

Abstract
Proteomic analysis of membrane proteins is challenged by the proteins solubility and detergent incompatibility with MS analysis. No single perfect protocol can be used to comprehensively characterize the proteome of membrane fraction. Here, we used cow milk fat globule membrane (MFGM) proteome analysis to assess six sample preparation procedures including one in-gel and five in-solution digestion approaches prior to LC-MS/MS analysis. The largest number of MFGM proteins were identified by suspension trapping (S-Trap) and filter-aided sample preparation (FASP) methods, followed by acetone precipitation without clean-up of tryptic peptides method. Protein identifications with highest average coverage was achieved by Chloroform/MeOH, in-gel and S-Trap methods. Most distinct proteins were identified by FASP method, followed by S-Trap. Analyses by Venn diagram, principal-component analysis, hierarchical clustering and the abundance ranking of quantitative proteins highlight differences in the MFGM fraction by the all sample preparation procedures. These results reveal the biased proteins/peptides loss occurred in each protocol. In this study, we found several novel proteins that were not observed previously by in-depth proteomics characterization of MFGM fraction in milk. Thus, a combination of multiple procedures with orthologous properties of sample preparation was demonstrated to improve the protein sequence coverage and expression level accuracy of membrane samples.
Keywords: Filter-aided sample preparation, In-solution digestion, Milk fat globule membrane, Suspension trapping
1 Introduction
Milk fat is a primary component of milk. Milk fat globule membrane (MFGM) is composed of an inner phospholipid monolayer and a complex lipid bilayer. In general, MFGM components contain neutral lipids, cholesterol, and polar lipids, as well as a protein mixture [1]. Protein content of the isolated cow MFGM fraction was estimated to be 22% and lipid fraction was 72% [2]. MFGM structure and composition, and their functional and nutritional properties have been investigated [3, 4]. Especially, MFGM protein components and complexity from human and dairy animals have recently drawn more attention [5, 6]. To characterize the MFGM proteome, several sample preparation procedures were applied in previous studies, and each method has both advantages and disadvantages in terms of sample compatibility with subsequent LC-MS/MS analysis [5,7,8].
Shotgun proteomic experiments generally consist of protein extraction, solubilization, digestion, and sample cleanup to remove compounds which interfere with LC-MS/MS analysis have been considered as important part of the integrity procedure. Of them, sample preparation is the most critical starting point to obtain high-quality large-scale proteomics data, which usually involves protein solubilization and subsequent denaturation, reduction, alkylation and digestion [9,10]. For protein solubilization and denaturation, SDS has been recognized as one of the most popular reagents and is especially useful for solubilizing hydrophobic membrane proteins [11]. Previous studies have concluded that SDS-assisted solutions provided potential benefits in helping to dissolve some difficult to solubilize proteins [11, 12]. However, SDS-containing solutions not only interfered with enzymatic protein digestion, but are also incompatible with subsequent LC-MS/MS analytical steps [10]. To overcome this issue, several methods were employed. Of them, in-gel and in-solution digestion, along with filter-aided sample preparation (FASP) were widely applied for the complete removal of SDS in protein samples prior to MS analysis [9, 13–15]. Comparative analysis of above mentioned sample preparation procedures for proteomics analysis were performed in previous studies [9,10,13]. Besides above methods, capillary electrophoresis is a highly attractive separation technique that could be directly coupled with tandem mass spectrometry for the proteome analysis [16–18]. Although these methods presented some drawbacks, such as, being labor intensive as well as some protein loss, these developments have provided important contributions to proteomic experiments. In addition, drawbacks of approaches could also promote the development in sample preparation methodology, a new method called Suspension trapping (S-Trap) for the samples preparation had been recently developed and implicated in saving time [19], and further for unbiased sample preparation protocols in proteomic analysis.
Thus, to explore the most effective approaches for analysis of membrane proteins, optimization of sample preparation workflows on the identification and quantitation of membrane proteins need to be evaluated for future research. In this study, a modified chloroform/methanol (Chl/MeOH) or acetone precipitation followed by in-solution digestion without clean-up of tryptic peptides for direct LC-MS/MS analysis were developed, and applied to characterization cow MFGM proteome in comparison with S-Trap, FASP, in-gel, and traditional Chl/MeOH workflows. The results show differences in components of the identified proteins discussed among the above methods and provide novel information to optimize samples preparation procedures.
2 Materials and methods
2.1 MFGM protein extraction
Three bottles of the same brand of pasteurized whole cow milk (Grade A) was purchased from the supermarket. Each bottle of milk served as a biological sample. Whole cow milk (900 mL) was centrifuged at 3000 × g at 4°C for 30 min and the fat layer was recovered. The recovered fat was washed with PBS for 20 min at 37°C, and the resuspened mixture was centrifuged again to obtain the milk fat. The wash procedure was repeated three times. Finally, milk fat was washed with MilliQ water to recover the fat globules. These wash procedures were used to remove residual caseins and whey proteins. Milk fat globules were solubilized with a SDS solution (100 mM Tris-HCl, pH 7.4, 100 mM DTT and 4% SDS) and then incubated in 95°C water bath for 10 min. The mixtures were centrifuged at 14 000 × g for 15 min, and the protein samples were collected. Protein concentrations were determined by SDS-PAGE with a known concentration E.coli proteins as a standard.
