly to the pyrazinone (e.g. PZN8, phevalin; Fig. 6e). Some products identified in the study were peptide aldehydes stabilized by N- acylation, supporting the proposition that the pyrazinones are active in their aldehyde form (Fig. 6f). Additionally, an in vitro panel of protease inhibition assays and chemical proteomics experiments revealed that the peptide aldehydes selectively target a subset of cathepsins in human cell proteomes with single-digit nanomolar activity. These results additionally suggest that these peptides may serve the producing commensal gut microbes by acting in the host lysosome to block host immune recognition and/or provide an intracellular niche in the phagolysosome.72 This class of compounds has yet to be identified in the human gut, but great interest exists for further study.
4.5. Azinomycin B / carzinophilin A
Azinomycin B, also known as carzinophilin A, is a hybrid NRPS/PKS antibiotic and anticancer natural product isolated from Streptomyces spp. that is produced by an iterative type I PKS and five NRPS modules.73,76 Azinomycin B is a potent anticancer compound, exerting cytotoxic activity against leukemic cell lines and mouse model tumors. The mechanism involves sequence-selective, covalent inter-strand crosslinking within the major groove of DNA by electrophilic attack of the epoxide and aziridine moieties onto purine bases.75, 76
The azinomycin B NRPS biosynthetic pathway sequentially condenses three unusual derivatized building blocks (the polyketide-derived 3-methoxy-5-methyl-naphthoic acid, α- ketoisovaleric acid modified from valine, and aziridino[1, 2a]pyrrolidinyl amino acid extensively derivatized from glutamic acid) and a C-terminal threonine. This backbone then undergoes numerous post-NRPS modifications (oxidation, epoxidation, and acetylation) that ultimately yield an exceptionally unique product. Adding to these modifications is the reductive release of the pre-azinomycin B aldehyde intermediate that converts to an enol tautomer (Fig. 7). This enol tautomer of the aldehyde is stabilized through conjugation when AziD2, a putative acyl-CoA dehydrogenase, oxidizes the adjacent hydroxyl group of threonine to give the carbonyl.
Fig. 7.
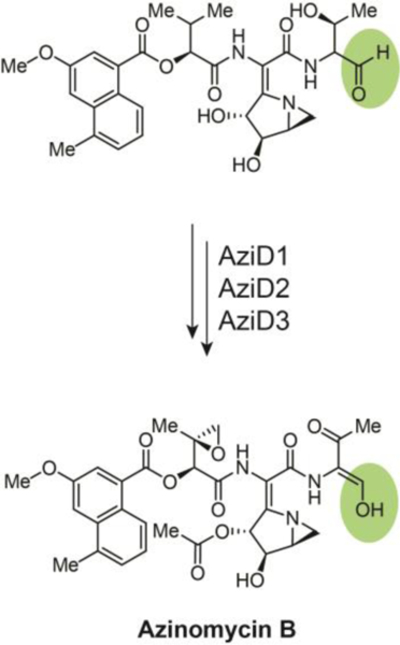
A two-electron reductive release of the aldehyde pre-azinomycin B precedes tautomerization to an enol that is stabilized through conjugation with the adjacent carbonyl, the product of threonine alcohol oxidation by AziD2, a putative acyl-CoA dehydrogenase.
5. Macrocyclic iminopeptides
Imine-containing natural products are commonly derived from NRPS pathways that terminate with thioester reductase domains (Fig. 8a). Whereas the peptide aldehyde natural products have primarily been found to inhibit proteases, cyclic imine natural products have demonstrated a wider range of biological activities (see Section 12). All currently known members of this NP class originate from linear heptapeptide aldehydes, with free N-termini that undergo an enzyme independent macrocyclization to yield an imine. This spontaneous imine macrocyclization was recently utilized in the synthesis of large macrocycles.77
Fig. 8.
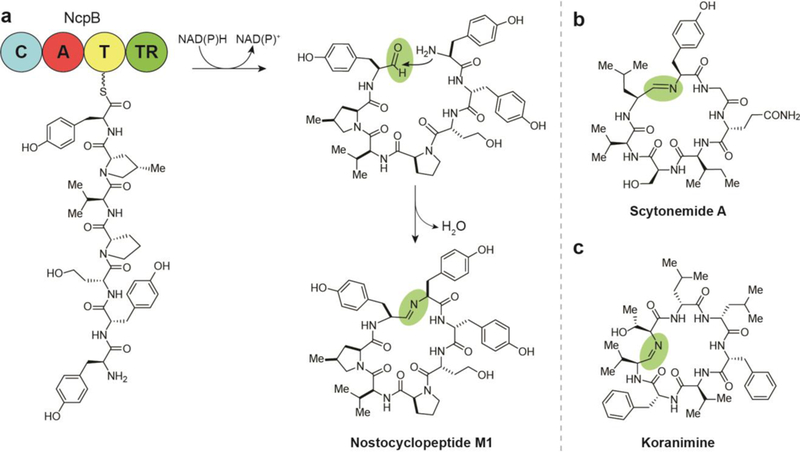
Biosynthesis of macrocyclic iminopeptide natural products by two electron reductive release, (a) Termination and cyclization steps in the biosynthesis of nostocyclopeptide M1. (b) Structure of scytonemide A. (c) Structure of koranimine.
5.1. Nostocyclopeptides
The first cyclic imino peptides discovered were the nostocyclopeptides from the terrestrial cyanobacterium Nostoc sp. ATCC-53789 by the Moore group.78 Characteristic of this family of compounds are the imino macrocycle linkage and incorporation of nonproteinogenic amino acids. While several members of nostocyclopeptides have been discovered, nostocyclopeptide Ml has received the most research focus. This NP was found to inhibit the uptake of microcystins and nodularin in hepatocytes, abating their toxicity.12 This activity extends to blockage of organic anion transporters in cancer cells, thus potentially nullifying some aggressive cancers’ tendency toward apoptosis resistance.
The NRPS-containing BGC terminates with a thioester reductase domain; however, during the isolation and structure elucidation of these compounds, no linear aldehyde-containing species was identified. Studies of the termination steps of the biosynthetic pathway suggest that the linear aldehyde is formed during release from NcpB before it is captured by the N-terminal amine and condenses to form the more stable macrocyclic imine (Fig. 8a).79 Investigations into the mechanism of macrocyclization of nostocyclopeptides revealed that the process is entirely enzyme independent and amino acid sequences were found to greatly affect the equilibrium of macrocyclization - replacement of some amino acids abolished cyclization completely.28
5.2. Scytonemide A
In 2010, the Orjala lab at the University of Illinois at Chicago isolated scytonemide A (Fig. 8b) from the freshwater cyanobacterium Scytonema hofmannii,14 The BGC for production of the scytonemide A has not yet been identified, but its structural similarity to nostocyclopeptides A1 and A2, including the characteristic imine linkage, suggests that it shares a common biosynthetic route that involves a terminal reductase domain. When screened for bioactivity, scytonemide A was found to potently inhibit the human 20S proteasome with an IC50 of 96 nM. Interestingly, cyclic peptides without the imine also serve as proteasome inhibitors,80 suggesting that this feature is not required for the inhibition mechanism of active cyclic peptides, which for some compounds has been found to entail a tight, yet reversible interaction with the canonical substrate-binding site of the proteasome β-ring.81
5.3. Koranimine
A Bacillus soil isolate from Koran, Louisiana was found to produce the macrocyclic imine, koranimine (Fig. 8c). This cyclic heptapeptide was discovered by the Kelleher lab using the same proteomics-based NP discovery approach that led to the isolation of the flavopeptins.29 Design of primers for DNA amplification of a novel BGC was based on peptide sequences gleaned from tandem mass spectrometry experiments of high molecular weight NP biosynthetic proteins. DNA sequencing of the resulting BGC facilitated prediction of functional biosynthetic domains, which in turn allowed for prediction of the amino acid monomers and detection of koranimine. Gene cluster-metabolite association was confirmed by heterologous expression, while biosynthesis was only confidently established following isolation and elucidation of the NP due to anomalies in the BGC organization. The koranimine gene cluster encodes four NRPS proteins with only five adenylation domains yet koranimine is realized as a heptapeptide. The presence of leucine as both the second and third amino acids in the chemical structure suggests that the A domain from KorA loads leucine monomers from two distinct C-T didomains. Using a proteomics technique known as the PPant ejection assay, the thiolation domains of a T-T-C tridomain in KorC were found to both be acted on by the A domain from the subsequent KorD.
Cyclization formed by the imine is initiated by termination by a TR domain on KorD and occurs in an enzyme independent fashion as shown in the nostocyclopeptides biosynthesis. The enzyme-independent macrocyclization was corroborated by the synthesis of the aldehyde precursor which spontaneously forms the mature koranimine.77 Koranimine, like the nostocyclopeptides and scytonemide A, contains a number of D-amino acids, including two iteratively incorporated D- leucines, which are hypothesized to be epimerized by a single epimerase domain. The presence of D-amino acids in these NPs may be necessary for the spontaneous macrocyclization to form imines. Presently, the biological activity of koranimine is unknown.
5.4. Lugdunin
A 2016 investigation provided evidence for the role of the antimicrobial NP of a Gram-positive human commensal, Staphylococcus lugdunensis, in preventing S. aureus colonization of the nasal cavity without significant toxicity to human cells.82 This compound, lugdunin, does not contain an imine in the final product, but instead uses the imine for macrocyclization and then converts it to a five-membered thiazolidine ring (Fig. 9), establishing it as the first representative ofthe macrocyclicthiazolidine peptide antibiotic class. Lugdunin exhibited pronounced bactericidal activity against a panel of nasal and clinical S. aureus isolates and gave evidence for an ability to evade resistance when no spontaneously resistant S. aureus mutants were generated after 30 days of continuous serial passaging in the presence of subinhibitory concentrations of the antibiotic. Studies suggest that lugdunin treatment leads to rapid cessation of bacterial metabolic pathways resembling the enigmatic mechanism of action of daptomycin.82
Fig. 9.
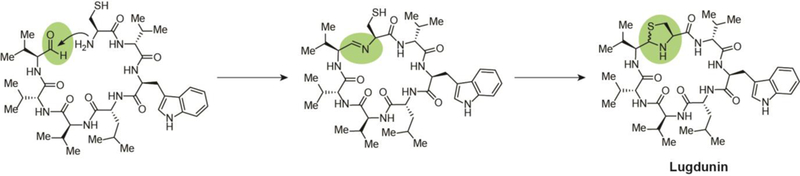
Unique terminal biosynthetic steps in the biosynthesis of lugdunin, in which two-electron reductive release to an aldehyde and subsequent macrocyclic imine formation leads to attack of the imine carbon by the sulfur on the adjacent cysteine for thiazolidine cyclization.
Four NRPS genes, lugA, B, C, and D, comprise the lugdunin operon. Interestingly for a heptapeptide, these genes encode only five amino acid adenylation domains. The LugC module is made up of one valine-incorporating adenylation domain but two condensation and three thiolation domains downstream for peptide bond formation and amino acid transfer, respectively. This suggests a biosynthesis partially similar to that of yersiniabactin and koranimine, wherein the single LugC adenylation domain activates three consecutive valine units for incorporation in alternating L- and D-configurations. As with the previous imine examples, the lugdunin reductase domain, LugC, cleaves the thioester of the bound peptide chain, releasing a linear heptapeptide with a C-terminal aldehyde. The N-terminal cysteine amine then attacks this aldehyde, condensing the linear peptide as a macrocyclic imine. Subsequent nucleophilic attack of the imine carbon by the cysteine thiol generates yields the five-membered thiazolidine heterocycle of the mature lugdunin. Interestingly, sufficient production of lugdunin under laboratory conditions could only be achieved through replacement of the native tetR-like regulator gene, lugR, with a xylose-based expression system.
6. Amine-containing natural products
The biosynthesis of amine functional groups in NPs by way of TRs is relatively rare. Reductase involvement in amine formation initially results in release of an aldehyde via two- electron reduction. A second, amine-donating compound then condenses onto the aldehyde and, following the action of a reductase and/or aminotransferase, a product is generated with a primary amine (Fig. 10a). As with the previously mentioned alcohol-containing NPs, amine functionality does not afford NPs with a distinct biological activity and therefore these molecules exhibit varying activities that may be influenced by other structural features.
Fig. 10.
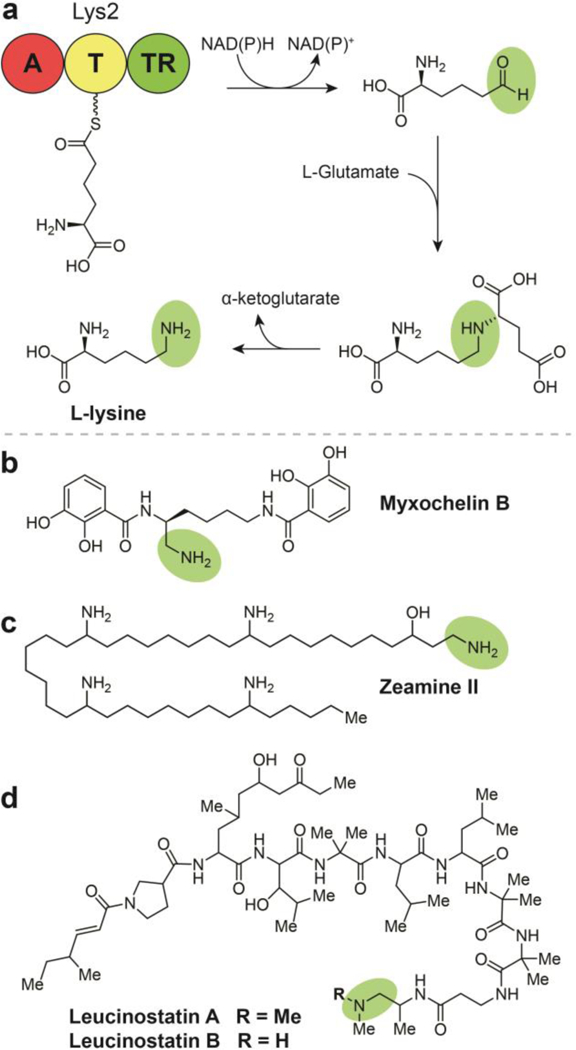
Biosynthesis of amine-containing natural products by two electron reductive release, (a) Termination and modification steps in the synthesis of L-Lysine in Saccharomyces cerevisiae. (b) Structure of myxochelin B. (c) Structure of zeamine II. (d) Structures of leucinostatins A and B.
6.1. Lysine in Saccharomyces cerevisiae
Along with saframycin in the late 1990’s (see Section 10.1), the biosynthesis of the essential amino acid L-lysine in Saccharomyces cerevisiae (Fig. 10a) reported by the Walsh group represents one of the earliest elucidated reductase- containing pathways.83 L-Lysine biosynthesis in lower eukaryotes is completely distinct from that of prokaryotes. In bacteria, lysine is synthesized via decarboxylation of 2,6- diaminopimelate. Conversely, investigations into lysine biosynthesis in higher fungi have revealed an NRPS-like protein, Lys2, which contains an adenylation domain, a PCP (thiolation) domain, and a TR domain to produce the amino acid. The biosynthesis includes reductive off-loading of α-aminoadipate semialdehyde from the TR domain of Lys2 prior to condensation with glutamate to form saccharopine, which is then hydrolyzed to generate L-lysine and α-ketoglutarate.
6.2. Myxochelin B
Myxochelin B (Fig. 10b) is a catecholate-type siderophore with weak anti-microbial activity that shares a BGC with myxochelin A (Section 3.1; Fig. 5a).32, 84 In contrast to the consecutive reduction steps that yield myxochelin A, myxochelin B is formed when the compounds’ shared aldehyde precursor is transaminated following reductive off-loading in a manner analogous to the polyketide alkaloids (Section 6.4) and L-lysine synthesis in S. cerevisiae. Biochemical studies revealed that the MxcG reductase and MxcL transaminase compete for a common free aldehyde intermediate rather than the diverging final steps being coordinated by substrate sequestration or configuration of the enzyme complex - relative amounts of each protein domain in the cell correspond to the amounts of myxochelin A or B produced.
