

Abstract
FIH (Factor Inhibiting HIF [Hypoxia Inducible Factor]) is an α-ketoglutarate (αKG)-dependent non-heme iron enzyme that catalyzes the hydroxylation of the C-terminal transactivation domain (CAD) asparagine residue in HIF-1α to regulate cellular oxygen levels. The role of the facial triad carboxylate ligand in O2 activation and catalysis was evaluated by replacing the Asp201 residue with Gly (D201G), Ala (D201A) and Glu (D201E). Magnetic circular dichroism (MCD) spectroscopy showed that the (FeII)FIH variants were all 6-coordinate (6C) and the αKG plus CAD bound FIH variants were all 5-coordinate (5C), mirroring the behavior of the wild-type (wt) enzyme. When only αKG is bound, all FIH variants exhibited weaker FeII-OH2 bonds for the sixth ligand compared to wt, and αKG bound D201E was found to be 5C, demonstrating that the Asp201 residue plays an important role in the wt enzyme in ensuring that the (FeII/αKG)FIH site remains 6C. Variable temperature, variable field (VTVH) MCD spectroscopy showed that all the αKG and CAD bound FIH variants, though 5C, have different ground state geometric and electronic structures, which impact their oxygen activation rates. Comparison of O2 consumption to substrate hydroxylation kinetics revealed uncoupling between the two half reactions in the variants. Thus, the Asp201 residue also ensures fidelity between CAD substrate binding and oxygen activation, enabling tightly coupled turnover.
Authors are required to submit a graphic entry for the Table of Contents (TOC) that, in conjunction with the manuscript title, should give the reader a representative idea of one of the following: A key structure, reaction, equation, concept, or theorem, etc., that is discussed in the manuscript. Consult the journal’s Instructions for Authors for TOC graphic specifications.
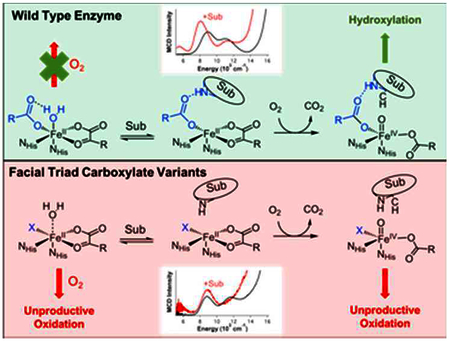
INTRODUCTION
Mononuclear non-heme iron enzymes play important roles in many biological processes including antibiotic, natural product and neurotransmitter biosynthesis, DNA and O2 regulation and bioremediation.1–6 These enzymes are divided into different subclasses depending on the cofactor they use, the reactive iron-oxygen intermediates that they proceed through and the type of reaction that they catalyze.7 These O2 activating FeII enzymes generally share a common 2-His-1-carboxylate facial triad motif to bind the Fe center but 3-His and 2-His-1-halide triads also exist.8 The α-ketoglutarate dependent hydroxylases utilize an FeII center and α-ketoglutarate (αKG) cofactor to facilitate the 4 electron reduction of dioxygen to perform hydroxylation, halogenation, de-saturation, ring closure or ring expansion on various different substrates.9 FIH (Factor Inhibiting HIF [Hypoxia Inducible Factor]) is an αKG-dependent asparaginyl hydroxylase that regulates global responses to O2 levels in mammalian cells by hydroxylating the β-carbon on the Asn-803 residue of the HIF-1α protein (key in shutting off downstream cellular responses).10,11 Under conditions of low O2 (i.e. hypoxia), the non-hydroxylated HIF-1α dimerizes with the HIF-1β protein, binds transcriptional machinery and activates genes leading to angiogenesis and erythropoiesis, which results in an increase in vascularization and cellular O2 levels.5,12 Studying the tightly regulated function of this enzyme provides insight into the factors that govern its highly selective reactivity and the design of better therapeutic strategies.13,14
αKG-dependent enzymes follow a general mechanistic strategy where the active site remains 6-coordinate (6C) until both cofactor and substrate are bound, resulting in a coordinatively unsaturated 5-coordinate (5C) site capable of reacting with dioxygen.7,9,15 These 5C sites activate O2 to form reactive FeIV=O moieties that then attack the associated substrate to form product and regenerate the FeII site (Scheme 1). αKG-dependent enzymes can also carry out uncoupled O2 reactivity, where the αKG is oxidized but product is not formed.16,17 This would be highly unfavorable in an enzyme like FIH as this would dysregulate gene expression from cellular O2 levels, leading to oxidative damage and apoptosis.5 Our previous study of wt-FIH computationally showed that the facial triad carboxylate plays a key role in ensuring that the FIH active site remains 6C when only αKG is bound by hydrogen bonding with a H2O ligand (see scheme 1, top middle),18–20 which is weakly bound due to the strong donor interaction of the αKG with the FeII. This is important because this αKG bound site has all 4 electrons needed for O2 activation and loss of the water ligand can lead to premature FeIV=O formation, which would result in autooxidation or enzyme inactivation.16,17
Scheme 1.