6.3. Zeamines
The zeamines are a class of potent, broad-spectrum antibiotics from Gram-negative bacteria Serratia plymuthica RVH1 and Dickeya zeoe.85–87 These hybrid NPs have a common pentaamino-hydroxyalkyl chain with appended peptide and polyketide subunits of varying composition (Fig. 10c). The mechanism of action for the anti-bacterial properties of the zeamines has not been elucidated, therefore it is undetermined if the amine installed as a byproduct of the thioester reductase is critical for activity. The functions of several key zeamine biosynthetic enzymes were defined by targeted gene deletions, LC-MS analyses, in vitro biochemical assays, and biosynthetic precursor feeding studies.85,87 These experiments revealed that zeamine biosynthesis is comprised of the combined action of NRPS, type I PKS, and polyunsaturated fatty acid synthase-like enzymatic machinery. As a part of this system, a standalone TR catalyzes the NADH-dependent release of a tetraamino- hydroxyalkyl thioester as an aldehyde from a type I iterative FAS/PKS hybrid complex. This reductase domain was confirmed to have the ability to catalyze the further reduction of the aldehyde to an alcohol, but the aldehyde intermediate preferentially undergoes a transamination to produce zeamine II, the precursor to zeamine and zeamine I.88 These latter two compounds share the long chain structure of zeamine II amide- bonded to a polyketide with a terminal valine. This terminal residue is proposed to be a remnant of an NRP leader sequence that is cleaved during a post-assembly processing step.
6.4. Leucinostatins
Leucinostatins are a class of 24 lipopeptide antibiotics produced by a variety of fungal genera. Leucinostatin A and the N- methylated leucinostatin B are comprised of nine amino acid residues, five of which are non-canonical, as well as an N- terminal 4-methylhex-2-enoic acid and a C-terminal dimethylamine (leucinostatin A) or methylamine (leucinostatin B) (Fig. 10d).89 Leucinostatin A exhibits a wide range of bioactivities, including inhibition of prostate cancer cell growth, anthelmintic activity toward trypanosomes (the causative agent of the neglected tropical disease trypanosomiasis), P. infestans inhibition (the causative agent of potato blight), nematicidal properties, and broad spectrum antimicrobial activity.90, 91 The leucinostatin antibiotic mechanism of action involves the inhibition of ATP synthesis in the mitochondria and various phosphorylation pathways.
In a study aimed at exploring leucinostatins role in the biocontrol function of its producer, Purpureocillium lilacinum, the leucinostatin gene cluster was identified by genomic analysis and genetic manipulation experiments.89 A C- domain in the first module catalyzes the condensation of 4-methylhex-2-enoic acid with 4-methyl-L-proline, followed by successive chain elongation by the remaining nine adenylation domains in the leucinostatin BGC. The mature peptide is then released from the assembly line and undergoes a putative transamination followed by a single methylation to afford leucinostatin B and an additional methylation to afford leucinostatin A. 89
6.5. Polyketide alkaloids
Compounds with piperidine, indolizidine, and related alkaloid substructures represent a wide range of scaffolds. Many of the NPs presented here were discovered as a result of chemical screenings targeting these lesser-occurring bacterial-derived piperidine-type alkaloids.92–101 The diverse scaffolds exhibit a similarly wide range of biological activities - while coelimycin P1 has no activity reported (Fig. 11a), cyclizidines have been found to have antifungal and anticancer properties (Fig. 11b),100–102 nigrifactin exhibits antihistamine and antihypertensive activity (Fig. 11c),92, 93 streptazolin exhibits limited antifungal and antibacterial activity (Fig. 11d),94 latumcidin (AKA: abikoviromycin) is an antiviral compound (Fig. 11e),103–105 pyrindicin is a weak antimicrobial with analgesic and antiplatelet properties,97 streptazones have exhibited antibiotic and cytotoxic activity,98, 99 and the complex set of argimycins PI—IX are weakly antibiotic.106,107
Fig. 11.
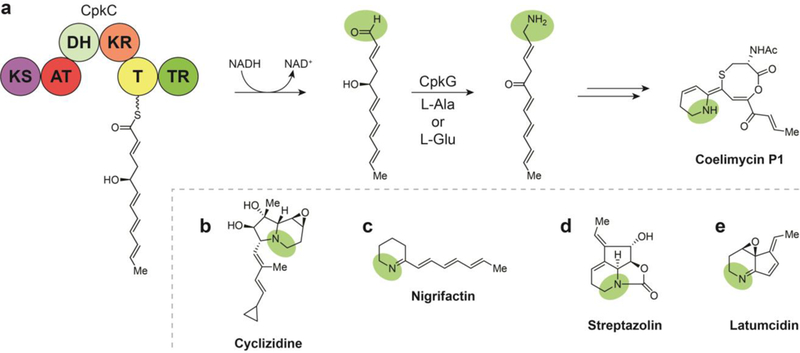
Polyketide alkaloid biosynthesis involves a TR-mediated two-electron reductive release and subsequent transamination, (a) Termination and transamination steps in the biosynthesis of coelimycin P1. (b) Structure of cyclizidine. (c) Structure of nigrifactin. (d) Structure of streptazolin. (e) Structure of latumcidin.
Piperidines, indolizidines, and related alkaloid NPs tend to arise from a conserved biosynthesis based on the incorporation of lysine cyclization products. Piperidines are often formed following lysine cyclization into 2-pipecolinic acid, while indolizidine is most often formed from the condensation of 2- pipecolinic acid and malonyl-CoA.31 However, there are examples of piperidines, indolizidines, and other alkaloids that are the products of modifications to reductively-released polyketide chains. Following decades of efforts by multiple groups to identify a yellow pigmented gene product, the Challis group reported it from Streptomyces coelicolor M145 as the alkaloid coelimycin PI with its biosynthetic identification, isolation, and structure elucidation, revealing an unprecedented 1,5-oxathiocane in its core.108 A subsequent study revealed details of a TR-mediated reductive chain release preceding a transamination that converts the offloaded PKS I product to coelimycin P1.30 In its biosynthesis, a growing polyketide chain is reductively off-loaded as a transient aldehyde, which is trapped for reductive amination by a co- transaminase for subsequent cyclization and additional tailoring. In searching publicly available genomic data for additional BGCs that utilized similar reductive and transamination steps, 22 were identified that were known or predicted to direct the biosynthesis of diverse polyketide alkaloids.30 Gene clusters were putatively assigned to alkaloid biosynthesis by matching the length of the polyketide chain encoded in the BGC to corresponding compound carbon skeletons in combination with logical tailoring enzymes. In this fashion, BGCs were putatively assigned to cyclizidines, 100–102, 109 nigrifactin,92, 93 streptazolin, streptazone E,110 pyrindicin,95–97 and latumcidin. Some of these NPs had received supporting biosynthetic characterization, such as cyclizidine,31 while others with consistent biosynthetic logic, such as argimycins PI—IX, were discovered later.106, 107 In addition to these known compounds, nine clusters remained for the production of putatively novel polyketide alkaloids. Thus, a common polyketide alkaloid biosynthetic logic has been established involving a modular PKS that terminates in homologs of the coelimycin P1 TR and a transaminase.30
7. Pyrrolobenzodiazepines
The tricyclic pyrrolobenzodiazepine core structure is comprised of an anthranilate-derived A-ring, a central diazepine B-ring, and a modified proline C-ring. Compounds in this class have been shown to affect antibiotic, antiviral and antiprotazoal activity, but they are primarily known for their potent anticancer properties (Fig. 12).111, 112
Fig. 12.
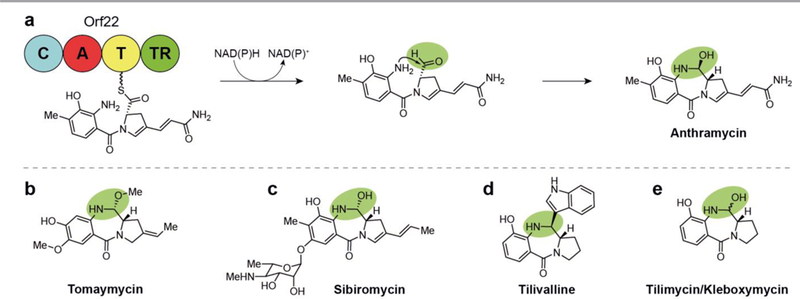
Termination and cyclization steps in the biosynthesis of pyrrolobenzodiazepine natural products involves the TR-mediated two-electron reductive release of an aldehyde, (a) Reductive release and attack of the resulting aldehyde by the aromatic amine in anthramycin biosynthesis, (b) Structure of tomaymycin. (c) Structure of sibiromycin. (d) Structure of tilivalline. (e) Structure of tilimycin/kleboxymycin.
The mechanism of anthramycin, tomaymycin, and sibiromycin cytotoxicity was initially elucidated through multiple studies in the 1970s as reversible covalent binding of the aldehyde- derived hemiaminal with DNA bases in the minor groove. It was later discovered that C-ring unsaturation patterns of these compounds was responsible for their exceptional potency.113 Recognition of the DNA sequence selectivity of this class and the increased affinity of anthranilate O-glycosylation (as in sibiromycin) came about in the early 2000’s.112, 114 Pyrrolobenzodiazepines such as sibiromycin and anthramycin with a C-9 hydroxylation have the detrimental effect of cardiotoxicity, though many efforts have been undertaken to generate analogs that omit this substructural feature.115 The potent and selective mechanism of these compounds have made them attractive targets for drug development, with extensive synthetic efforts aimed at generating dimers and various substitutions on the anthranilate and hydropyrrole moieties.112 A pyrrolobenzodiazepine dimer synthesized in the 1990s, SJG-136, has the additional ability to affect interstrand and intrastrand DNA crosslinking and completed Phase II clinical trials for the treatment of leukemia and ovarian cancer in 2015. Related dimers are also being used as antibody-drug conjugate payloads to target tumors, multiple of which are also in clinical trials or preclinical development.116
Biosynthesis of hemiaminal-containing products by reductase terminating NRPS/PKS pathways is rare and all known examples contain an aromatic amine that attacks the reductively offloaded aldehyde. Approximately a dozen highly similar pyrrolobenzodiazepines have been identified, all produced by homologous pathways.112, 117–120 While additional hemiaminal- containing secondary metabolites of the tetrahydroisoquinoline alkaloid class are reviewed later (Section 10), the distinct structures and concise biosynthesis of the pyrrolobenzodiazepine class will be discussed here.
7.1. Anthramycin
The anthramycin structure is characterized by methylation and hydroxylation on the A-ring and an acrylamide substitution on the C-ring. This benzodiazepine alkaloid was discovered from in 1968 and exhibits potent anti-tumor and antibiotic activity.15, 121, 122 The interesting structure and biological mechanism of anthramycin has led to extensive investigations into the applicability of the pyrrolobenzodiazepines in oncology. An early study of the biosynthesis uncovered the monomers from which anthramycin was formed,123 but direct characterization of the biosynthesis was not uncovered until the responsible gene cluster was sequenced, validated, and analyzed by the Bachmann lab in 2007.16 The terminal steps of the anthramycin biosynthesis involves reductive release from an NRPS to yield an aldehyde. The proximal anthranilate-derived arylamine adds to the to the dehydroproline acrylamide aldehyde resulting in the tetrahedral center.
7.2. Tomaymycin
Tomaymycin is produced by Streptomyces achromogenes and exhibits structural variation from anthramycin in its substitution pattern (Fig. 12b).124, 125 These differences include the positions of the methyl and hydroxyl groups on ring A and a ethylidene substitution on ring C, the latter of which has been identified as critical for this compound’s potency.113, 126 These substructural features are similar to those on the monomer of the pyrrolobenzodiazepine dimer drug candidate SJG-136 and accordingly, the two exhibit similarly potent cytotoxicity. Tomaymycin is active in ovarian, plasmacytoma, and leukemia cancer cell lines with IC50 values of 0.13, 1.8, and 3.7 nM, respectively.113
Sequencing and analysis of the tomaymycin BGC in 2009 illuminated the biosynthesis of pyrrolobenzodiazepine A-ring formation,118 while a subsequent study proved the full biosynthesis using mass-spectrometric analysis of whole proteins from the in vitro reconstitution of the tomaymycin NRPS pathway. 127 In this approach, LC-MS analysis of NRPS thiolation domains bound to tomaymycin intermediates and detection of free tomaymycin intermediates facilitated the elucidation of individual biosynthetic steps.
7.3. Sibiromycin
The discovery and structure elucidation of sibiromycin from the actinomycete Streptosporangium sibiricum was reported in the early 1970’s.128–130 The compound contains a methylation, a hydroxylation, and a glycosylation on the A-ring, with a propylidene substitution on the C-ring (Fig. 12c). In addition to its antibiotic activity, sibiromycin exhibits the greatest DNA binding affinity and the most potent antitumor activity of the NP pyrrolobenzodiazepines.112
In 2009, the Gerratana lab identified the sibiromycin BGC using comparative genomics analysis, cloning, gene replacement, and chemical complementation experiments.120 Though the study does not describe direct characterization of a TR or any other individual NRPS domains, the two putative sibiromycin NRPS genes, sibD and sibE, reportedly encode the same domain organization as the anthramycin NRPSs.
7.4. Tilivalline
In 2014, tilivalline was discovered from the human enteric Gram-negative bacterium Klebsiella oxytoca, an unexpected source considering all other pyrrolobenzodiazepines have been discovered in soil-derived Gram-positive actinomycete bacteria.119 The tilivalline chemical structure includes a single A-ring hydroxylation and a B-ring indole substitution rather than the hemiacetal hydroxy group found in anthramycin and sibiromycin (Fig. 12d). Tilivalline was initially implicated as the primary cytotoxin causing disruption of epithelial barrier function in antibiotic-associated hemorrhagic colitis (AAHC).119 The influence of tilivalline on this bioactivity was soon brought into question in nearly simultaneous publications because of the compound’s lack of a hemiaminal, which is known to be critical for pyrrolobenzodiazepine cytotoxicity,. 131, 132 It was instead suggested that an indole-free tilivalline precursor newly discovered in Klebsiella oxytoca, named both tilimycin and kleboxymycin (Fig. 12e), was the more relevant enterotoxin and was confirmed to be orders of magnitude more potent. Using genomic analysis, chemical synthesis, and mutagenesis studies, the tilivalline NRPS was shown to initially produce tilimycin/kleboxymycin, with tilivalline arising from a non- enzymatic, spontaneous reaction between the TR-domain released precursor and indole.132
8. Le-pyrrolopyrazines
Le-pyrrolopyrazines A and C are cyclized dipeptides reported by Li and coworkers in 2017.133 The study was also the first report of a third analog, Le-pyrrolopyrazine B, as a NP.134 The structures of Le-pyrrolopyrazines A, B, and C are differentiated based on whether valine, leucine, or isoleucine, respectively, are incorporated with a conserved proline during biosynthesis (Fig. 13). In order to explore the biosynthetic capacity of the lesser-investigated Lysobacter genus of proteobacteria, the weak promoter preceding a cryptic, novel, three-gene, L. enzymogenes NRPS BGC was replaced with the strongest known NP BGC promoter from the same strain (PHSAF from biosynthesis of HSAF, a polycyclic tetramate macrolactam). The newly incorporated promoter activated the latter two of three targeted NRPS ORFs (ledD, IedE, ledF) and allowed subsequent identification of the novel Le-pyrrolopyrazines A-C (Fig. 13). Though only the second and third ORF were sufficiently expressed, both the first and third in this biosynthetic complex terminate in thioester reductases. In elucidating the biosynthesis, diketopiperazine intermediates were proposed to arise from condensations between proline and valine, leucine, or isoleucine followed by reductive release and dehydration, with a putative non-enzymatic dehydrogenation as the final step in generating the stable pyrazines of the pyrrolopyrazine products. In a unique enzymatic process with precedent only in natural mutation-driven domain skipping (bleomycin,135 myxochromide S136), the proline is loaded interchangeably between one of two redundant A-T-C tri-domain complexes. Targeted gene inactivation experiments revealed that while the LedE A domain contributed significantly to proline incorporation, the downstream LedF A domain was required for pyrrolopyrazine biosynthesis.
Fig. 13.
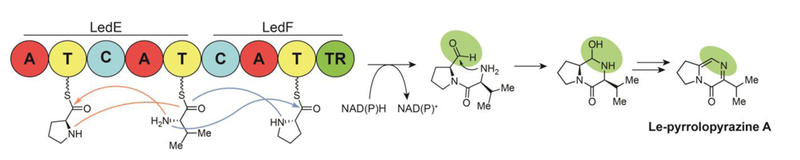
Biosynthesis of pyrrolopyrazine natural products by a unique alternating proline incorporation mechanism. The tethered dipept ide is formed by one of two pathways indicated by the orange and the blue arrows. The final product is formed from putatively non-enzymatic dehydration and dehydrogenation steps that follow two electron reductive release from the enzymatic assembly complex.
9. Nonreducing iterative polyketide synthase natural products
Extensive investigations into fungal NPs have revealed a diversity of small molecules with biosyntheses that rely on TR domain-terminating nonreducing iterative polyketide synthases. Due to the diversity of their chemical structures, these secondary metabolites are reported to exhibit the full breadth of antimicrobial, antiviral, cytotoxic, anticancer, and anti-inflammatory activities. This class commonly arises from a hybrid nonreducing iterative PKS/PKS complex and can be subdivided into the tropolones (Fig. 14a-e), the azaphilones (Fig. 14f-h), and the sorbicillins (Fig. 15).8, 137–139 Fundamental to these compounds’ biosynthesis are fungal enzyme complexes known as nonreducing iterative PKSs, which orchestrate chain extension by iterative incorporation of acyl- CoA with chain lengths determined by a KS domain. Unlike more commonly studied bacterial PKS machinery, this is accomplished without processing at the β-carbon during elongation. Two domains within the nonreducing iterative PKS module are unique among PKS systems: a starter unitacyl- carrier protein transferase (SAT) responsible for recruiting a chain initiating component onto the enzyme and a product template (PT) domain that orchestrates cyclization of the fully elongated polyketide chain intermediate to aromatic products.8, 140
Fig. 14.
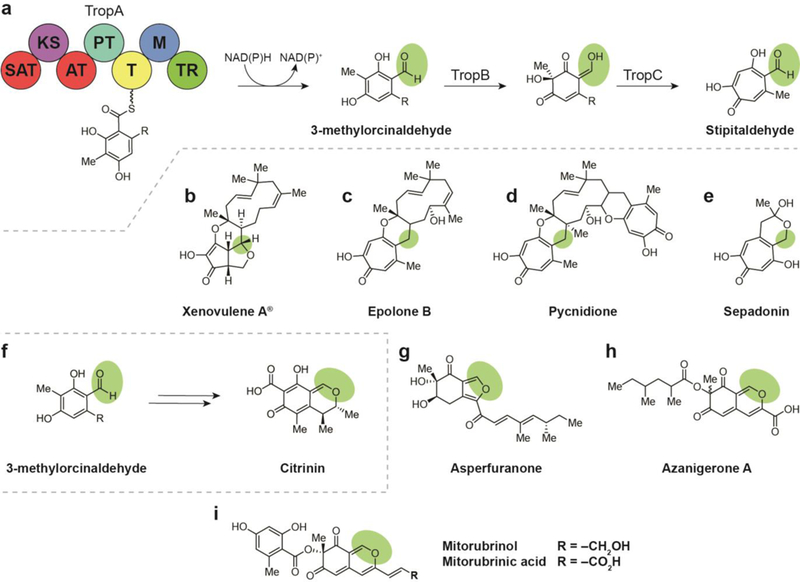
(a) Biosyntheses of natural products arising from a reductively released orcinaldehyde intermediate include the tropolones, which are biosynthesized via a tropolone intermediate (a-e), and the azaphilones, which arise from the cyclization of reductively released 3- methylorcinaldehyde to the bicyclic isochromene (f-h). (a) Terminal thioester reductase release of 3-methylorcinaldehyde leads to formation of stipitaldehyde. (b) Structure of xenovulene A. (c) Structure of epolone B. (d) Structure of pycnidione. (e) Structure of sepadonin. (f) Terminal thioester reductase release of 3-methylorcinaldehyde leads to formation of citrinin. (g) Structure of asperfuranone. (h) Structure of azanigerone A. (i) Structures of mitorubrinol and mitorubrinic acid.
Fig. 15.
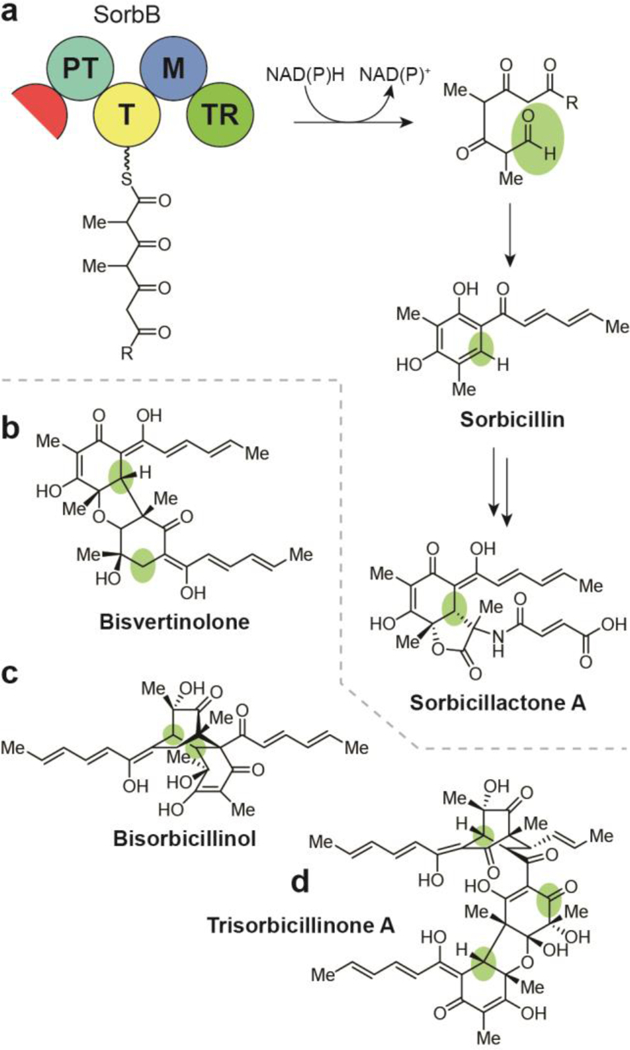
(a) Biosynthesis of the sorbicillin natural products, including sorbicillactone A, involves cyclization of a hexaketide to the sorbicillin intermediate following TR-mediated release from the nonreducing iterative PKS complex, (b) Structure of bisvertinolone. (c) Structure of bisorbicillinol. (d) Structure of trisorbicillinone A.
9.1. Tropolones
Most compounds produced by nonreducing iterative PKS- containing pathways arise from an orcinaldehyde intermediate. In the case of the tropolones, 3-methylorcinaldehyde is released by a TR domain and undergoes three oxidations and a ring expansion to form a tropolone core (Fig. 14a).
In 2012, gene knockout experiments aimed at characterizing the activity of these oxidase genes yielded the tropolone derivatives talaroenamine (the condensation product of 3-hydroxy-3- methylorcinaldehyde with anthranilic acid), leptosphaerdione (the oxidation product of 3-hydroxy-3-methylorcinaldehyde), and stipitaldehyde (Fig. 14a), which is also an intermediate in many tropolone NPs.137
9.1.1. Xenovulene A
Discovered in the 1990s, xenovulene A is a modified tropolone- humulene product that exhibits nanomolar inhibition of benzodiazepine binding to the GABA-benzodiazepine receptor (Fig. 14b).137, 141, 142 During a study of a tropolone pathway, a gene homology search yielded ten strains with 3- methylorcinaldehyde biosynthetic genes.137 One of these was a cluster from the xenovulene-producing fungal strain, Acremonium strictum. Further investigation supported assignment of xenovulene A biosynthesis when tropolone- related biosynthetic genes were observed surrounding a 3- methylorcinaldehyde cluster. Similar biochemistry is suggested to lead to analogous tropolone-humulene erythropoietin stimulators epolones A and B (Fig. 14c),143 the antitumor ditropolones eupenifeldin144 and pycnidione (Fig. 14d),145 as well as the antibiotic sepadonin (Fig. 14e).146, 147
9.2. Azaphilones
Results from the tropolone biosynthetic studies have also shed light on the biosynthesis of other important fungal NPs that arise from cyclization of the 3-methylorcinaldehyde intermediate to isochromene or furan-containing bicyclic cores instead of a tropolone. This class of nearly 400 compounds is classified as the azaphilones and exhibits a diversity of biological activities, including antibacterial, antifungal, and antitumor.148 The azaphilones include compounds such as citrinin (Fig. 14f),26, 148 asperfuranone (Fig. 14g),137, 149 sclerotiorin,150–152 azanigerones,153 and mitorubrins (Fig. 14i). 154, 155 Limited biosynthetic investigations of a host of other azaphilone fungal NPs involving stable isotope-labeled precursor feeding studies suggest a shared biosynthetic logic. Compounds including berkelic acid, spiciferinone, perinadine A, and austdiol, among others, are likely biosynthesized via a TR-domain released orcinaldehyde-like intermediate by nonreducing iterative PKS/PKS hybrid enzyme complexes.152
9.2.1. Citrinin
Citrinin is a mycotoxin that was first discovered in the 1930s from Penicillium citrinum.156 In years since, the compound has also been detected in several species of the genera Penicillium, Monascus, and Aspergillus. Citrinin is among the first discovered polyketides. It is characterized by an isochromene core and a lack of the carbon side chains often found on other azaphilones (Fig. 14f).26, 157 Citrinin is a potent mycotoxin, affecting nephrotoxicity by inhibition of respiration complex III.158 Its biosynthesis in Monascus ruber M7 was elucidated in 2016 by targeted gene knockout experiments and heterologous gene expression in Aspergillus oryzae.26 The pathway involves the nonreducing PKS incorporation of an unreduced, trimethylated pentaketide that is cyclized and offloaded by a TR domain to yield a substituted, aldehyde-bearing 3-methylorcinaldehyde intermediate. This is followed by a series of oxidations and cyclization to yield citrinin. High-yielding heterologous expression of the compound’s biosynthetic genes unambiguously confirmed assignment of this biosynthesis.
9.2.2. Asperfuranone
Asperfuranone is a bioactive fungal azaphilone that contains a bicyclic furan-containing core (Fig. 14g) and exhibits antiproliferative activity toward human non-small A549 cancer cells.137, 149 The biosynthesis of asperfuranone BGC from Aspergillus nidulans was analyzed in 2009.159 This NP was discovered when an inducible promoter was swapped in for an endogenous promoter with homology to citrinin. Promoter- enabled production of asperfuranone allowed isolation and structure elucidation. A series of gene deletions confirmed hybrid nonreducing iterative PKS/PKS genes responsible for 3- methylorcinaldehyde intermediate production and five additional genes comprising the asperfuranone biosynthetic pathway. As a part of the study, the aldehyde-containing intermediate was also purified, providing direct evidence for TR release from the PKS machinery.159
9.2.3. Azanigerones
Azanigerones A-F, highly oxygenated isochromene azaphilones that arise from an orcinaldehyde intermediate, were discovered by the Zabala group in 2012 following activation of the silent aza BGC in Aspergillus niger ATCC 1015 (Fig. 14h).153 The bioactivity of these compounds has not been reported. Azanigerone biosynthesis was confirmed using transcriptional analysis and gene deletion experiments. Assembly of these compounds is initiated when a methylated hexaketide bound to an nonreducing iterative PKS, AzaA, is subjected to product template (PT)-mediated cyclization and TR-domain release to yield a 3-methylorcinaldehyde with a 1,3-diketo side chain. Hydroxylation-mediated pyran ring formation by flavin- dependent monooxygenase, AzaH, converts this to the bicyclic core of the azanigerones. Parallel processing of a 2,4- dimethylhexanoyl chain from the AzaB PKS complex encoded in the cluster is converted into the corresponding acyl-CoA by AzaF, which is then used as a substrate for the acyltransferase, AzaD, to form azanigerones A—D.
9.2.4. Mitorubrins
Analogous biosynthesis has been described for the mitorubrins, which incorporate 3-methylorcinaldehyde into their core azaphilone isochromene scaffold and are modified with a benzoyl group (Fig. 14i). The first of these compounds, mitorubrinol and mitorubrinic acid, were discovered in 1965 when they were targeted for isolation as pigments from Penicillium rubrum.154 In 2012, these same compounds were identified as virulence factors in Penicillium marneffei, a thermally dimorphic fungus that is known to cause mycosis in immunocompromised patients in China and Southeast Asia.155
This same study used gene knockdown experiments to identify the nonreducing PKS BGCs responsible for production of the compounds and predict functions for each gene. Two PKS genes were identified, the first, PKS12, putatively serving to synthesize a mature orsellinic acid starter unit for PKS11, the second PKS, which also contains the terminal TR domain for product release.
9.3. Sorbicillins
The sorbicillins, also known as vertinoids, represent immense structural diversity due to unique cyxlizations of the hexaketide- derived sorbicillin core that serves as a foundation for subclasses of modified monomers, dimers, trimers, and other polycyles.160 Due to the large number of reported sorbicillin NPs, we limit this review to a summary of a select few members of the class. Sorbicillactones A (Fig. 15a) and B were isolated as a part of a program to identify bioactive compounds from marine sponge-associated microbes.161 Sorbicillactone A was identified as a potent and selective anti-leukemic and anti-HIV agent, while no significant sorbicillactone B activity was identified. Bisvertinolone has been isolated based on pigment from the fungi Verticillium intertextum and Acremonium stricturn in two key studies (Fig. 15b).162, 163 The dimeric compound was found to inhibit β-l,6-glucan biosynthesis in the fungus-like oomycete plant pathogen Phytophthora capsid.162 Bisorbicillinol was isolated with other sorbicillins in the late 1990’s based on its antioxidant activity as a DPPH (2,2-diphenyl- 1-picrylhydrazyl) radical scavenger (Fig. 15c).164 Trisorbicillinones A, B, C, and D were isolated from a deep- ocean fungus based on their various levels of cytotoxicity against cancer cell lines.165, 166 Trisorbicillinone A (Fig. 15d) exhibited the most potent activity with an IC50 of 3.14 μM against the HL60 acute myeloid leukemia cell line.
As with the tropolones and the azaphilones, the sorbicillin family of NPs are biosynthesized by an nonreducing iterative PKS/PKS hybrid cluster, though without orcinaldehyde and tropolone intermediates.139 Following the TR-mediated release of a mature linear hexaketide, a putatively non-PT-mediated Knoevenagel cyclization of the free aldehyde results in sorbicillin (Fig. 15a), the aromatic precursor to members of this class.139 Oxidative dearomatization followed by additional cyclization and dimerization patterns afford the full diversity of the sorbicillin family of NPs. Though there are no genomic investigations into the complete bisorbicillinol or trisorbicillinone biosyntheses, feeding studies using isotopically labeled precursors have established sorbicillin incorporation and those for trisorbicillinone suggest convergent pathways, one involving bisvertinolone as an intermediate.160,167
9.4. Nocardia polyketides
A rare example of bacterial orcinaldehyde-containing NPs was reported in 2016 by the Khosla group at Stanford University when they reconstituted five of eight Nocardia spp. polyketide catalytic modules in vitro.166 These strains were targeted because they contained a family of orphan PKS BGCs that were unique to nine clinical Nocardia strains from patients diagnosed with nocardiosis. A pentamodular PKS system in the presence of octanoyl-CoA, malonyl-CoA, NADPH, and S-adenosyl methionine yielded orcinaldehydes with extended polyketide side chains (Fig. 16). Interestingly, the biosynthesis includes a “stuttering,” iterative synthase for the production of a suite of varying ketide lengths. It should be noted that due to the incomplete reconstitution and in vitro nature of these experiments, the compounds characterized were non-natural intermediates of NPs that might arise from the biosynthetic machinery in the native host. These compounds were not evaluated for bioactivity.