Reaction mechanism for the αKG-dependent hydroxylases
The FIH D201X (D201G/D201A/D201E) enzymes are the first reported facial triad variants in an αKG-dependent enzyme that are capable of binding FeII.21 Furthermore, D201G is able to hydroxylate the C-terminal transactivation domain (CAD) of a truncated form of the HIF-1α substrate at up to half the efficiency of the native enzyme in the presence of added chloride (referred to as D201GCl).20 However, the other variants are comparatively unreactive in substrate hydroxylation. This loss in activity can be due to the inability to form the 5C site required for O2 activation or the inability of the resultant FeIV=O moiety to properly react with the substrate. Previous studies of these enzyme variants focused on substrate hydroxylation and not coordination changes at the FeII center.20 However, these variants reveal perturbations that provide mechanistic insight into the role of the carboxylate in controlling the FeII site, thus the timing of O2 activation. In this study, we have used magnetic circular dichroism (MCD) spectroscopy to define the coordination number and geometry of the different D201X-FeII variants and their interaction with αKG and CAD cosubstrates and correlated these structural changes with O2 consumption kinetics to characterize the role of the facial triad carboxylate in O2 activation for enzyme function.
MATERIALS AND METHODS
Materials and Enzyme Purification.
All reagents were purchased from commercial vendors and used as received, with the exception of CAD substrate. CAD was purchased (EZBiolab, Carmel, IN, USA) as a desalted peptide and further purified by reverse phase HPLC methods. Point mutations to Asp201 were made using the QuickChange mutagenesis kit (Stratagene) in the pET28a-FIH construct. The resulting plasmid DNA was sequenced (Genewiz, NJ, USA) to confirm the point mutation. His6-WT FIH and the His6-D201X variants were overexpressed in BL21-DE3 E. coli and purified as previously described.18 Briefly, the cell pellet was lysed using sonication, centrifuged, then dialyzed into 10 mM Tris pH 8.00 to remove EDTA from the lysis buffer. Centrifugation of the supernatant further clarified the lysate prior to loading on to a Ni-NTA column. After loading the lysate, the column was washed with 5 column volumes each of 100% A buffer (50 mM Tris pH 8.0, 300 mM NaCl, 15 mM Imidazole) and 15% B buffer (50 mM Tris pH 8.0, 300 mM NaCl, 250 mM Imidazole) before eluting the His6-FIH protein with 100% B buffer. Thrombin was then added to the eluent to cleave the His6 tag for 36 hours at 4 °C. The cleaved FIH was loaded on to the Ni-NTA again to remove the His6 tag and any remaining uncleaved protein. Overnight incubation with 50 mM EDTA removed exogenous metals. Size exclusion chromatography was then used to separate the protein from the thrombin and EDTA. Purified protein was aliquotted and stored at 20 °C in 50 mM HEPES pH 7.00. Protein purity (>95%) was evaluated using SDS-PAGE.
MCD Spectroscopy.
Apo-D201X(FIH) variants were buffer exchanged into deuterated 50mM HEPES buffer (pD 7.5) at 4°C. For the D201GCl samples, 100mM NaCl was added to the buffering system. Protein was degassed in an ice bath, transferred into a nitrogen atmosphere maintained in a Vacuum Atmosphere Nexus-1 glovebox (<2 ppm oxygen) and stirred at 4°C. 1 eq of ferrous ammonium sulfate, 10 eq αKG and 2.5 eq CAD were added and equilibrated for 15 mins each. Samples were then saturated with deuterated sucrose and loaded into an MCD cell. Near-IR (600nm – 200nm) MCD spectroscopy was performed using a JASCO J-730 spectropo larimeter equipped with an InSb photodiode detector cooled with liquid nitrogen and fitted with an Oxford Instruments SM4000–7T superconducting magnet. UV-Vis MCD (300nm – 900nm) spectroscopy was performed using a JASCO J-810 spectropolarimeter equipped with a photomultiplier tube and fitted with an Oxford Instruments SM4000–7T superconducting magnet. The sample temperature was measured using a calibrated Cernox resistor from Lakeshore Cryotronics. MCD spectra were corrected for zero-field baseline effects by subtracting the corresponding 0T scan from the data. Variable temperature, variable field (VTVH) MCD isotherms were collected at 6–8 temperatures between 2 and 25K and at 12 fields between 0 and 7T.
Kinetic Methods – O2 Consumption and CAD hydroxylation assays.