Fig. 16.
Structure of the Nocardia polyketides. Structural elements in light blue are predicted and the ‘R’ group represents the unknown primer unit of the first PKS module.
10. Tetrahydroisoquinoline alkaloids
The metabolites that constitute the tetrahydroisoquinoline alkaloid NP group contain hemiaminals (or similarly active cyano residues) that are derived from the TR-released aldehyde intermediate similar to those found in the pyrrolobenzodiazepines. In both classes, dehydration of this hemiaminal in situ results in an iminium intermediate that forms a covalent adduct with DNA. However, this activity has been achieved by distinct biosynthetic means that translates into gross differences in scaffolds between the classes. Biosynthetically, tetrahydroisoquinoline alkaloid NPs are formed by a Pictet-Spengler reaction rather than the internal aryl amines found in the hemiaminals. Additionally, biosynthesis of this class requires multiple transformations, including multiple steps of reductive release from the enzyme complex (Fig. 17a), yielding structurally similar compounds often exhibiting anti-tumor activity.
Fig. 17.
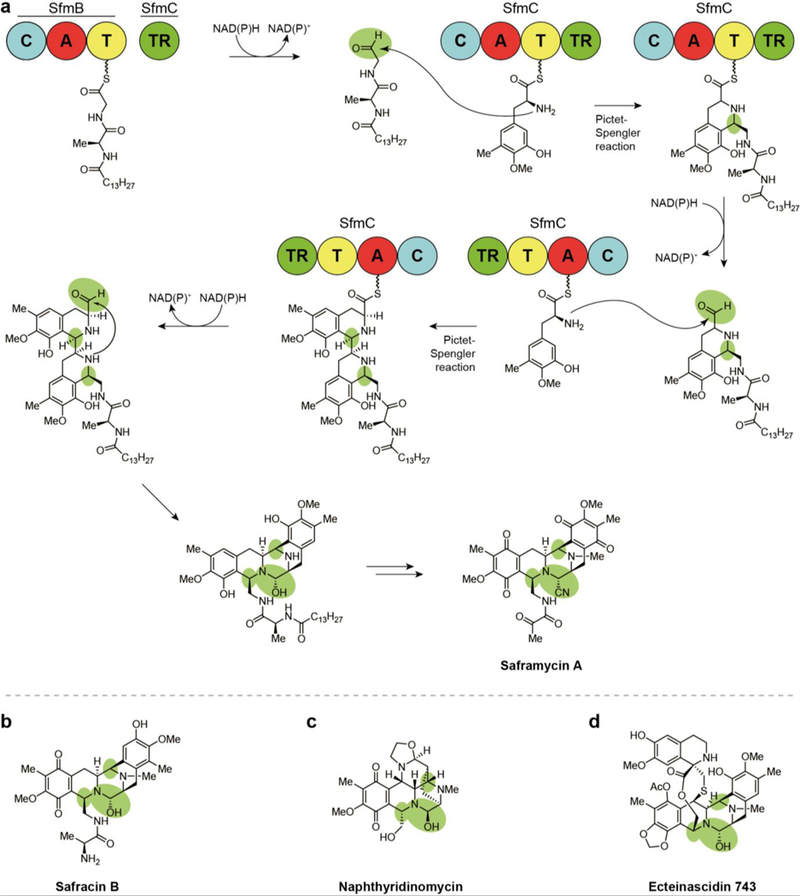
Biosynthesis of tetrahydroisoquinoline alkaloid natural products assembled via Pictet-Spengler reaction following two-electron reductive release, (a) Termination and final steps in the biosynthesis of saframycin A - the ‘R’ group indicates a leader peptide, a different form of which is involved in naphthyridinomycin biosynthesis, (b) Structure of safracin B. (c) Structure of naphthyridinomycin. (d) Structure of ecteinascidin 743.
10.1. Saframycins
The first thioester reductase-terminating pathway was discovered in 1996 when combined mutagenesis, intermediate feeding studies, and heterologous expression experiments confirmed the biosynthesis of the saframycin family of NPs (Fig. 17a).169, 170 This class also represents the safracins (Fig. 17b) and includes more than a dozen compounds that have been isolated since the late 1970s from Pseudomonas fiuorescens strains, Streptomyces lavendulae NRRL 11002, and Myxococcus xanthus.33, 171–174 Structurally, this class includes the complex pentacyclic ring systems of the saframycins and safracins as well as the larger hexacyclic naphthyridinomycin and nonacyclic ecteinascidin 743, all variously methylated and oxidized. Biological activity testing has revealed that all saframycins exhibit antitumor and antimicrobial activity. Their cytotoxicity involves inhibition of DNA and RNA synthesis via multiple modes of action including direct, sequence-specific binding to GC-rich triplets on the DNA template and redox-mediated DNA scission. 173,175–177 DNA binding is reported to occur at either a nitrile or hydroxyl group at the hemiaminal that originates from the released aldehyde. This substructural feature allows for the formation of an electrophilic iminium species that covalently alkylates DNA at the N-2 residue of guanine in the minor groove.178
Two NRPSs that incorporate glycine, alanine, and two tyrosine monomers are involved in the seven-step biosynthesis of these NPs, with an O-methyltransferase responsible for installation of the five methyl groups. In a logic divergent from other reductively released NPs and confirmed through domain- deletion mutants, a single reductase domain affects three rounds of reductive release on saframycin intermediates with two C-domain-mediated Pictet-Spengler reactions occurring following the first two releases (Fig. 17a).174, 178, 179 Surprisingly, the saframycin BGC has been found to require an acylated leader dipeptide for substrate recognition and successful conversion of intermediates by the Pictet-Spengler reaction.179 Feeding of synthetic intermediates to a heterologous mutant strain allowed for the detection of leader peptide-bound late stage saframycin intermediates.
10.2. Naphthyridinomycin
Naphthyridinomycin (Fig. 17c) is an antimicrobial and anticancer agent that was isolated from Streptomyces lusitanus by the Vezina lab.180, 181 Despite its alternative ring arrangement, naphthyridinomycin exhibits cytotoxicity similar to saframycin A and safracin B — contributing to evidence that these compounds owe their DNA binding activity to the hemiaminal residues that result from reductive release and subsequent Pictet-Spengler transformation.180, 182, 183 Naphthyridinomycin’s structurally complex contiguous stereocenters and fused ring system in addition to extensive non-collinearity of its BGC with the product has inspired multiple biosynthetic studies.40
Of these, analysis by the Tang group revealed that four naphthyridinomycin NRPS modules do not correspond with the structural units in the final product.183 Similar to saframycin biosynthesis, genetic and biochemical investigation of the BGC suggested a requirement for an N-terminal acylated leader peptide in core peptide intermediate assembly, a biosynthetic logic common in ribosomally synthesized and post- translationally modified peptides (RiPPs) but very rare in nonribosomal peptides.179 Deletion of the leader peptide genes completely ablated naphthyridinomycin biosynthesis while replacement of the genes severely diminished its production, supporting the importance of the encoded leader peptide. The terminal biosynthetic module, NapJ, reductively releases the biosynthetic intermediate as an aldehyde which subsequently undergoes a putative leader-peptide-dependent Pictet- Spengler reaction to form the core fused ring systems.183 Many of the other NPs in this class follow a similar biosynthetic logic.
10.3. Ecteinascidin 743
Ecteinascidin 743 (Fig. 17d), also known as trabectedin and used clinically for the treatment of soft tissue sarcoma and ovarian cancer, is produced by PharmaMar in Spain under the trade name Yondelis. It is the first marine-derived drug used clinically for the treatment of cancers. The anti-tumor activity of ecteinascidin 743 led to its discovery during a screening of the Caribbean mangrove tunicate Ecteinascidia turbinate in 1969.184, 185 Fortunately, ecteinascidin 743 shares a similar core to the saframycins. To fulfill supply that would be impossible from the scarcely available source tunicate, ecteinascidin 743 is prepared by a multi-step semi-synthetic route using cyanosafracin B from fermentation of its producer, Pseudomonas fluorescens.186 The specific ecteinascidin 743 mechanism of action includes the same sequence-specific DNA minor groove-alkylating action exhibited by the hemiaminal residue of similar compounds covered here.187 An additive benefit is achieved when a second tetrahydroisoquinoline ring beyond the typical core of others in this class protrudes from the bound DNA backbone to interfere with proteins involved in transcription-coupled nucleotide excision repair.
The BGC for the production of ecteinascidin 743 is similar to those of the saframycins and naphthyridinomycin,188, 189 differing only in the extensive post-reductive release modifications to the core. Due in part to its similarity to others in this class, the biosynthesis of ecteinascidin 743 has long been suspected to arise from a bacterial endosymbiont of the source tunicate. This suspicion was not confirmed until 2015 due to the inability to culture a producing organism from tunicate tissues in laboratory conditions. When the David Sherman group at the University of Michigan finally sequenced the full genome of Candidatus Endoecteinascidia frumentensis from metagenomic DNA isolated from the source organism, they discovered that nearly a third of the ~631 kb genome was devoted to production of the anti-cancer drug, suggesting a crucial role for the metabolite in the symbiont-host interaction. 190
11. Thioester reductase-catalyzed Dieckmann cyclization products
A subclass of TR domains catalyze a Dieckmann condensation- type release mechanism from NRPS and PKS systems instead of the typical reductive release (Fig. 18). Thioester reductase (TR) domains from these BGCs show sequence similarities to canonical enzymes in the SDR family but are NAD(P)H- independent, lack the Ser-Tyr-Lys catalytic triad, and do not catalyze a redox reaction.191 The type of release mechanism employed by these domains can lead to the formation of five- and six-membered heterocyclic rings.
Fig. 18.
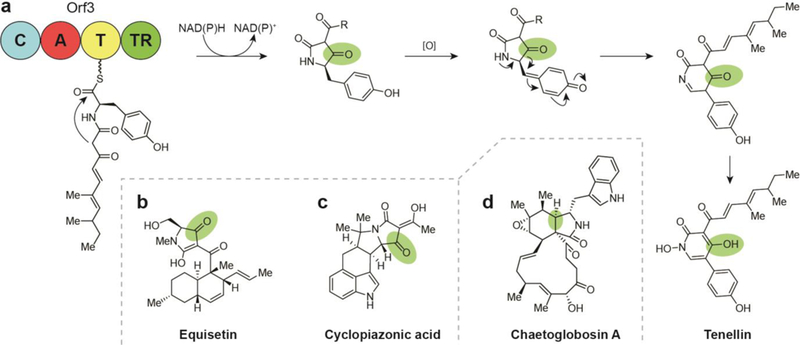
Biosynthesis of natural products by TR-domain directed Dieckmann condensation. (a) TR-domain directed Dieckmann condensation on tenellin. (b) Structure of equisetin. (c) Structure of cyclopiazonic acid. (d) Structure of chaetoglobosin of the cytochalasan class, which is structurally related to the tetramic acids, but undergoes cofactor-dependent reductive release as an aldehyde and spontaneously cyclizes.
11.1. Tenellin
The 2-pyridone, dienone side chain-containing tenellin was isolated in 1977 from the fungal insect pathogen Beauveria bassiana (Fig. 18a).37 Despite its source, it is not involved in pathogenicity. Instead, tenellin acts as an iron chelator, preventing reactive oxygen species generated from iron from causing toxicity in B. bassiana.192 The compound has also been reported to inhibit erythrocyte membrane ATPase activity, promoting cell lysis by membrane disruption. Additionally, the family of NPs that includes the tenellins has been shown to exhibit cytotoxicity in a range of cell lines and possess antibiotic and antifungal properties.192,193
Tenellin and several related compounds (bassianin from Beauveria tenella and farinosone B from Paecilomyces farinosus) are synthesized by a TR-terminating hybrid NRPS/PKS pathway. The NRPS module can accommodate incorporation of either tyrosine or phenylalanine, with a P450 monooxygenase putatively assigned to hydroxylation of phenylalanine so that selection of either amino acid results in the same final product.36 A condensation domain then links the amino acid to a doubly methylated pentaketide produced by the PKS machinery. Until 2008 it was proposed that the biosynthesis involved an aldehyde-containing tenellin precursor which underwent a non-enzymatic ring closing and a series of oxidation steps to eventually afford the 2-pyridone.34, 35 It was later proven through heterologous expression of the entire tenellin BGC that the TR domain in the biosynthetic pathway does not lead to the reductive release of the substrate, but rather behaves as a Dieckmann cyclase to perform the condensation that generates the tetramic acid-containing pretenellin A as shown in Fig. 18a.36 In the case of tenellins, a Dieckmann-type cyclization would be biosynthetically favorable over the reduction pathway because a reductive release would provide a product at a lower oxidation level than in the downstream pretenellin, whereas the Dieckmann pathway would not.
11.2. Tetramic acid natural products
Tetramic acids such as the equisetins can result from TR- catalyzed Dieckmann condensation release.194, 195 Equisetin and cyclopiazonic acid (Fig. 18b-c) were isolated from the fungal plant pathogen Fusarium equiseti and found to exhibit antibiotic activities against Gram-positive bacteria. Additional studies into equisetin bioactivity have determined that the compound is capable of inhibiting an HIV integrase and though it is broadly toxic, it is being investigated as an antiviral agent.196 The compound’s biosynthesis was identified upon the realization that it shared polyketide structural features with lovastatin, the blockbuster marketed drug to treat high cholesterol. From this observation, degenerate primers based on the lovastatin PKS sequences were used to amplify equisetin biosynthetic genes from a Fusarium producing strain. Notably, three equisetin PKSs share homology with lovastatin, with an identical domain order. A portion of the equisetin NRPS BGC resembles that of lovastatin, but the sequence of the latter is truncated and mostly inactive save for a hypothesized role for a C-domain in lactone formation - there is no known TR domain involvement in lovastatin. Similarities between the N-termini of lovastatin and equisetin suggest that the former has evolved from a degraded tetramic acid biosynthetic gene. Taken together, the biosynthesis of equisetin involves an iterative NRPS/PKS hybrid that produces a mature polyketide covalently linked to an amino acid for TR-mediated Dieckmann cyclization that forms a pyrrolidinone precursor to the tetramic acid. The Dieckmann condensation release in the biosyntheses of equisetin, cyclopiazonic acid, and other tetramic acids follow a similar mechanism (Fig. 18).197 It is suggested that point mutations and deletions within NRPS/PKS hybrids has evolved the vast diversity of NPs in fungi rather than by module swaps as is typical in bacteria. By this mechanism compounds such as cyclopiazonic acid and the cytochalasans class (Fig. 18d) have evolved as structurally dissimilar structures with similar biosyntheses.197–199
Cyclopiazonic acid is an indole-tetramic acid neurotoxin produced by a variety of Aspergillus species (Fig. 18b). Similar to equisetin, cyclopiazonic acid biosynthesis involves linking an NRPS-incorporated single amino acid (in this case a tryptophan) with a PKS-loaded polyketide for TR Dieckmann cyclization release and subsequent formation of a tetramic acid.198
Biosynthesis of cytochalasans by Aspergillus spp. may represent an example of the aforementioned divergence in fungal NRPS/PKS hybrid evolution via point mutations and deletions. The mature linear precursor is closely related to others in this section but retains the Tyr-containing catalytic triad and is released by the canonical cofactor-dependent reduction to a free aldehyde that spontaneously cyclizes.200 The assembly of cytochalasan occurs very similarly to cyclopiazonic acid in its NRPS incorporation of a single amino acid, but its PKS assembles a much longer polyketide chain that later becomes a macrocycle. Thioester reductase-mediated release leads into a Diels-Alder cyclization and a series of oxidations to yield a unique isoindolone within a macrocyclic, three-ring core skeleton, as in chaetoglobosin A (Fig. 18d).199 Biologically, the cytochalasan family affects distinctive antibiotic, antiviral, antiinflammatory, and antitumor activities and exhibits a marked affinity for actin filament binding.