An Oxygraph Plus System (Hansatech) was used to monitor the amount of oxygen consumed by the enzyme. This oxygen sensor contains a central reaction vessel surrounded by a water jacket, with a Clark-type electrode disc at the bottom of the reaction vessel. The electrode disc utilized a KCl bridge and a PTFE membrane that is selectively permeable to oxygen molecules. A new membrane was prepared and calibrated each day with air saturated water and dithionite. 400 μL reactions were equilibrated at atmospheric O2 in the reaction vessel at 37 °C until a stable baseline was achieved. Assays were initiated with cold purified FIH (10 μM) using a Hamilton gas-tight syringe. The rate of O2 consumption was monitored over time until the rate of O2 consumption resembled the baseline slope. Assays contained 100 μM αKG, 50 μM FeSO4, 80 μM CTAD and 10 μM FIH in 50 mM HEPES pH 7.00, and 50 μM ascorbate. Controls were performed to determine the optimal amount of ascorbate that could be used to minimize baseline slope while retaining maximal FIH activity. 5 μL of each reaction was then quenched in 20 μL of matrix (3,5-dimethoxy-4-hydroxycinnamic acid saturated in 75% ACN/0.2% TFA) and analyzed by MALDI-TOF-MS (Ultra-flex, Bruker) monitoring the +1 charge state of the substrate (CAD) and the product (hydroxylated CAD).
RESULTS AND ANALYSIS
MCD Spectroscopy.
Low temperature (LT) MCD spectroscopy in the near-infrared (NIR) region is a powerful tool for studying FeII active sites as it probes 5T2 → 5E ligand field (LF) transitions, which reflect coordination geometry. LF transitions, though weak in absorption spectroscopy, are intense in MCD at low temperatures due to different selection rules.22 We have calibrated this methodology by assigning the transition energies and excited state splittings (Δ5E, see Fig. 1A top) of a variety of 6C, 5C and 4C model complexes with different geometries.22–24 Briefly, two transitions centered around 10,000 cm−1 split by ~2,000 cm−1 is indicative of a distorted 6C site. Removal of an axial ligand leads to a 5C square pyramidal (SP) structure with one transition above 10,000 cm−1 and one around ~5,000 cm−1. Alternatively, a 5C trigonal bipyramidal (TBP) site exhibits one transition around or less than 10,000 cm−1 and one <5,000 cm−1 (often undetectable). Finally, 4C distorted tetrahedral sites exhibit two transitions around ~6,000–7,000 cm−1.
Figure 1.

5K, 7T MCD spectra of wt-FIH(black), D201GCl(red), D201G(blue), D201E(green) and D201A(purple). A) FeII-loaded resting sites, B) FeII/αKG sites and C) FeII/αKG/CAD ternary sites. In A, B and C, the Gaussianresolved band energies and Δ5E splittings are summarized in Table 1. The arrows indicate the energies at which VTVH data were collected for each sample.
The LT MCD spectra of the resting FeII-bound sites (Fig. 1A) of all these variants including D201GCl show two transitions centered around 10,000 cm−1 with Δ5E ~ 1,800 cm−1, thus all are 6C distorted octahedral sites. There are some small differences in the excited state splittings (Table 1) reflecting small changes in the ligand field due to the variation of the D201 replacement ligand. In the absence of the carboxylate ligand, D201A and D201G likely have a water-derived ligand, i.e. H2O or OH6, bound to the FeII center. The excited state splitting of D201GCl relative to D201G is greater by 150 cm−1 and intensity ratios of the two transitions are slightly perturbed (Fig. S1). This evidence, combined with X-ray absorption spectroscopy performed on the (FeII/N-oxalylglycine)D201GCl site,25 argues for a Cl− ion binding to the FeII center in the D201GCl resting site. Finally, these resting FeII spectra are also similar to that of the wt enzyme, thus all have FeII bound with 2 His and 3 H2O ligands with the 6th ligand position being a monodentate carboxylate or a Cl6 or another water-derived ligand.
Table 1.
Gaussian resolved bands for the observed transitions reported in Figure 1 along with the values of their excited state splittings. All values are in cm−1. aThe second transition associated with this site is not observed and likely ~10,000 cm−1 from VTVH MCD spectroscopy. bThree transitions have been observed but the bands have not been assigned to a single FeII site.