12. Biological activity and therapeutic relevance of reductively off-loaded natural products
A chart of the activities exhibited by classes of thioester reductase tailored NPs is shown in Table 1. Anti-cancer (cytotoxic, anti-tumor, antiproliferative) activities were exhibited by a predominance of the compound families, while anti-microbial (anti-bacterial, anti-fungal) activities are less often observed. Among the remaining activities are an inhibitor of HIV-1 integrase, a family of iron-binding compounds (siderophores), a family of skin irritants, and an antihistamine. Of the reductively released compounds discussed here, those with the greatest therapeutic potential and those with evidenced clinical success tend to fall in the peptide aldehyde (Fig. 19a), pyrrolobenzodiazepine (Fig. 19b), or tetrahydroisoquinoline alkaloid classes and all are reversible covalent inhibitors of their targets. Despite these examples, only recently has covalent binding of targets become appreciated.
Table 1.
Biological activity of natural products from biosynthetic gene clusters with thioester reductase domains. Compounds are sorted based on structure and biosynthesis.
| Natural product | Isolation source | Discovery approach | Biological activity | Reference |
|---|---|---|---|---|
| primary alcohol-containing natural products | ||||
| myxochelin A | Angiococcus disciformis An d30 | bioactivity-guided | siderophore, tumor cell invasion inhibitor, Gram-positive antibiotic |
27, 32, 40, 41 |
| (−)-indolactam V (lyngbyatoxins/teleocidins, olivoretins) |
Moorea producens, Streptomyces mediocidicus |
bioactivity-guided | skin irritant, toxin, tumor promoter |
10, 42–49 |
| myxalamids A-D | Myxococcus xanthus | bioactivity-guided | antifungal, Gram-positive antibiotic |
24, 50–53 |
| non-tuberculosis mycobacterial glycopeptidolipids |
non-tuberculosis mycobacteria (NTM) |
mutagenesis | cell wall integrity, pathogenicity |
22, 23, 54–56, 58 |
| aldehyde-containing natural products | ||||
| leupeptins Ac-LL, Pr-LL | Actinobacteria | bioactivity-guided | antiproliferative (proteasome inhibition) |
61–64 |
| ureido tetrapeptide aldehydes [antipains, elastatinal, chymostatin, microbial alkaline protease inhibitor (β MAPI)] |
Streptomyces spp. | reactivity-based screening | antiproliferative (proteasome inhibition) |
64 |
| fellutamides A-F |
Penicillium fellutanum Biourge, Aspergillus nidulans |
bioactivity-guided | antiproliferative (proteasome inhibition) |
18, 68, 70 |
| flavopeptins 1–4 | Streptomyces flavogriseus | Proteomic Investigation of Secondary Metabolism (PrISM) |
antiproliferative (proteasome inhibition) |
17 |
| pyrazinones | human gut commensals (Clostridia, Bacteroides, Desulfovibrio) |
metagenomic mining | unknown | 72 |
| dipeptide aldehydes | human gut commensals (Clostridia, Bacteroides, Desulfovibrio) |
metagenomic mining | antiproliferative (proteasome inhibition) |
72 |
| azinomycin B / carzinophilin A | Streptomyces spp. | bioactivity-guided | broad-spectrum antibiotic, cytotoxic, DNA alkylating |
73–76 |
| macrocyciic iminopeptides | ||||
| nostocyclopeptides A1–3, Ml | Nostoc sp. ATCC-53789 | bioactivity-guided | cytotoxic, organic anion transporter inhibiting, microcystin antitoxins |
12, 13, 28, 78, 79 |
| scytonemide A | Scytonema hofmannii | bioactivity-guided | antiproliferative (proteasome inhibition) |
14 |
| Koranimine | Bacillus sp. | Proteomic Investigation of Secondary Metabolism (PrISM) |
unknown | 29 |
| Lugdunin | Staphylococcus lugdunensis | metagenome mining | Gram-positive antibiotic | 82 |
| amine-containing natural products | ||||
| L-lysine | Saccharomyces cerevisiae | essential amino acid, biochemical assays |
essential amino acid | 83 |
| myxochelin B | Angiococcus disciformis An d30 | genetic/biochemical screening | siderophore, tumor cell invasion inhibitor, Gram-positive antibiotic |
32, 84 |
| zeamine, zeamine I, II |
Serratia plymuthica RVH1, Dickeya zeae |
bioactivity-guided | antibiotic | 85–87 |
| leucinostatins A–B | Purpureocillium lilacinum | bioactivity-guided | anthelmintic, anti-oomycete, antimicrobial, |
89–91 |
| polyketide alkaloids | ||||
| latumcidin (AKA: abikoviromycin) |
Streptomyces reticuli var. latumcidicus |
bioactivity-guided | antiviral | 103–105 |
| coelimycin P1 | Streptomyces coelicolor M145 | genome mining | unknown | 30, 108 |
| nigrifactin |
Streptomyces nigrifaciens var. FFD-101 |
chemical screening | antihistamine, antihypertensive |
92, 93 |
| pyrindicin | Streptomyces NA-15 | chemical screening | weak antimicrobial, analgesic, antiplatelet |
95–97 |
| indolizomycin | Streptomyces SK2–52 | bioactivity-guided | unknown | 206 |
| streptazolin |
Streptomyces viridochromogenes Tü-1678 |
chemical screening | antimicrobial | 94 |
| streptazones A, B1, B2, C-E |
Streptomyces sp. FORM5, Streptomyces sp. A1, Streptomyces sp. HS-NF-853, Streptomyces sp. MSC090213JE08 |
chemical screening | cytotoxic, antibiotic | 98, 99 |
| cyclizidines A-l, (+)-ent-cyclizidine |
Streptomyces sp. NCIB 11649, Streptomyces sp. HNA39, Saccharopolyspora RL78 |
bioactivity-guided, chemical screening |
antifungal, anticancer | 100–102 |
| argimycins PI–IX | Streptomyces argillaceus | genome mining | weak antibiotics | 106, 107 |
| pyrrolobenzodiazepines | ||||
| anthramycin, mazethramycin, porothramycin, tomaymycin, sibiromycin |
Streptomyces spp., Streptosporangium sibiricum |
bioactivity-guided | anti-tumor, antibiotic, antiviral, antiprotazoal |
15, 16, 112, 114, 117–123, 207– 210 |
| tilivalline, tilimycin/kleboxymycin |
Klebsiella oxytoca | mutagenesis, gene deletion | enterotoxic | 119, 131 |
| pyrrolopyrazines | ||||
| Le-pyrrolopyrazines A–C | Lysobacter enzymogenes | genome mining | unknown | 133 |
| nonreducing iterative polyketide synthase natural products | ||||
| tropolones | ||||
| 3-methylorcinaldehyde, talaroenamine, leptosphaerdione, stipitaldehyde |
Talaromyces stipitatus (Penicillium stipitatum) |
gene knockout experiments | unknown | 137 |
| xenovulene A | Acremonium strictum | bioactivity-guided | GABA-benzodiazepine receptor inhibitor |
137, 141, 142 |
| epolones A and B | Fungus OS-F69284 | bioactivity-guided | erythropoietin stimulators | 137, 143 |
| eupenifeldin |
Eupenicillium brefeldianum ATCC 74184 |
bioactivity-guided | antitumor | 137, 144 |
| pycnidone | Phoma sp. MF 5726 | bioactivity-guided | antiarthritic, erythropoietin stimulators |
137, 143, 145 |
| sepadonin | Sepedonium chrysospermum Fries | bioactivity-guided | broad-spectrum antibiotic, antifungal |
137, 146, 147 |
| azaphilones | ||||
| citrinin |
Monascus spp., Aspergillus spp., Penicillium spp. |
pigment-guided | nephrotoxin | 26, 137 |
| asperfuranone | Aspergillus nidulans | genome mining | antiproliferative | 137, 149, 159 |
| sclerotiorin |
Cephalotheca faveolata, Penicillium frequentans CFTRI A-24 |
bioactivity-guided | antifungal, antioxidant | 137, 150, 151 |
| azanigerones A–F | Aspergillus niger ATCC 1015 | genome mining | unknown | 153 |
| mitorubrinol, mitorubrinic acid |
Penicillium rubrum, Penicillium marneffei |
pigment-guided | virulence factors, | 154, 155 |
| sorbicittins | ||||
| sorbicillactones A and B | Penicillium chrysogenum | bioactivity-guided | antileukemic, antiviral |
139, 161 |
| bisvertinolone |
Penicillium sp., Verticillium intertextum, Acremonium stricturn |
bioactivity-guided | anti-oomycete, cytotoxic |
139, 211, 212 |
| bisorbicillinol | Trichoderma sp. USF-2690 | chemical screening | antioxidant | 164 |
| trisorbicillinone A–D | Phialocephala sp. FL30r | bioactivity-guided | cytotoxic | 139, 165, 166 |
| tetrahydroisoquinoline alkaloids | ||||
| safracin A and B; saframycins A–G, Y3, and Mxl; cyanosafracin B (AKA: cyanoquinonamine) |
Streptomyces lavendulae | bioactivity-guided, spectroscopic screening |
anti-tumor |
33, 169–173, 175–177, 213– 215 |
| naphthyridinomycins A and B | Streptomyces lusitanus | bioactivity-guided | anti-tumor | 180–183 |
| ecteinascidins 729, 743, 759B, 759C, 770, 786 |
Ecteinascidia turbinate (Candidatus Endoecteinascidia frumentensis) |
bioactivity-guided | anti-tumor |
184, 185, 187– 189 |
| thioester reductase-catalyzed Dieckmann cyclization products | ||||
| tenellins (tenellin, pretenellin A and B,25-hydroxytenellin, Ilicicolin FI) |
Beauveria bassiana | bioactivity-guided, pigment-guided |
antiproliferative | 34–37, 193 |
| equisetins (equisetin, trichosetin, pallidorosetin A, ophiosetin, cyclopiazonic acid, aspyridone A) |
Fusarium equiseti | bioactivity-guided, genome mining |
antibiotic, cytotoxic, HIV integrase inhibition |
194–197 |
| chaetoglobosin A |
Chaetomium globosum, Aspergillus spp. |
bioactivity-guided | antibiotic, antiviral, anti-inflammatory, antitumor |
199, 216, 217 |
Fig. 19.
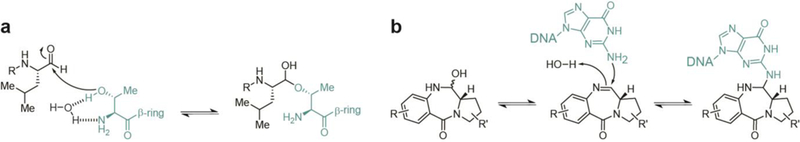
Select mechanisms of action for reductively released natural products, (a) The peptide aldehyde proteasome inhibition mechanism of action involves formation of a hemiacetal with the threonine hydroxyl in one of the (3-ring active sites, (b) The pyrrolobenzodiazepine cytotoxic mechanism of action involves sequence-specific DNA crosslinking to guanidine in the minor groove. Inhibitors are depicted in black while target residues are cyan.
Insights about toxicity reported in the 1970s and 80s deterred those in drug discovery from developing reversible and irreversible covalent inhibitors, which were thought to pose a risk of promiscuously modifying critical off-target biomolecules.201 Despite this concern, 30% of current drugs, including multiple blockbusters, act by a covalent mechanism.201 Many of these were intentionally designed for non-covalent inhibition and were only later discovered to act by a covalent mechanism.202 Covalent binding has been linked to greater biochemical efficiency in competing with endogenous substrates202 and a longer duration of action, which permits less-frequent administration at lower doses to result in wide therapeutic margins. In addition, NPs and other small molecules with this property hold the promise of targeting formerly ‘undruggable’ targets, while sustained target suppression amounts to a reduced risk for the development of resistance, growing challenges in both oncology and infectious disease. 201 These realizations have resulted in a recent, significant increase in the number of covalent inhibitor drug candidates progressing through clinical trials or being approved by the FDA.
Of the covalent inhibitors reviewed, the peptide aldehydes are the most varied and numerous. Among the peptide aldehydes discovered primarily from bacteria, chymostatin and leupeptin are commonly found as components in commercially available protease inhibitor cocktails for biochemical research. In a rare case within NP discovery, biomedical application of the peptide aldehydes mimics their evolved role as protease inhibitors as evidenced by the discovery of gut commensal dipeptide aldehydes by the Fischbach group.72 What’s more, the compounds they discovered are of utmost relevance to human health - having evolved within humans, the dipeptide aldehydes have direct applicability to and may have been evolved specifically to modulate human health. Of note, the earliest discovery of tripeptide aldehydes provided the inspiration for bortezomib, a peptide boronic acid and covalent proteasome inhibitor, with approvals for the treatment of multiple myeloma and mantle cell lymphoma since 2006.203
Targeting of proteases is of particular interest for the treatment of cancers. The 26S proteasome is a protease in eukaryotic cells with multiple catalytic sites that is responsible for breaking down and recycling transient, damaged, oxidized, and misfolded proteins. In addition to its essential physiological role in healthy cells, the proteasome can act inappropriately in some cancers, promoting proliferation.204 Myelomas are a particularly attractive disease target as their dysregulated production of large immunoglobulin proteins confers a heightened sensitivity to protease inhibitors, therefore allowing for effective treatment with less toxicity to healthy cells.38 Covalent inhibitors of the proteasome tend to exert their activity by forming adducts with the hydroxy group of the N-terminal threonine at the protease catalytic sites (Fig. 19a).
As discussed in Section 7, the potent and selective reversible covalent DNA alkylating mechanism (Fig. 19b) of synthetically optimized NP pyrrolobenzodiazepines has led to efforts aimed at generating dimers, most notably SJG-136, for inter- and intrastrand binding in oncology.112 This compound completed phase II clinical trials for cancers in 2015 and a related pyrrolobenzodiazepine dimer antibody-drug conjugate named rovalpituzumab tesirine (Rova-T; SC16LD6.5) and developed by Stemcentrx Inc. (AbbVie) finished phase clinical trials for the treatment of lung cancer in mid-2017.116, 205
An overview of ecteinascidin 743 (trabectedin) was given in Section 10.3. This tetrahydroisoquinoline alkaloid drug is clinically administered as Yondelis for the treatment of soft tissue sarcoma and ovarian cancer and is notable as the first marine-derived drug used clinically to treat cancers. Additionally, ecteinascidin 743 exhibits unique potency via a dual-mode mechanism of action involving covalent and specific reversible DNA binding in tandem with transcription protein interferance.184, 186, 187 Based on the investigations and numerous drug development accounts summarized here, it is clear that new natural products from reductase domain containing biosynthetic pathways would offer significant prospects as molecular probes and drug-leads.
13. Conclusions
As presented, natural products from thioester reductase terminating biosynthetic gene clusters represent a wide range of structural classes. It follows that the biological sources of thioester reductase domains are similarly broad, occurring in NP biosynthesis across bacterial and fungal taxa. The wealth of unexplored TR-containing biosynthetic modules with associated NPs awaiting discovery from microbes is worthy of repeated emphasis. The majority of genomic research in NP discovery has focused on bacteria, specifically from the phylum Actinobacteria, since they devote approximately 10% of their total genome to secondary metabolite production.218 Filamentous fungi are also prolific producers of secondary metabolites, often containing >50 BGCs per genome.219 Surprisingly, fungi of the genera Aspergillus and Pencillium have about a ten times greater prevalence of reductase-terminating NRPSs than bacteria of the genus Streptomyces, which are among the most prolific producers of bioactive compounds since the dawn of the golden age of antibiotics. A search of all UniProt 2017 entries reveals average TR-domain frequencies of 13.5,10.2, and 0.7 per Aspergillus, Pencillium, and Streptomyces genome, respectively (Table 2). This amounts to 19 of 63 BGCs for the model fungus Aspergillus nidulans,219, 220 and 15 of 47 BGCs for the industrial penicillin producer Penicillium chrysogenum,221 compared to a frequency of <1 per genome for the model bacterial organisms Streptomyces coelicolor and Streptomyces lividans 222 The most common biological sources of microbial NPs analyzed here represent promising sources for continued discovery efforts targeting reductively modified metabolites, with the most remarkable potential found within fungal genomes.
Table 2.
Reductase-terminated biosynthetic gene cluster frequency of occurrence across microbiology.
| NRPS and NRPS hybrid BGCs | PKS and PKS hybrid BGCs> | |||||
|---|---|---|---|---|---|---|
| Taxa | Number of whole genome sequencesa |
A+T+TR/A+T (%) |
A+T+TR per genome* |
Number of whole genome sequencesa |
KS+AT+TR/KS+AT (%) |
KS+AT+TR per genome |
| Proteobacteria | 6969 | 5.9 | 0.12 | 6969 | 0.84 | 0.00 |
| Actinobacteria | 2193 | 9.8 | 0.72 | 2193 | 0.66 | 0.03 |
| Streptomyces | 359 | 4.2 | 0.70 | 359 | 1.08 | 0.14 |
| Cyanobacteria | 249 | 5.8 | 0.56 | 249 | 0.30 | 0.01 |
| Firmicutes | 3847 | 11.3 | 0.16 | 3847 | 5.14 | 0.01 |
| Ascomycota | 615 | 46.4 | 5.27 | 615 | 18.56 | 1.94 |
| Aspergillus | 40 | 44.6 | 13.50 | 40 | 18.28 | 4.90 |
| Penicillium | 29 | 43.2 | 10.24 | 29 | 19.12 | 3.76 |
| Basidiomycota | 190 | 51.8 | 1.88 | 190 | 18.53 | 0.23 |
A, adenylation domain; T, thiolation domain; TR, thioester reductase domain; KS, ketosynthase domain; AT, acyltransferase domain; A+T+TR, total number of adenylation-thiolation didomains with an associated thioester reductase domain; A+T, total number of adenylation-thiolation didomains; KS+AT+TR, total number of ketosynthase-acyltransferase di-domains with an associated thioester reductase domain; A+T, total number of ketosynthase-acyltransferase di-domains
genomes available from the 2017 release of UniProt
The compounds arising from these BGCs affect a variety of useful biological activities. A relatively newfound appreciation for covalent inhibitors in oncology and other disease treatments has renewed research interest in reductively released products with reactive DNA crosslinking and protease inhibition mechanisms. The mounting genomic evidence of a vast, untapped microbial reductive natural product chemical space combined with an apparent privilege for potent bioactivity justifies continued discovery efforts targeting thioester reductase domains for novel natural products.
15. Acknowledgements
This publication was supported by the National Cancer Institute (NCI) of the National Institutes of Health (NIH) under Award Number F32CA221327 (MWM), the National Institute of General Medical Sciences (NIGMS) of the NIH through the Chemistry of Life Processes (CLP) Training Grant under Award Number T32GM105538 (RAM), and the National Center for Complementary and Integrative Health (NCCIH) of the NIH under Award Number R01AT009143 (RJT, NLK). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Biography

Michael Mullowney studied chemistry and biology at DePaul Univerity while conducting research in the lab of Dr. Justin J. Maresh. He earned his Ph.D. in 2016 as an AFPE and NIH NCCIH T32 predoctoral fellow in the lab of Prof. Brian T. Murphy at the University of Illinois at Chicago, where he discovered novel anti-tuberculosis drug-leads from acquatic-derived actinomycete bacteria. Dr. Mullowney is currently at Northwestern University as an NIH NRSA Postdoctoral Fellow, working under the co-mentorship of Prof. Neil L. Kelleher and Prof. Regan J. Thomson to advance correlative metabolomic and genomic approaches toward natural products discovery.

Ryan McClure obtained his B.S. in Chemistry from the Illinois Institute of Technology in 2011. He then earned his Ph.D. in Chemistry from Northwestern University in 2016 under the joint mentorship of Prof. Neil Kelleher and Prof. Regan Thomson as an NIH NIGMS T32 Predoctoral Fellow, where he worked on the discovery, synthesis, and/or bioactivity evaluation of seven natural products including the flavopeptins as well as the development of the metabologenomics platform. Dr. McClure is currently a Senior Scientist at AbbVie, Inc. as a member of the Proteomics and Chemical Biology groups.

Matthew Robey obtained his B.S. in Biochemistry and Molecular Biology from Bethel University in Minnesota and is currently undertaking PhD research with Prof. Neil Kelleher. His current research is focused on the discovery of fungal specialized metabolites and characterization of their biosynthetic gene clusters.

Neil Kelleher received his B.S. in Chemistry from Pacific Lutheran University in 1992. He then obtained his Ph.D. in Chemistry from Cornell University in 1997 where he worked in the labs of Prof. Fred McLafferty and Prof. Tadhg Begley studying the characterization of proteins by electrospray ionization fourier- transform mass spectrometry. Neil completed his postdoctoral fellowship in natural products biosynthesis with Prof. Christopher Walsh at Harvard Medical School in 1999. He is currently a Professor of Chemistry at Northwestern University where he leads his research group in top-down proteomics, chromatin biology, and natural products biosynthesis/discovery and is the Director of the Proteomics Center of Excellence.

Regan J. Thomson was born in New Zealand in 1976 and received his Ph.D. in 2003 at The Australian National University working with Prof. Lewis N. Mander. Following postdoctoral studies with Prof. David A. Evans at Harvard University, he joined the faculty at Northwestern University in 2006 where he is currently a Professor of Chemistry. Regan’s research interests include reaction development, total synthesis, natural product discovery and biosynthesis, and atmospheric chemistry. He is the recipient of an NSF CAREER Award (2009), an Amgen Young Investigator Award (2010), an Illinois Division American Cancer Society Research Scholar Award (2012), and a Novartis Chemistry Lectureship (2015— 2016).
Footnotes
Conflicts of interest
There are no conflicts to declare.
16. References
- 1.Donadio S, Monciardini P and Sosio M, Nat. Prod. Rep, 2007, 24, 1073–1109. [DOI] [PubMed] [Google Scholar]
- 2.Fischbach MA and Walsh CT, Chem. Rev, 2006, 106, 3468–3496. [DOI] [PubMed] [Google Scholar]
- 3.Newman DJ and Cragg GM, J. Nat. Prod, 2016, 79, 629–661. [DOI] [PubMed] [Google Scholar]
- 4.Alberts AW, Chen J, Kuron G, Hunt V, Huff J, Hoffman C, Rothrock J, Lopez M, Joshua H, Harris E, Patchett A, Monaghan R, Currie S, Stapley E, Albers-Schonberg G, Hensens O, Hirshfield J, Hoogsteen K, Liesch J and Springer J, Proc. Natl. Acad. Sci. U. S. A, 1980, 77, 3957–3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hajar R, Heart Views, 2011, 12, 121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whicher JR, Dutta S, Hansen DA, Hale WA,Chemler JA, Dosey AM, Narayan ARH, Håkansson K, Sherman DH, Smith JL and Skiniotis G, Nature, 2014, 510, 560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dutta S, Whicher JR, Hansen DA, Hale WA, Chemler JA, Congdon GR, Narayan AR, Håkansson K, Sherman DH, Smith JL and Skiniotis G, Nature, 2014, 510, 512–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crawford JM and Townsend CA, Nat. Rev. Microbiol,8, 879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du L and Lou L, Nat. Prod. Rep, 2010, 27, 255–278. [DOI] [PubMed] [Google Scholar]
- 10.Read JA and Walsh CT, J. Am. Chem. Soc,2007,129, 15762–15763. [DOI] [PubMed] [Google Scholar]
- 11.Kavanagh KL, Jörnvall H, Persson B and Oppermann U, Cell. Mol. Life Sci, 2008, 65, 3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herfindal L, Myhren L, Kleppe R, Krakstad C, Selheim F,Jokela J, Sivonen K and Doskeland SO, Mol. Pharm,2010, 8, 360–367. [DOI] [PubMed] [Google Scholar]
- 13.Jokela J, Herfindal L, Wahlsten M, Permi P, Selheim F, Vasconcelos V, Doskeland SO and Sivonen K, ChemBioChem, 2010, 11, 1594–1599. [DOI] [PubMed] [Google Scholar]
- 14.Krunic A, Vallat A, Mo S, Lantvit DD, Swanson SM and Orjala J, J. Nat. Prod, 2010, 73, 1927–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leimgrub W, Stefanov V, Schenker F, Karr A and Berger J, J. Am. Chem. Soc, 1965, 87, 5791. [DOI] [PubMed] [Google Scholar]
- 16.Hu Y, Phelan V, Ntai I, Farnet CM, Zazopoulos E and Bachmann BO, Chem. Biol, 2007, 14, 691–701. [DOI] [PubMed] [Google Scholar]
- 17.Chen YQ, McClure RA, Zheng YP, Thomson RJ and Kelleher NL, J. Am. Chem. Soc, 2013, 135, 10449–10456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hines J, Groll M, Fahnestock M and Crews CM, Chem. Biol, 2008, 15, 501–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rentsch A, Landsberg D, Brodmann T, Buelow L, Girbig A-K and Kalesse M, Angewandte Chemie-International Edition, 2013, 52, 5450–5488. [DOI] [PubMed] [Google Scholar]
- 20.Teicher BA, Ara G, Herbst R, Palombella VJ and Adams J, Clin. Cancer. Res, 1999, 5, 2638–2645. [PubMed] [Google Scholar]
- 21.D’Incalci M and Galmarini CM, Mol. Cancer Ther, 2010, 9, 2157–2163. [DOI] [PubMed] [Google Scholar]
- 22.Chhabra A, Haque AS, Pal RK, Goyal A, Rai R, Joshi S, Panjikar S, Pasha S, Sankaranarayanan R and Gokhale RS, Proc. Natl. Acad. Sci. U.S.A, 2012, 109, 5681–5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kinatukara P, Patel KD, Haque AS, Singh R, Gokhale RS and Sankaranarayananan R, J. Struct. Biol, 2016, 194, 368–374. [DOI] [PubMed] [Google Scholar]
- 24.Barajas JF, Phelan RM, Schaub AJ, Kliewer JT, Kelly PJ, Jackson DR, Luo R, Keasling JD and Tsai SC, Chem. Biol, 2015, 22, 1018–1029. [DOI] [PubMed] [Google Scholar]
- 25.Gahloth D, Dunstan MS, Quaglia D, Klumbys E, Lockhart-Cairns MP, Hill AM, Derrington SR, Scrutton NS, 50 Turner NJ and Leys D, Nat. Chem. Biol, 2017, 13, 975–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He Y and Cox RJ, Chem. Sci, 2016, 7, 2119–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kunze B, Bedorf N, Kohl W, Hofle G and Reichenbach H, J. Antibiot. (Tokyo), 1989, 42, 14–17. [DOI] [PubMed] [Google Scholar]
- 28.Kopp F, Mahlert C, Grunewald J and Marahiel MA, J. Am. Chem. Soc, 2006, 128, 16478–16479. [DOI] [PubMed] [Google Scholar]
- 29.Evans BS, Ntai I, Chen Y, Robinson SJ and Kelleher NL, J. Am. Chem. Soc, 2011, 133, 7316–7319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Awodi UR, Ronan JL, Masschelein J, de los Santos ELC and Challis GL, Chem. Sci, 2017, 8, 411–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng H, Wei E, Wang J, Zhang Y, Cheng L, Ma H, Deng Z and Qu X, ACS Chem. Biol, 2016, 11, 3278–3283. [DOI] [PubMed] [Google Scholar]
- 32.Li Y, Weissman KJ and Muller R, J. Am. Chem. Soc, 2008, 130, 7554–7555. [DOI] [PubMed] [Google Scholar]
- 33.Mikami Y, Takahashi K, Yazawa K, Arai T, Namikoshi M, Iwasaki S and Okuda S, J. Biol. Chem, 1985, 260, 344–348. [PubMed] [Google Scholar]
- 34.Eley KL, Halo LM, Song Z, Powles H, Cox RJ, Bailey AM, Lazarus CM and Simpson TJ, ChemBioChem, 2007, 8, 289–297. [DOI] [PubMed] [Google Scholar]
- 35.Halo LM, Heneghan MN, Yakasai AA, Song Z, Williams K, Bailey AM, Cox RJ, Lazarus CM and T. J. 60 Simpson, J. Am. Chem. Soc, 2008, 130, 17988–17996. [DOI] [PubMed] [Google Scholar]
- 36.Halo LM, Marshall JW, Yakasai AA, Song Z, Butts CP, Crump MP, Heneghan M, Bailey AM, Simpson TJ, C. 61 M. Lazarus and R. J. Cox, ChemBioChem, 2008, 9, 585–594. [DOI] [PubMed] [Google Scholar]
- 37.Wat CK, Mcinnes AG, Smith DG, Wright JLC and Vining LC, Can. J. Chem, 1977, 55, 4090–4098. [Google Scholar]
- 38.Kisselev AF, van der Linden WA and Overkleeft HS, Chem. Biol, 2012, 19, 99–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Manavalan B, Murugapiran SK, Lee G and Choi S, BMC Struct. Biol, 2010, 10, 1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miyanaga S, Obata T, Onaka H, Fujita T, Saito N, Sakurai H, Saiki I, Furumai T and Igarashi Y, J. Antibiot, 2006, 59, 698–703. [DOI] [PubMed] [Google Scholar]
- 41.Gaitatzis N, Kunze B and Muller R, Proc. Natl. Acad. Sci. 66 U.S.A., 2001, 98, 11136–11141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cardellina JH 2nd, Marner FJ and Moore RE, Science, 1979, 204, 193–195. [DOI] [PubMed] [Google Scholar]
- 43.Aimi N, Odaka H, Sakai S-I, Fujiki H, Suganuma M, Moore RE and Patterson GML, J. Nat. Prod, 1990, 53, 1593–1596. [DOI] [PubMed] [Google Scholar]
- 44.Jiang W, Zhou W, Uchida H, Kikumori M, Irie K, Watanabe R, Suzuki T, Sakamoto B, Kamio M and Nagai H, Mar. Drugs, 2014, 12, 2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sakai S, Aimi N, Yamaguchi K, Hitotsuyanagi Y, Watanabe C, Yokose K, Koyama Y, Shudo K and Itai A, Chem. Pharm. Bull. (Tokyo), 1984, 32, 354–357. 71 [DOI] [PubMed] [Google Scholar]
- 46.Hitotsuyanagi Y, Yamaguchi K, Ogata K, Aimi N, Sakai S, Koyama Y, Endo Y, Shudo K, Itai A and Iitaka Y, Chem. Pharm. Bull. (Tokyo), 1984, 32, 3774–3778. [DOI] [PubMed] [Google Scholar]