| Enzyme | Band 1 | Band 2 | Δ5E |
|---|---|---|---|
| FeII Sites | |||
| wt-FIH | 8950 | 10700 | 1750 |
| D201GCl | 9200 | 11200 | 2000 |
| D201G | 9100 | 10950 | 1850 |
| D201E | 9050 | 10800 | 1750 |
| D201A | 9150 | 10950 | 1800 |
| FeII/αKG Sites | |||
| wt-FIH | 8900 | 11100 | 2300 |
| D201GCl | 8950 | 11550 | 2600 |
| 7400 | a | ||
| D201G | 8900 | 11300 | 2400 |
| D201E | 8850 | 11550 | 2700 |
| D201A | 9100 | 11400 | 2300 |
| FeII/αKG/CAD Sites | |||
| wt-FIH | <5000 | 7950 | >3000 |
| 9000 | 11150 | 2150 | |
| D201GCl | <5000 | 9250 | >4,250 |
| 6200 | 10900 | 4,700 | |
| D201G | <5000 | 8800 | >3800 |
| 6100 | 11350 | 5,250 | |
| D201Eb | 8950 | 11500 | >2,500 |
| 14000 | |||
| D201Ab | <5000 | 9100 | >2,500 |
| 11600 | |||
In the FeII/αKG site for wt-FIH (Fig. 1B, top), there are two transitions centered around 10,000 cm−1 with Δ5E of 2,300 cm−1.19 This splitting is larger than that observed for many other 6C sites in the literature and is attributed to the weakening of the FeII-OH2 bond of the 6th ligand due to the strong donor interaction associated with the αKG-bound to the FeII.26,27 The FeII/αKG sites of D201A and D201G are both similar to wt-FIH and also have two transitions centered around 10,000 cm−1 with Δ5E splittings of 2,300 and 2,400 cm−1 respectively (Fig. 1B, purple and blue respectively & Table 1), indicating that they are 6C but with a weak water ligand. (FeII/αKG)D201E has two transitions centered at 10,000 cm−1 with Δ5E of ~2,700 cm−1 (Fig. 1B, green, Table 1). This observed splitting is large compared to the other 6C sites observed in αKG-dependent enzymes19,26 and likely indicates a site with a very weakly bound/unbound 6th ligand. (FeII/αKG)D201GCl has two transitions centered around 10,000 cm−1 and a third transition at ~7,400 cm−1 (Fig. 1B, red, Table 1), indicating a mixture of two different FeII sites (note that a single FeII site can only have two LF transitions in this energy region). The ground state associated with the low energy transition has been investigated using VTVH MCD to determine if it arises from a 5C or 6C site (vide infra). All the (FeII/αKG)D201X spectra show a transition tailing into the NIR spectral region from higher energy that is due to the FeII-to-αKG metal-to-ligand charge transfer transitions observed around 20,000 cm−1 (Figure S2), which are also present in αKG bound wt-FIH and other αKG-dependent enzymes. 19,26,27 This demonstrates that the αKG is bound to the FeII site in a similar bidentate manner to the wt enzyme in the variants and has replaced two water ligands from the resting sites.
The ternary (FeII/αKG/CAD)D201X spectra all have more than 2 LF transitions, thus are all mixtures of two different FeII sites and all show substantial differences in the energy positions and excited state splittings of their LF transitions (Fig. 1C & Table 1). Additionally, they all have FeII-to-αKG charge transfer transitions at similar energies (~20,000 cm−1), demonstrating that αKG is analogously bound in a bidentate manner in these active sites (Fig. S2). wt-FIH has 4 transitions at <5,000, 7,950, 9,000 and 11,150 cm−1; the presence of one band around 5,000 cm−1 indicates that it is a 5C/6C mixture.19 For the glycine variants, D201GCl (Fig. 1C, red) has four observed transitions at <5,000, 6,200, 9,250 and 10,900 cm−1 whereas D201G (Fig. 1C, blue) has four transitions at <5,000, 6,100, 8,800 and 11,350 cm−1. These sites have different energy LF transitions and geometries due to the presence of the Cl− ligand in D201GCl. Importantly, both sites have two transitions at <5,000 and ~ 6,000 cm−1, which demonstrate that they are both 5C/5C mixtures with one square pyramidal component (from the >10,000 cm−1 feature). The D201E site (Fig. 1C, green) has three observed transitions at ~9,000, 11,500 and 14,000 cm−1. The high energy of the 14,000 cm−1 LF transition requires one of the sites to be square pyramidal. Finally, D201A (Fig. 1C, purple) shows a low energy transition at <5,000 cm−1, along with two bands around 10,000 cm−1 demonstrating that it has at least one 5C component as well. Together, these data demonstrate that when both cofactor and substrate are bound all the variants have coordinatively unsaturated 5C FeII sites with an open position for the O2 reaction.
VTVH MCD Spectroscopy.
Variable temperature, variable field (VTVH) MCD spectroscopy uses an excited state transition to probe the ground state. This involves measuring the temperature and field dependence of the MCD intensity of a particular transition. For FeII systems, fitting these data using a non-Kramers doublet model22,23 provides detailed information on the S=2 ground state zero-field splitting (ZFS), which in turn gives the sign and magnitude of the tetragonal splitting (Δ) between the dxz,yz and dxy orbitals and the rhombic splitting (V) between dxz and dyz of the FeII ground state. These parameters, coupled with the LF excited state data obtained from the LT MCD spectra, give a complete picture of the ligand field around the iron center and allow for the detailed description of the electronic and geometric structure of the FeII active site. The analysis presented below focuses on trends in the values of Δ, which correlate with coordination number and π interactions with the ligands. Note that a positive value of Δ is reflected in the saturation magnetization data through a large nesting (i.e. spread) of the isotherms (Fig. 2, top right), while a negative value of Δ is reflected in a small nesting (Fig. 2, top left).22
Figure 2.

VTVH MCD isotherms of D201GCl (orange, collected at 7,100 cm−1) and D201G (light blue, collected at 11,100 cm−1) FeII/αKG complexes and D201GCl (red, collected at 6,430 cm−1) and D201G (blue, collected at 5,500 cm−1) FeII/αKG/CAD complexes.