- 47.Fujiki H, Mori M, Nakayasu M, Terada M, Sugimura T and Moore RE, Proc. Natl. Acad. Sci. U. S. A, 1981, 78, 3872–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fujiki H, Tanaka Y, Miyake R, Kikkawa U, Nishizuka Y and Sugimura T, Biochem. Biophys. Res. Commun, 1984, 120, 339–343. [DOI] [PubMed] [Google Scholar]
- 49.Edwards DJ and Gerwick WH, J. Am. Chem. Soc, 2004, 126, 11432–11433. [DOI] [PubMed] [Google Scholar]
- 50.Gerth K, Jansen R, Reifenstahl G, Hofle G, Irschik H, Kunze B, Reichenbach H and Thierbach G, J. Antibiot, 1983, 36, 1150–1156. [DOI] [PubMed] [Google Scholar]
- 51.Jansen R, Reifenstahl G, Gerth K, Reichenbach H and Hofle G, Liebigs Ann. Chem, 1983, 1081–1095. [Google Scholar]
- 52.Beyer S, Kunze B, Silakowski B and Muller R, Biochim. Biophys. Acta, 1999, 1445, 185–195. [DOI] [PubMed] [Google Scholar]
- 53.Silakowski B, Nordsiek G, Kunze B, Blocker H and Muller R, Chem. Biol, 2001, 8, 59–69. [DOI] [PubMed] [Google Scholar]
- 54.Ripoll F, Deshayes C, Pasek S, Laval F, Beretti J-L, Biet F,Risler J-L, Daffé M, Etienne G, Gaillard J-L and Reyrat J-M, BMC Genomics, 2007, 8, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Billman-Jacobe H, McConville MJ, Haites RE, Kovacevic S and Coppel RL, Mol. Microbiol, 1999, 33, 1244–1253. [DOI] [PubMed] [Google Scholar]
- 56.Fujiwara N, Naka T, Ogawa M, Yamamoto R, Ogura H and Taniguchi H, Tuberculosis, 2012, 92, 187–192. [DOI] [PubMed] [Google Scholar]
- 57.Brennan PJ and Nikaido H, Annu. Rev. Biochem, 1995, 64, 29–63. [DOI] [PubMed] [Google Scholar]
- 58.Pang L, Tian X, Pan W and Xie J, J. Cell. Biochem, 2013, 114, 1705–1713. [DOI] [PubMed] [Google Scholar]
- 59.Chiou KC, PhD thesis, University of Michigan, 2013. [Google Scholar]
- 60.Iizaka Y, Higashi N, Ishida M, Oiwa R, Ichikawa Y, Takeda M, Anzai Y and Kato F, Antimicrob. Agents Chemother, 2013, 57, 1529–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aoyagi T, Takeuchi T, Matsuzaki A, Kawamura K and Kondo S, J. Antibiot. (Tokyo), 1969, 22, 283–286. [DOI] [PubMed] [Google Scholar]
- 62.Hori M, Hemmi H, Suzukake K, Hayashi H, Uehara Y, Takeuchi T and Umezawa H, J. Antibiot. (Tokyo), 1978, 31, 95–98. [DOI] [PubMed] [Google Scholar]
- 63.Suzukake K, Hori M, Tamemasa O and Umezawa H, Biochim. Biophys. Acta, 1981, 661, 175–181. [DOI] [PubMed] [Google Scholar]
- 64.Maxson T, Tietz JI, Hudson GA, Guo XR, Tai H-C and Mitchell DA, J. Am. Chem. Soc, 2016, 138, 15157–15166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Suda H, Takeuchi T, Umezawa H, Aoyagi T and Hamada M, J. Antibiot. (Tokyo), 1972, 25, 263–266. [DOI] [PubMed] [Google Scholar]
- 66.Umezawa S, Tatsuta K, Fujimoto K, Tsuchiya T and Umezawa HJ, J. Antibiot. (Tokyo), 1972, 25, 267–270. [DOI] [PubMed] [Google Scholar]
- 67.Imker HJ, Walsh CT and Wuest WM, J. Am. Chem. Soc, 2009, 131, 18263–18265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shigemori H, Wakuri S, Yazawa K, Nakamura T, Sasaki T and Kobayashi J, Tetrahedron, 1991, 47, 8529–8534. [Google Scholar]
- 69.Albright JC, Henke MT, Soukup AA, McClure RA, Thomson RJ, Keller NP and Kelleher NL, ACS Chem. Biol, 2015, 10, 1535–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yeh HH, Ahuja M, Chiang YM, Oakley CE, Moore S, Yoon O, Hajovsky H, Bok JW, Keller NP, Wang CC and Oakley BR, ACS Chem. Biol, 2016, 11, 2275–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Imker HJ, Krahn D, Clerc J, Kaiser M and Walsh CT, Chem. Biol, 2010, 17, 1077–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guo C-J, Chang F-Y, Wyche TP, Backus KM, Acker TM, Funabashi M, Taketani M, Donia MS, Nayfach S, Pollard KS, Craik CS, Cravatt BF, Clardy J, Voigt CA and Fischbach MA, Cell, 2017,168, 517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yokoi K, Nagaoka K and Nakashima T, Chem. Pharm. Bull. (Tokyo), 1986, 34, 4554–4561. [DOI] [PubMed] [Google Scholar]
- 74.Nagaoka K, Matsumoto M, Oono J, Yokoi K, Ishizeki S and Nakashima T, J. Antibiot. (Tokyo), 1986, 39, 1527–1532. [DOI] [PubMed] [Google Scholar]
- 75.Kelly GT, Sharma V and Watanabe CMH, Bioorg. Chem, 2008, 36, 4–15. [DOI] [PubMed] [Google Scholar]
- 76.Zhao Q, He Q, Ding W, Tang M, Kang Q, Yu Y, Deng W, Zhang Q, Fang J, Tang G and Liu W, Chem. Biol, 2008,15, 693–705. [DOI] [PubMed] [Google Scholar]
- 77.Malins LR, deGruyter JN, Robbins KJ, Scola PM, Eastgate MD, Ghadiri MR and Baran PS, J. Am. Chem. Soc, 2017, 139, 5233–5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Golakoti T, Yoshida WY, Chaganty S and Moore RE, J. Nat. Prod., 2001, 64, 54–59. [DOI] [PubMed] [Google Scholar]
- 79.Becker JE, Moore RE and Moore BS, Gene, 2004, 325, 35–42. [DOI] [PubMed] [Google Scholar]
- 80.Welker M and Von Dohren H, FEMS Microbiol. Rev, 2006, 30, 530–563. [DOI] [PubMed] [Google Scholar]
- 81.Stauch B, Simon B, Basile T, Schneider G, Malek NP, Kalesse M and Carlomagno T, Angew. Chem. Int. Ed, 2010, 49, 3934–3938. [DOI] [PubMed] [Google Scholar]
- 82.Zipperer A, Konnerth MC, Laux C, Berscheid A, Janek D, Weidenmaier C, Burian M, Schilling NA, Slavetinsky C, Marschal M, Willmann M, Kalbacher H, Schittek B, Brotz-Oesterhelt H, Grand S, Peschel A and Krismer B, Nature, 2016, 535, 511–516. [DOI] [PubMed] [Google Scholar]
- 83.Ehmann DE, Gehring AM and Walsh CT, Biochem, 1999, 38, 6171–6177. [DOI] [PubMed] [Google Scholar]
- 84.Silakowski B, Kunze B, Nordsiek G, Blöcker H, Höfle G and Müller R, Eur. J. Biochem, 2000, 267, 6476–6485. [DOI] [PubMed] [Google Scholar]
- 85.Wu J, Zhang H-B, Xu J-L, Cox RJ, Simpson TJ and Zhang L-H, Chem. Commun, 2010, 46, 333–335. [DOI] [PubMed] [Google Scholar]
- 86.Zhou J, Zhang H, Wu J, Liu Q, Xi P, Lee J, Liao J, Jiang Z and Zhang L-H, Mol. Plant-Microbe Interact, 2011, 24, 1156–1164. [DOI] [PubMed] [Google Scholar]
- 87.Masschelein J, Mattheus W, Gao L-J, Moons P, Van Houdt R, Uytterhoeven B, Lamberigts C, Lescrinier E, Rozenski J, Herdewijn P, Aertsen A, Michiels C and Lavigne R, PLoS One, 2013, 8, e54143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Masschelein J, Clauwers C, Awodi UR, Stalmans K, Vermaelen W, Lescrinier E, Aertsen A, Michiels C, Challis GL and Lavigne R, Chem. Sci, 2015, 6, 923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang G, Liu Z, Lin R, Li E, Mao Z, Ling J, Yang Y, Yin W-B and Xie B, PLoS Path, 2016,12, el005685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Isogai A, Suzuki A, Tamura S, Higashikawa S and Kuyama S, J. Chem. Soc, Perkin Trans. 1, 1984, DOI: 10.1039/P19840001405, 1405–1411. [DOI] [Google Scholar]
- 91.Park J0, Hargreaves JR, McConville EJ, Stirling GR, Ghisalberti EL and Sivasithamparam K, Lett. Appl. Microbiol, 2004, 38, 271–276. [DOI] [PubMed] [Google Scholar]
- 92.Kaneko Y, Terashima T and Kuroda Y, Agric. Biol. Chem,32, 783–785. [Google Scholar]
- 93.Terashima T, Kuroda Y and Kaneko Y, Tetrahedron Lett,10, 2535–2537. [DOI] [PubMed] [Google Scholar]
- 94.Drautz H, Zähner H, Kupfer E and Keller-Schierlein W, Helv. Chim. Acta, 1981, 64, 1752–1765. [Google Scholar]
- 95.Onda M, Konda Y, Narimatsu Y, Omura S and Hata T, Chem. Pharm. Bull. (Tokyo), 1973, 21, 2048–2050. [Google Scholar]
- 96.Omura S, Tanaka H, Awaya J, Narimatsu Y, Konda Y and Hata T, Agric. Biol. Chem, 1974, 38, 899–906. [Google Scholar]
- 97.Omura S, Tanaka H, Awaya J, Narimatsu Y, Onda Y and Hata T, Agric. Biol. Chem, 1974, 38, 899–906. [Google Scholar]
- 98.Puder C, Krastel P and Zeeck A, J. Nat. Prod, 2000, 63, 1258–1260. [DOI] [PubMed] [Google Scholar]
- 99.Liu Q-F, Wang J-D, Wang X-J, Liu C-X, Zhang J, Pang Y-W, Yu C and Xiang W-S, J. Asian Nat. Prod. Res, 2013, 15, 221–224. [DOI] [PubMed] [Google Scholar]
- 100.Freer AA, Gardner D, Greatbanks D, Poyser JP and Sim GA, J. Chem. Soc., Chem. Commun, 1982, DOI: 10.1039/C39820001160, 1160–1162. [DOI] [Google Scholar]
- 101.Jiang Y-J, Li J-Q, Zhang H-J, Ding W-J and Ma Z-J, J. Nat. Prod., 2018, 81, 394–399. [DOI] [PubMed] [Google Scholar]
- 102.Izumikawa M, Hosoya T, Takagi M and Shin-ya K, J. Antibiot, 2011, 65, 41. [DOI] [PubMed] [Google Scholar]
- 103.Sakagami Y, Yamaguchi I, Yonehara H, Okimoto Y, Yamanouchi S, Takiguchi K and Sakai H,!. Antibiot. (Tokyo), 1958,11, 6–13. [PubMed] [Google Scholar]
- 104.Gurevich AI, Kolosov MN, Korobko VG and Onoprienko VV, Tetrahedron Lett, 1968, 9, 2209–2212. [DOI] [PubMed] [Google Scholar]
- 105.Seto H, Sato T, Yonehara H and Jankowski WC, J. Antibiot. (Tokyo), 1973, 26, 609–611. [DOI] [PubMed] [Google Scholar]
- 106.Ye S, Molloy B, Braña AF, Zabala D, Olano C, Cortés J, Morís F, Salas JA and Méndez C, Front. Microbiol, 2017, 8,194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ye S, Braña AF, González-Sabíin J, Morís F, Olano C, Salas JA and Méndez C, Frontiers in Microbiology, 2018, 9, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gomez-Escribano JP, Song L, Fox DJ, Yeo V, Bibb MJ and Challis GL, Chem. Sci, 2012, 3, 2716–2720. [Google Scholar]
- 109.Huang W, Kim SJ, Liu J and Zhang W, Org. Lett, 2015, 17, 5344–5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ohno S, Katsuyama Y, Tajima Y, Izumikawa M, Takagi M,Fujie M, Satoh N, Shin-ya K and Ohnishi Y, ChemBioChem, 2015,16, 2385–2391. [DOI] [PubMed] [Google Scholar]
- 111.Hurley LH, J. Antibiot. (Tokyo), 1977, 30, 349–370. [DOI] [PubMed] [Google Scholar]
- 112.Gerratana B, Med. Res. Rev, 2012, 32, 254–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Thurston DE, Bose DS, Howard PW, Jenkins TC, Leoni A, Baraldi PG, Guiotto A, Cacciari B, Kelland LR, Foloppe MP and Rault S, J. Med. Chem, 1999, 42, 1951–1964. [DOI] [PubMed] [Google Scholar]
- 114.Hurley LH, Gairola C and Zmijewski M, Biochim. Biophys. Acta, Nucleic Acids Protein Synth, 1977, 475, 521–535. [DOI] [PubMed] [Google Scholar]
- 115.Yonemoto IT, Li W, Khullar A, Reixach N and Gerratana B, ACS Chem. Biol, 2012, 7, 973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mantaj J, Jackson PJM, Rahman KM and Thurston DE, Angew. Chem. Int. Ed, 2017, 56, 462–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Arima K, Kosaka M, Tamura G, Imanaka H and Sakai H, J. Antibiot. (Tokyo), 1972, 25, 437–444. [DOI] [PubMed] [Google Scholar]
- 118.Li W, Chou S, Khullar A and Gerratana B, Appl. Environ. Microbiol, 2009, 75, 2958–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schneditz G, Rentner J, Roier S, Pletz J, Herzog KAT, Biicker R, Troeger H, Schild S, Weber H, Breinbauer R, Gorkiewicz G, Högenauer C and Zechner EL, Proc. Natl. Acad. Sci. U.S.A, 2014, 111, 13181–13186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li W, Khullar A, Chou S, Sacramo A and Gerratana B, Appl. Environ. Microbiol, 2009, 75, 2869–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Adamson RH, Hart LG, DeVita VT and Oliverio VT, Cancer Res, 1968, 28, 343–347. [PubMed] [Google Scholar]
- 122.Leimgruber W, Batcho AD and Schenker F, J. Am. Chem. Soc, 1965, 87, 5793–5795. [DOI] [PubMed] [Google Scholar]
- 123.Hurley LH, Zmijewski M and Chang CJ, J. Am. Chem. Soc, 1975, 97, 4372–4378. [DOI] [PubMed] [Google Scholar]
- 124.Tozuka Z and Takaya T, J. Antibiot. (Tokyo), 1983, 36,142–146. [DOI] [PubMed] [Google Scholar]
- 125.Tozuka Z, Yazawa H, Murata M and Takaya T, J. Antibiot. (Tokyo), 1983, 36, 1699–1708. [DOI] [PubMed] [Google Scholar]
- 126.Kariyone K, Yazawa H and Kohsaka M, Chem. Pharm. Bull. (Tokyo), 1971, 19, 2289–2293. [Google Scholar]
- 127.von Tesmar A, Hoffmann M, Pippel J, Fayad AA, Dausend-Werner S, Bauer A, Blankenfeldt W and Muller R, Cell Chem. Biol, 24,1216–1227.el218. [DOI] [PubMed] [Google Scholar]
- 128.Gause GF, Preobrazhenskaya TP, Ivanitskaya LP and Sveshniko MA, Antibiotiki, 1969,14, 963–969. [PubMed] [Google Scholar]
- 129.Brazhnikova MG, Konstantinova HV and Mesentsev AS, J. Antibiot, 1972, 25, 668–673. [DOI] [PubMed] [Google Scholar]
- 130.Mesentsev AS, Kuljaeva VV and Rubasheva LM, J. Antibiot, 1974, 27, 866–873. [DOI] [PubMed] [Google Scholar]
- 131.Tse H, Yang D, Sze K-H, Chu IK, Kao RYT, Lee K-C, Lam C-W, Gu Q, Tai SS-C, Ke Y, Chan E, Chan W-M, Dai J, Leung S-P, Leung S-Y and Yuen K-Y, J. Biol. Chem, 2017, 292, 19503–19520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dornisch E, Pletz J, Glabonjat RA, Martin F, Lembacher-Fadum C, Neger M, Högenauer C, Francesconi K, Kroutil W, Zangger K, Breinbauer R and Zechner EL, Angew. Chem. Int. Ed, 2017, 56,14753–14757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Li S, Wu X, Zhang L, Shen Y and Du L, Org. Lett, 2017,19 5010–5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jin S and Liebscher J, Journal fur Praktische Chemie/Chemiker-Zeitung, 2004, 340, 390–392. [Google Scholar]