MCD spectroscopy on the resting FeII-bound sites showed that they are all similar and VTVH MCD data previously collected for the wt enzyme at 8,200 cm−1 (Figure S3A) were fit with Δ = −275 cm−1, which is typical for 6C distorted octahedral sites.22 The arrows in Fig. 1 show the energies at which the VTVH MCD data were collected and Table 2 (middle) summarizes the ground state parameters obtained for the αKG bound FeII complexes. Data collected at 8,200 cm−1 for the αKG bound wt enzyme (Fig. S3B), a 6C site from the excited state splitting (Δ5E), show an increase in magnitude of Δ to −950 cm−1 relative to the resting site. This increase in magnitude is due to the back-bonding of the FeII into the π* orbital of the αKG and is well documented in the αKG-dependent hydroxylases.19,26 From the MCD spectroscopy on the (FeII/αKG)D201GCl site, there were three transitions present, with one at low energy (Fig. 1B). To address the coordination number of the low energy component of this mixture of FeII sites, VTVH MCD data were collected at 7,100 cm−1 (Fig. 2, top left) and fit with Δ = −950 cm−1, similar to the wt enzyme, suggesting that this band arises from a 6C site. Thus, the second transition associated with this low energy feature should be at higher energy and overlapped by the additional components present. The lower energy for these transitions associated with the second site could be due to the presence of the chloride ligand. VTVH MCD data were not measured for the other bands in D201GCl due to spectral overlap. Data for (FeII/αKG)D201E collected at 8,560 cm−1 (Figure S4) were fit with Δ = −1,100 cm−1. This larger |Δ| is similar to the values observed for the 5C ternary complexes (vide infra) and, combined with the relatively large value for Δ5E (2,700 cm−1), suggests that this site is either 6C with a very weak axial ligand or 5C square pyramidal with unbound axial ligand (i.e. loss of H2O). Data collected for αKG bound D201G at 11,100 cm−1 and D201A at 11,430 cm−1 (Fig. 2, top right and Fig. S5) were fit with positive values of U (due to their large nesting behavior) of 950 and 700 cm−1 respectively, consistent with the 6C geometry observed from their excited state splittings (Table 1). The VTVH MCD data on the αKG only bound FeII sites show that, apart from D201E, the variants are 6C, though with different ground states and larger excited state splittings compared to the wt enzyme.
Table 2.
Ground state parameters of the different αKG only and αKG+CAD bound complexes of the D201X variants. *All values in cm−1. VTVH parameters for the wt enzyme are taken from ref. 19.
| Variant | δ* | g|| | Δ* | |V|* |
|---|---|---|---|---|
| FeII Sites | ||||
| wt-FIH | 3.9 | 9.2 | −275 | 100 |
| FeII/αKG Sites | ||||
| wt-FIH | 2.8 | 8.9 | −950 | 530 |
| D201GCl | 2.5 | 9.05 | −950 ± 100 | 475 |
| D201G | 5.0 | 8.0 | 950 ± 40 | 210 |
| D201A | 5.0 | 8.0 | 700 ± 80 | 330 |
| D201E | 2.4 | 8.9 | −1100 ± 60 | 570 |
| FeII/αKG/CAD Sites | ||||
| wt-FIH | 1.8 | 9.2 | −1200 | 410 |
| D201GCl | 3.7 | 8.0 | 1150 ± 50 | 480 |
| D201G | 4.0 | 8.0 | 1000 ± 80 | 340 |
| D201A | 5.3 | 8.0 | 700 ± 40 | 360 |
The bottom panel in Figure 2 (along with Fig. S3C and S6) present the saturation magnetization behavior observed among the ternary (cofactor and substrate bound) complexes of wt-FIH, D201GCl, D201G and D201A and the ground state parameters for these ternary complexes are summarized in Table 2 (bottom). Due to overlapping bands and sample concentration issues, useful VTVH MCD data on the ternary complex of D201E could not be obtained. Data on the wt enzyme collected at 7,550 cm−1 (Figure S2C) were fit with Δ = −1,200 cm−1. This increase in the magnitude of U relative to the αKG only bound site (where Δ = −950 cm−1) is consistent with a 5C structure. 19 Unlike the wt enzyme, the ternary complexes of the variants were all fit with positive values of Δ due to their large nesting behaviors. Since all these variants are 5C, this change in sign of Δ correlates with the replacement of the carboxylate equatorial ligand (in the wt enzyme) with the chloride or water-derived ligand and has implications with respect to the observed O2 reactivity (vide infra). Data on D201GCl collected at 6,430 cm−1 (Figure 2, bottom left) were fit with Δ = 1,150 cm−1, similar in magnitude to the wt-FIH ternary site, and the difference between the binary and ternary complex Δ values is also consistent with a 6C → 5C conversion. Data collected for the ternary complexes of D201G at 5,500 cm−1 (Figure 2, bottom right) and D201A at 5,200 cm−1 (Fig. S6) were fit with Δ values of 1,000 cm−1 and 700 cm−1 respectively. Though the differences in Δ between the αKG bound and the ternary complex (Table 2) for these variants are small, the presence of low energy transitions in the MCD spectra (in Fig. 1C) show that they are 5C. VTVH MCD on the ternary complexes of the variants show that, though they are all 5C, they have different coordination environments and geometries, and this is reflected in their observed O2 reactivities (vide infra).
O2 Consumption and Substrate Hydroxylation Kinetics.
From the LT MCD and VTVH MCD spectroscopy presented above, 5C sites are present in all the ternary complexes and possibly in the (FeII/αKG)D201E complex. The presence of a 5C site with αKG bound should enable O2 activation and this was tested using a Clark O2 sensor to track O2 consumption. Table 3 summarizes the O2 consumption rates of all the ternary and the (FeII/αKG)D201E complex. The other αKG only bound wt and variant enzymes that are 6C from MCD did not show measurable O2 consumption, confirming that a 5C site correlates with O2 activation. Compared to the wt-FIH ternary complex, all the variants react with O2 at >10-fold slower rates. Furthermore, there are differences in the O2 consumption rates among the variants. The (FeII/αKG)D201E, (FeII/αKG/CAD)D201E and (FeII/αKG/CAD)D201GCl sites react with O2 at similar rates of 1.7 min−1, 1.7 min−1 and 1.9 min−1 respectively. The D201G ternary complex, which is also a 5C/5C mixture like (FeII/αKG/CAD)D201GCl, consumes O2 at a rate of 0.3 min−1, while the D201A ternary complex does not show consumption of O2 above the background (ascorbate reacting with O2), i.e. < 0.01 min−1. Thus, the differences in 5C geometries from MCD (Table 1 & 2) lead to greater than 2 orders of magnitude variation in the O2 consumption rates.
Table 3.
Initial rates of O2 consumption and CAD hydroxylation for the different D201X variants. All values are in min−1 units. None of the other (FeII/αKG) variants consume O2.
| wt-FIH | 16.3 ± 0.7 | 15.4 ± 0.6 |
| D201GCl | 1.9 ± 0.4 | 0.41 ± 0.03 |
| D201G | 0.3 ± 0.1 | 0.16 ± 0.01 |
| D201E | 1.7 ± 0.1 | 0.16 ± 0.02 |
| D201A | <0.01 | Not Detectable |
| D201E (No CAD) | 1.67 ± 0.08 | N/A |
Comparing the rates of substrate hydroxylation to O2 consumption in Table 3 shows that, in contrast to wt where both rates are the same within error, substrate hydroxylation is slower than O2 consumption for all the variants. This demonstrates that the two half reactions, i.e. O2 activation and substrate hydroxylation, are uncoupled, which is confirmed through the end point analysis assays presented in Table S1. It is also important to note that these reactions only produce marginal amounts of H2O2 as a byproduct (Table S1). This, coupled with the observation that succinate is produced20, suggest that most of the O2 equivalents used by these variants produce an FeIV=O intermediate capable of reacting with substrate. Furthermore, since the rates of substrate hydroxylation are an order of magnitude lower than the O2 consumption, the second half of the reaction involving substrate hydroxylation is far more impaired by the D201X variants.17,18,28 Together, these data show that the 5C sites do consume O2 (albeit at different and lower rates than wt) and that the variants show a decrease in their coupling ratios demonstrating that there is also a loss in substrate hydroxylating efficiency due to the mutation of the D201 facial triad ligand.
DISCUSSION
This study presents new insights into the geometric and electronic structures and O2 consumption reactivity of the FeII active sites in the facial triad carboxylate variants of FIH. MCD spectroscopy on the resting sites shows that all the variants bind FeII and are all 6C with similar Δ5E splittings of ~1,800 cm−1. All the variants thus have 2 histidines, 3 waters with a 6th Cl6 (D201GCl), carboxylate (D201E) or water-derived (D201A, D201G) ligand bound to the FeII center.
The structures of all the variants substantially change when αKG is bound to the active site. From the MCD data, the FeII-to-αKG metal to ligand charge transfer transitions are observed at similar energies as wt, suggesting that αKG is analogously bound bidentate with the replacement of two water ligands in all the (FeII/αKG)D201X sites. All the variants, except D201E, are 6C but with larger Δ5E splittings (Table 1) than the wild-type enzyme. This supports the predicted role of the facial triad carboxylate in stabilizing the binding of the sixth water ligand through a hydrogen bond to its distal oxygen, as mutations eliminating this carboxylate lead to the weakening of this FeII-OH2 bond.
VTVH MCD on (FeII/αKG)D201E showed that it has a ground state splitting (Δ) of 1,200 cm−1 (Table 2), similar to the ternary 5C active sites, and the largest Δ5E splitting in αKG only bound sites of 2,700 cm−1. It is also the only αKG bound FeII variant that reacts with O2 in the absence of substrate (Table 3). From crystallography performed on this variant, the presence of the extra methyl group on the carboxylate affects its orientation relative to the FeII center such that it has a steric clash between its unbound O and the position where the sixth water ligand would bind, resulting in the 5C site with a monodentate carboxylate or a 6C site with a very weakly bound distal O of the glutamate residue.20 From computational studies on wt, a similar steric clash between asparagine on the CAD substrate and the water ligand bound to the FeII center leads to H2O dissociation and an open coordination position for O2 reactivity.29 Coupled with the MCD results on the (FeII/αKG)D201E site, the steric interaction with the sixth water ligand is an important factor along with the donation of the αKG in determining the 6C → 5C conversion in the FIH active site.