- 135.Du L, Chen M, Zhang Y and Shen B, Biochem, 2003, 42, 9731–9740. [DOI] [PubMed] [Google Scholar]
- 136.Wenzel SC, Meiser P, Binz TM, Mahmud T and Muller R, Angew. Chem. Int. Ed., 2006, 45, 2296–2301. [DOI] [PubMed] [Google Scholar]
- 137.Davison J, al Fahad A, Cai M, Song Z, Yehia SY, Lazarus CM, Bailey AM, Simpson TJ and Cox RJ, Proc. Natl. Acad. Sci. U. S. A, 2012, 109, 7642–7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fisch KM, Skellam E, Ivison D, Cox RJ, Bailey AM, Lazarus CM and Simpson TJ, Chem. Commun, 2010, 46, 5331–5333. [DOI] [PubMed] [Google Scholar]
- 139.al Fahad A, Abood A, Fisch KM, Osipow A, Davison J, Avramovic M, Butts CP, Piel J, Simpson TJ and Cox RJ, Chem. Sci, 2014, 5, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Crawford JM, Korman TP, Labonte JW, Vagstad AL, Hill A, Kamari-Bidkorpeh O, Tsai S-C and Townsend CA, Nature, 2009, 461,1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Raggatt ME, Simpson TJ, Raggatt ME and Ines Chicarelli-Robinson M, Chem. Commun, 1997, 0, 2245–2247. [Google Scholar]
- 142.Ainsworth AM, Chicarelli-Robinson MI, Copp BR, Fauth U, Hylands PJ, Holloway JA, Latif M, O’Beirne GB, Porter N, Renno DV and et al. , J. Antibiot. (Tokyo), 1995, 48, 568–573. [DOI] [PubMed] [Google Scholar]
- 143.Cai P, Smith D, Cunningham B, Brown-Shimer S, Katz B, Pearce C, Venables D and Houck D, J. Nat. Prod, 1998, 61, 791–795. [DOI] [PubMed] [Google Scholar]
- 144.Mayerl F, Gao Q, Huang S, Klohr SE, Matson JA, Gustavson DR, Pirnik DM, B. R. L., Fairchild C and Rose WC, J. Antibiot. (Tokyo), 1993, 46, 1082–1088. [DOI] [PubMed] [Google Scholar]
- 145.Harris GH, Hoogsteen K, Silverman KC, Raghoobar SL, Bills GF, Lingham RB, Smith JL, Dougherty HW, Cascales C and Pelaez F, Tetrahedron, 1993, 49, 2139–2144. [Google Scholar]
- 146.Wright JLC, Mclnnes AG, Smith DG and Vining LC, Can. J. Chem, 1970, 48, 2702–2708. [Google Scholar]
- 147.Nagao K, Yoshida N, Iwai K, Sakai T, Tanaka M and Miyahara T, Environ. Sci, 2006,13, 251–256. [PubMed] [Google Scholar]
- 148.Osmanova N, Schultze W and Ayoub N, Phytochem. Rev, 2010, 9, 315–342. [Google Scholar]
- 149.Wang CCC, Chiang Y-M, Praseuth MB, Kuo P-L, Liang H-L and Hsu Y-L, Basic Clin. Pharmacol. Toxicol, 2010, 107, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Maccurin T and Reilly J, Nature, 1940,146, 335. [Google Scholar]
- 151.Chidananda C and Sattur AP, J. Agric. Food. Chem, 2007, 55, 2879–2883. [DOI] [PubMed] [Google Scholar]
- 152.Gao J-M, Yang S-X and Qin J-C, Chem. Rev, 2013, 113, 4755–4811. [DOI] [PubMed] [Google Scholar]
- 153.Angelica 0. Zabala, W. Xu, Y.-H. Chooi and Y. Tang, Chem. Biol, 2012, 19, 1049–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Buchi G, White JD and Wogan GN, J. Am. Chem. Soc, 1965, 87, 3484–3489. [DOI] [PubMed] [Google Scholar]
- 155.Woo PCY, Lam C-W, Tam EWT, Leung CKF, Wong SSY, Lau SKP and Yuen K-Y, PLoS Negl. Trop. Dis, 2012, 6, el871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hetherington AC and Raistrick H, Philosophical Transactions of the Royal Society of London. Series B, Containing Papers of a Biological Character, 1931, 220, 269–295. [Google Scholar]
- 157.Brown JP, Cartwright NJ, Robertson A and Whalley WB, Nature, 1948,162, 72. [DOI] [PubMed] [Google Scholar]
- 158.Chagas GM, Campello AP and Kluppel ML, J. Appl. Toxicol, 1992, 12, 123–129. [DOI] [PubMed] [Google Scholar]
- 159.Chiang Y-M, Szewczyk E, Davidson AD, Keller N, Oakley BR and Wang CCC, J. Am. Chem. Soc, 2009, 131, 2965–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Harned AM and Volp KA, Nat. Prod. Rep, 2011, 28, 1790–1810. [DOI] [PubMed] [Google Scholar]
- 161.Bringmann G, Lang G, Gulder TAM, Tsuruta H, Miihlbacher J, Maksimenka K, Steffens S, Schaumann K, Stohr R, Wiese J, Imhoff JF, Perovic-Ottstadt S, Boreiko 0 and Muller WEG, Tetrahedron, 2005, 61, 7252–7265. [Google Scholar]
- 162.Kontani M, Sakagami Y and Marumo S, Tetrahedron Lett, 1994, 35, 2577–2580. [Google Scholar]
- 163.Trifonov LS, Hilpert H, Floersheim P, Dreiding AS, Rast DM, Skrivanova R and Hoesch L, Tetrahedron, 1986, 42, 3157–3179. [Google Scholar]
- 164.Abe N, Murata T and Hirota A, Biosci., Biotechnol., Biochem, 1998, 62, 661–666. [DOI] [PubMed] [Google Scholar]
- 165.Li D, Wang F, Xiao X, Fang Y, Zhu T, Gu Q and Zhu W, Tetrahedron Lett, 2007, 48, 5235–5238. [Google Scholar]
- 166.Li D, Cai S, Zhu T, Wang F, Xiao X and Gu Q, Tetrahedron, 2010, 66, 5101–5106. [Google Scholar]
- 167.Abe N, Arakawa T, Yamamoto K and Hirota A, Biosci. Biotechnol. Biochem, 2002, 66, 2090–2099. [DOI] [PubMed] [Google Scholar]
- 168.Kuo J, Lynch SR, Liu CW, Xiao X and Khosla C, ACS Chem. Biol, 2016, 11, 2636–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Pospiech A, Cluzel B, Bietenhader J and Schupp T, Microbiology, 1995,141,1793–1803. [DOI] [PubMed] [Google Scholar]
- 170.Pospiech A, Bietenhader J and Schupp T, Microbiology, 1996, 142, 741–746. [DOI] [PubMed] [Google Scholar]
- 171.Ikeda Y, Idemoto H, Hirayama F, Yamamoto K, Iwao K, Asao T and Munakata T, J. Antibiot. (Tokyo), 1983, 36, 1279–1283. [DOI] [PubMed] [Google Scholar]
- 172.Ikeda Y, Matsuki H, Ogawa T and Munakata T,!. Antibiot. (Tokyo), 1983, 36, 1284–1289. [DOI] [PubMed] [Google Scholar]
- 173.Velasco A, Acebo P, Gomez A, Schleissner C, Rodriguez P, Aparicio T, Conde S, Munoz R, de la Calle F, Garcia JL and Sanchez-Puelles JM, Mol. Microbiol, 2005, 56, 144–154. [DOI] [PubMed] [Google Scholar]
- 174.Li L, Deng W, Song J, Ding W, Zhao Q-F, Peng C, Song W-W, Tang G-L and Liu W, J. Bacteriol, 2008, 190, 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ishiguro K, Sakiyama S, Takahashi K and Arai T, Biochem, 1978, 17, 2545–2550. [DOI] [PubMed] [Google Scholar]
- 176.Okumoto T, Kawana M, Nakamura I, Ikeda Y and Isagai K, J. Antibiot. (Tokyo), 1985, 38, 767–771. [DOI] [PubMed] [Google Scholar]
- 177.Ishiguro K, Takahashi K, Yazawa K, Sakiyama S and Arai T, J. Biol. Chem, 1981, 256, 2162–2167. [PubMed] [Google Scholar]
- 178.Scott JD and Williams RM, Chem. Rev, 2002, 102, 1669–1730. [DOI] [PubMed] [Google Scholar]
- 179.Koketsu K, Watanabe K, Suda H, Oguri H and Oikawa H, Nat. Chem. Biol, 2010, 6, 408. [DOI] [PubMed] [Google Scholar]
- 180.Kluepfel D, Baker HA, Piattoni G, Sehgal SN, Sidorowicz A, Singh K and Vezina C, J. Antibiot. (Tokyo), 1975, 28, 497–502. [DOI] [PubMed] [Google Scholar]
- 181.Zmijewski MJ Jr., Miller-Hatch K and Goebel M, Antimicrob. Agents Chemother, 1982, 21, 787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zmijewski MJ Jr., J. Antibiot. (Tokyo), 1985, 38, 819–820. [DOI] [PubMed] [Google Scholar]
- 183.Pu JY, Peng C, Tang MC, Zhang Y, Guo JP, Song LQ,Hua Q and Tang GL, Org. Lett, 2013, 15, 3674–3677. [DOI] [PubMed] [Google Scholar]
- 184.Le VH, lnai M, Williams RM and Kan T, Nat. Prod. Rep, 2015, 32, 328–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Rinehart KL, Holt TG, Fregeau NL, Stroh JG, Keifer PA, Sun F, Li LH and Martin DG, J. Org. Chem, 1990, 55, 4512–4515. [Google Scholar]
- 186.Cuevas C and Francesch A, Nat. Prod. Rep, 2009, 26, 322–337. [DOI] [PubMed] [Google Scholar]
- 187.Aune GJ, Furuta T and Pommier Y, Anti-Cancer Drugs, 2002, 13, 545–555. [DOI] [PubMed] [Google Scholar]
- 188.Kerr RG and Miranda NF, J. Nat. Prod, 1995, 58, 1618–1621. [Google Scholar]
- 189.Sakai R, JaresErijman EA, Manzanares I, Elipe MVS and Rinehart KL, J. Am. Chem. Soc, 1996, 118, 9017–9023. [Google Scholar]
- 190.Schofield MM, Jain S, Porat D, Dick GJ and Sherman DH, Environ. Microbiol, 2015, 17, 3964–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Liu X and Walsh CT, Biochem, 2009, 48, 8746–8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Hu Q, Li F and Zhang Y, BioMed Res. Int, 2016, 2016, 3194321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hayakawa S, Minato H and Katagiri K, J. Antibiot. (Tokyo), 1971, 24, 653–654. [DOI] [PubMed] [Google Scholar]
- 194.Vesonder RF, Tjarks LW, Rohwedder WK, Burmeister HR and Laugal JA, J. Antibiot. (Tokyo), 1979, 32, 759–761. [DOI] [PubMed] [Google Scholar]
- 195.Quek NC, Matthews JH, Bloor SJ, Jones DA, Bircham PW, Heathcott RW and Atkinson PH, Mol. Biosyst, 2013, 9, 2125–2133. [DOI] [PubMed] [Google Scholar]
- 196.Jeong YC and Moloney MG, Future Med. Chem, 2015, 7, 1861–1877. [DOI] [PubMed] [Google Scholar]
- 197.Sims JW, Fillmore JP, Warner DD and Schmidt EW, Chem. Commun, 2005, 0, 186–188. [DOI] [PubMed] [Google Scholar]
- 198.Chang P-K, Ehrlich KC and Fujii I, Toxins (Basel), 2009, 1, 74–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Schumann J and Hertweck C, J. Am. Chem. Soc, 2007, 129, 9564–9565. [DOI] [PubMed] [Google Scholar]
- 200.Qiao K, Chooi Y-H and Tang Y, Metab. Eng, 2011, 13, 723–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Mah R, Thomas JR and Shafer CM, Bioorg. Med. Chem. Lett, 2014, 24, 33–39. [DOI] [PubMed] [Google Scholar]
- 202.Wang L, Zhao J, Yao Y, Wang C, Zhang J, Shu X, Sun X, Li Y, Liu K, Yuan H and Ma X, Eur. J. Med. Chem, 2017, 142, 493–505. [DOI] [PubMed] [Google Scholar]
- 203.Field-Smith A, Morgan GJ and Davies FE, Ther. Clin. Risk Manag, 2006, 2, 271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Adams J, Behnke M, Chen S, Cruickshank AA, Dick LR, Grenier L, Klunder JM, Ma Y-T, Plamondon L and Stein RL, Bioorg. Med. Chem. Lett, 1998, 8, 333–338. [DOI] [PubMed] [Google Scholar]
- 205.Rudin CM, Pietanza MC, Bauer TM, Ready N, Morgensztern D, Glisson BS, Byers LA, Johnson ML, Burris HA, Robert F, Han TH, Bheddah S, Theiss N, Watson S, Mathur D, Vennapusa B, Zayed H, Lally S, Strickland DK, Govindan R, Dylla SJ, Peng SL and Spigel DR, Lancet Oncol, 2017, 18, 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Gomi S, Ikeda D, Nakamura H, Naganawa H, Yamashita F, Hotta K, Kondo S, Okami Y, Umezawa H and Iitaka Y, J. Antibiot. (Tokyo), 1984, 37, 1491–1494. [DOI] [PubMed] [Google Scholar]
- 207.Kunimoto S MT, Kanbayashi N, Hamada M, Naganawa H, Miyamoto M, Takeuchi T, Umezawa H, J. Antibiot. (Tokyo), 1980, 33, 665–667. [DOI] [PubMed] [Google Scholar]
- 208.Barkley MD, Cheatham S, Thurston DE and Hurley LH, Biochem, 1986, 25, 3021–3031. [DOI] [PubMed] [Google Scholar]
- 209.Tsunakawa M KH, Konishi M, Miyaki T, Oki T, Kawaguchi H, J. Antibiot. (Tokyo), 1988, 41, 1366–1373. [DOI] [PubMed] [Google Scholar]
- 210.Najmanova L, Ulanova D, Jelinkova M, Kamenik Z, Kettnerova E, Koberska M, Gazak R, Radojevic B and Janata J, Folia Microbiol, 2014, 59, 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Nicolaou KC, Vassilikogiannakis G, Simonsen KB, Baran PS, Zhong Y-L, Vidali VP, Pitsinos EN and Couladouros EA, J. Am. Chem. Soc, 2000, 122, 3071–3079. [DOI] [PubMed] [Google Scholar]
- 212.Du L, Zhu T, Li L, Cai S, Zhao B and Gu Q, Chem. Pharm. Bull, 2009, 57, 220–223. [DOI] [PubMed] [Google Scholar]
- 213.Xing C, LaPorte JR, Barbay JK and Myers AG, Proc. Natl. Acad. Sci. U. S. A, 2004, 101, 5862–5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Japan Pat., JP 59225189, 1985.
- 215.Yazawa K TK, Mikami Y, Arai T, Saito N, Kubo A, J. Antibiot. (Tokyo), 1986, 39, 1639–1650. [DOI] [PubMed] [Google Scholar]
- 216.Ohtsubo K, Saito M, Sekita S, Yoshihira K and Natori S, Jpn. J. Exp. Med, 1978, 48, 105–110. [PubMed] [Google Scholar]
- 217.Sekita S, Yoshihira K, Natori S and Kuwano H, Tetrahedron Lett, 1973, 14, 2109–2112. [Google Scholar]
- 218.Udwary DW, Zeigler L, Asolkar RN, Singan V, Lapidus A, Fenical W, Jensen PR and Moore BS, Proc. Natl. Acad. Sci. U.S.A, 2007, 104, 10376–10381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Inglis DO, Binkley J, Skrzypek MS, Arnaud MB, Cerqueira GC, Shah P, Wymore F, Wortman JR and Sherlock G, BMC Microbiol, 2013, 13, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.von Döhren H, Fungal Genet. Biol, 2009, 46, S45–S52. [DOI] [PubMed] [Google Scholar]
- 221.van den Berg MA, Albang R, Albermann K, Badger JH, Daran J-M, Driessen AJM, Garcia-Estrada C, Fedorova ND, Harris DM, Heijne WHM, Joardar V, Kiel JAKW, Kovalchuk A, Martin JF, Nierman WC, Nijland JG, Pronk JT, Roubos JA, van der Klei IJ, van Peij NNME, Veenhuis M, von Dohren H, Wagner C, Wortman J and Bovenberg RAL, Nat. Biotechnol, 2008, 26, 1161–1168. [DOI] [PubMed] [Google Scholar]
- 222.Doroghazi JR, Albright JC, Goering AW, Ju KS, Haines RR, Tchalukov KA, Labeda DP, Kelleher NL and Metcalf WW, Nat. Chem. Biol, 2014, 10, 963–968. [DOI] [PMC free article] [PubMed] [Google Scholar]