VTVH MCD data on the ternary (FeII/αKG/CAD)D201X complexes show that their ground state splittings (Δ) are oppositely signed compared to the wt enzyme (Table 2). The change in sign of Δ, due to the replacement of the carboxylate ligand with a Cl− or a water derived ligand, is significant as it demonstrates that the unpaired electron in the d manifold of the highspin FeII center, that is crucial for O2 activation, is now in a different redox active molecular orbital. This change is reflected in the O2 consumption kinetics as all the variants consumed O2 at rates more than 10-fold slower than the wildtype enzyme. Furthermore, from MCD spectroscopy on these ternary sites, all the variants have 5C components with their LF transitions at different energies, indicating structurally inequivalent active sites. These structural changes are manifested in the O2 consumption rates as they vary by more than two orders of magnitude across the variants. Though these O2 consumption rates are significantly different, it is important to note that, in terms of the ΔΔG‡ for the O2 activation reaction, these rate differences only correspond to a change in activation barrier of <3 kcal/mol relative to the wt barrier of ~15 kcal/mol. Thus, the carboxylate ligand of the facial triad is crucial in tuning the electronics of the ternary active site, such that the redox active molecular orbital is properly oriented for O2 activation.
Comparing the O2 consumption rate to the CAD hydroxylation rate showed that all the variants performed uncoupled turnover. D201 plays an important role in docking the CAD substrate in the native reaction by hydrogen bonding with an amide group in the backbone of the substrate (Fig. 3).17,28 From these O2 consumption and substrate hydroxylation kinetics, the variants react with O2 to form an FeIV =O but appear to be poorly positioned to react with the substrate through hydrogen atom abstraction (HAA), followed by rebound hydroxylation. Since these variants have structurally distinct 5C sites, changes in the orientation of the CAD substrate with respect to the FeIV=O would result in dissipation of the high-valent intermediate through enzyme inactivation or reduction (normally by the excess ascorbate in the assays).16,18
Figure 3.

Crystal structure of FIH ternary complex active site (1H2N) showing the H-bond between D201 and the N803 backbone amide. The carbon involved in HAA is highlighted in magneta.
It is interesting to note that while D201GCl has a chloride bound perpendicular to the open position on the Fe, only hydroxylation is observed. We have previously shown that the HAA reaction performed by non-heme FeIV=O intermediates can proceed through two possible channels: a σ-channel that is active for HAA along the Fe-O bond and a π-channel that performs HAA perpendicular to the Fe-O bond. While the π-channel can hydroxylate or halogenate depending on the positioning of carbon radical product (formed after HAA) relative to the OH− or Cl− equatorial ligand, the σ HAA channel produces a radical oriented along the FeIII-OH bond and can only lead to substrate hydroxylation.30,31 From the crystal structure in Fig. 3, the CAD substrate docks directly above the open coordination position in the FeII active site. This is consistent with FIH being oriented to use the Fe=O σ-channel for HAA leading to hydroxylation but not halogenation.
In summary, steric interaction with the sixth water ligand through either the substrate or an active site residue is the factor that determines the 6C to 5C conversion in FIH. The facial triad carboxylate in FIH plays key roles in ensuring that the 5C FeII center has the proper electronics for facile O2 activation and that the substrate is bound in the correct orientation for coupled HAA.
Supplementary Material
ACKNOWLEDGMENT
This research was supported by the U.S. National Institutes of Health grants GM 77413 (M.J.K) and GM 40392 (E.I.S.).
ASSOCIATED CONTENT
Supporting Information
Kinetic endpoint assays, hydrogen peroxide production assays & spectroscopic data not shown in the main text are plotted in the SI. The Supporting Information is available free of charge on the ACS Publications website.
The authors declare no competing financial interest.
REFERENCES
- (1).Baldwin JE; Abraham E Nat. Prod. Rep 1988, 5 (2), 129. [DOI] [PubMed] [Google Scholar]
- (2).Vaillancourt FH; Yin J; Walsh CT Proc. Natl. Acad. Sci 2005, 102 (29), 10111–10116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Fitzpatrick PF Annu. Rev. Biochem 1999, 68 (1), 355–381. [DOI] [PubMed] [Google Scholar]
- (4).Gerken T; Girard CA; Tung Y-CL; Webby CJ; Saudek V; Hewitson KS; Yeo GSH; McDonough MA; Cunliffe S; McNeill LA; Galvanovskis J; Rorsman P; Robins P; Prieur X; Coll AP; Ma M; Jovanovic Z; Farooqi IS; Sedgwick B; Barroso I; Lindahl T; Ponting CP; Ashcroft FM; O’Rahilly S; Schofield CJ Science 2007, 318 (5855), 1469–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Wenger RH J. Exp. Biol 2000, 203, 1253–1263. [DOI] [PubMed] [Google Scholar]
- (6).Gibson DT; Parales RE Curr. Opin. Biotechnol 2000, 11 (3), 236–243. [DOI] [PubMed] [Google Scholar]
- (7).Solomon EI; Light KM; Liu LV; Srnec M; Wong SD Acc. Chem. Res 2013, 46, 2725–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Que L Jr. Nature 2000, 7 (3), 182–184. [DOI] [PubMed] [Google Scholar]
- (9).Solomon EI; Goudarzi S; Sutherlin KD Biochemistry 2016, 55 (46), 6363–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Hewitson KS; McNeill LA; Riordan MV; Tian Y-M; Bullock AN; Welford RW; Elkins JM; Oldham NJ; Bhattacharya S; Gleadle JM; Ratcliffe PJ; Pugh CW; Schofield CJ J. Biol. Chem 2002, 277, 26351–26355. [DOI] [PubMed] [Google Scholar]
- (11).Elkins JM; Hewitson KS; McNeill LA; Seibel JF; Schlemminger I; Pugh CW; Ratcliffe PJ; Schofield CJ J. Biol. Chem 2003, 278, 1802–1806. [DOI] [PubMed] [Google Scholar]
- (12).Semenza GL Genes Dev 2000, 14 (16), 1983–1991. [PubMed] [Google Scholar]
- (13).Nagel S; Talbot NP; Mecinovic J; Smith TG; Buchan AM; Schofield CJ Antioxid. Redox Signal 2010, 12, 481–501. [DOI] [PubMed] [Google Scholar]
- (14).Hewitson KS; Schofield CJ Drug Discov. Today 2004, 9, 704–711. [DOI] [PubMed] [Google Scholar]
- (15).Zhou J; Kelly WL; Bachmann BO; Gunsior M; Townsend CA; Solomon EI J. Am. Chem. Soc 2001, 123 (30), 7388–7398. [DOI] [PubMed] [Google Scholar]
- (16).Liu A; Ho RYN; Que L; Ryle MJ; Phinney BS; Hausinger RP J. Am. Chem. Soc 2001, 123 (21), 5126–5127. [DOI] [PubMed] [Google Scholar]
- (17).Chen Y-H; Comeaux LM; Eyles SJ; Knapp MJ Chem. Commun. (Camb) 2008, 4768–4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chen Y-H; Comeaux LM; Herbst RW; Saban E; Kennedy DC; Maroney MJ; Knapp MJ J. Inorg. Biochem 2008, 102, 2120–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Light KM; Hangasky JA; Knapp MJ; Solomon EI J. Am. Chem. Soc 2013, 135, 9665–9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hangasky JA; Taabazuing CY; Martin CB; Eron SJ; Knapp MJ J. Inorg. Biochem 2017, 166, 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Hewitson KS; Holmes SL; Ehrismann D; Hardy AP; Chowdhury R; Schofield CJ; McDonough MA J. Biol. Chem 2008, 283, 25971–25978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Solomon EI; Pavel EG; Loeb KE; Campochiaro C Coord. Chem. Rev 1995, 144, 369–460. [Google Scholar]
- (23).Solomon EI; Brunold TC; Davis MI; Kemsley JN; Lee S; Lehnert N; Neese F; Skulan AJ; Yang Y; Zhou J Chem. Rev 2000, 100, 235–349. [DOI] [PubMed] [Google Scholar]
- (24).Pavel EG; Kitajima N; Solomon EI J. Am. Chem. Soc 1998, 120 (6), 3949–3962. [Google Scholar]
- (25).This is manuscript is under review at Inorganic Chemistry: ic-2018-01736q “Ligand substitution supports O2 activation in facial triad variants of FIH, an αKG dependent oxygenase.” Chaplin, Vanessa; Hangasky, John; Huang, Hsin-Ting; Duan, Ran; Maroney, Michael; Knapp, Michael. [DOI] [PMC free article] [PubMed]
- (26).Pavel EG; Zhou J; Busby RW; Gunsior M; Townsend C. a.; Solomon EI J. Am. Chem. Soc 1998, 120 (4), 743–753. [Google Scholar]
- (27).Neidig ML; Brown CD; Light KM; Fujimori DG; Nolan EM; Price JC; Barr EW; Bollinger JM; Krebs C; Walsh CT; Solomon EI J. Am. Chem. Soc 2007, 129 (46), 14224–14231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28.) Saban E; Flagg SC; Knapp MJ J. Inorg. Biochem 2011, 105, 630–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Light KM; Hangasky JA; Knapp MJ; Solomon EI Dalt. Trans 2014, 43 (4), 1505–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Srnec M; Solomon EI J. Am. Chem. Soc 2017, 139 (6), 2396–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Srnec M; Wong SD; England J; Que L Jr.; Solomon EI Proc. Natl. Acad. Sci. U. S. A 2012, 109, 14326–14331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.