2.2 Sample preparation procedures for identification of MGFG proteins
FASP method: Fifty micrograms of cow MFGM protein mixtures were diluted with 200 μL UT buffer (8 M urea and 100 mM Tris-HCl, pH 8.0), loaded onto the ultrafiltration filter (10-kDa cutoff, Sartorius, Germany) and centrifuged at 14 000 × g for 25 min. After washed, 100 μL 50 mM iodoacetamide solution was added, incubated for 45 min at room temperature in the dark. Then, samples were washed twice with 200 μL UT buffer and three times with 200 μL 50 mM NH4HCO3 solution. Finally, 100 μL trypsin (Promega, USA) buffer (2.5 μg trypsin in 50 mM NH4HCO3) was added, placed in the thermo mixer for 1 min at 800 rpm, and the protein samples were digested at 37°C water bath for 16–18 h. The filter unit was centrifuged at 14 000 × g for 15 min and washed twice with 50 μL 50 mM NH4HCO3 solution. All three eluents containing tryptic peptides were pooled together and dried in Speed Vacuum.
S-Trap method: Sample preparation of S-Trap micro spin column (ProtiFi, Huntington, NY, USA) was referred the vendor’s protocol and Zougman et al [19]. Fifty micrograms of the protein samples were mixed with 50 mM iodoacetamide (final concentration) for 45 min in the dark at room temperature. 12% aqueous phosphoric acid was added at 1:10 for a final concentration of 1.2%, and S-Trap buffer (90% methanol in 100 mM triethylammonium bicarbonate (TEAB), pH 7.1) was also added to form colloidal protein particulate. Then, protein mixtures were transferred into the S-Trap micro column and centrifuged at 4 000 × g for 10 min, and washed with 150 μL S-Trap buffer. Finally, 60 μL trypsin buffer (2.5 μg trypsin in 50 mM TEAB) was added and digested at 37°C for 16–18 h. For peptide elution collection, 40 μL TEAB was added into micro spin column and centrifuged at 4 000 × g for 10 min and washed twice with 50 μL 50% ACN containing 0.2% formic acid solution. The digested peptides were dried in Speed Vacuum.
In-gel digestion method: Protein samples (50 μg) were mixed with 4 × SDS loading buffer and 100 mM DTT and then incubated in 95°C water for 10 min. The mixtures were loaded on the 10% NuPAGE (Thermo Fisher Scientific; Novex ®10% Bis-Tris Gels) and run at 100 V for 20 min until bromophenol blue migrated 2 cm into the gel. The gel was fixed with 40% MeOH/10% acetic acid and stained with colloidal Comassie blue. Subsequently, one fraction MGFM band of each lane was excised for in-gel digestion/extraction. Gel pieces were washed with MilliQ water, destained with 50% ACN and 50 mM NH4HCO3, and dehydrated with 100% ACN. Then, gels were reduced with 10 mM DTT at 56°C for 60 min and alkylated with 50 mM iodoacetamide for 45 min. Finally, repeated washing steps and trypsin buffer (2.5 μg trypsin in 50 mM NH4HCO3) was added and digested at 37°C for 16–18 h and then 5% formic acid was added. Gel pieces were extracted twice with 50% ACN and 5% formic acid and one time extraction with 75% ACN and 5% formic acid. Tryptic peptides were pooled and dried in Speed Vacuum.
Chloroform/methanol method (Chl/MeOH): Protein samples were precipitated on the basis of a protocol described by Wessel and Flügge [20]. Protein samples (50 μg) were mixed with methanol and chloroform (sample/water/methanol/chloroform 1:3:4:1 by volume), vortexed vigorously followed by centrifuged 14 000 × g for 15 min. The precipitated pellets were washed three times with methanol. Protein pellets were then dissolved with 50 μL 8 M urea in 50 mM NH4HCO3 pH 8.0. A final concentration of 50 mM iodoacetamide was added for 45 min in the dark at room temperature to alkylate the samples. Finally, 200 μL 50 mM NH4HCO3 pH 8.0 solution were added to dilute the urea concentration to less than 2 M, followed by trypsin buffer (2.5 μg trypsin in 50 mM NH4HCO3) and digestion at 37°C for 16–18 h. The digested peptides were desalted by C18 column (Sep-pack Cartridges, Waters) and then dried.
Chloroform/Methanol precipitation without clean-up of tryptic peptides method (Chl/MeOH-M): This method was performed following a protocol by Zhang et al with modification [21]. Briefly, samples of reduced proteins (50 μg) were alkylated with 50 mM iodoacetamide and precipitated with chloroform and methanol as described above. Subsequently, protein pellets were dissolved with 50 μL 50 mM NH4HCO3 pH 8.0 and digested with 50 μL trypsin buffer (2.5 μg trypsin in 50 mM NH4HCO3). Finally, digested peptides were dried in Speed Vacuum.
Acetone precipitation without clean-up of tryptic peptides method (Acetone-M): Protein samples (50 μg) were alkylated with 50 mM iodoacetamide for 45 min in the dark at room temperature. Subsequently, samples were precipitated with 5 volumes cold acetone at −20°C overnight and centrifuge 14 000 × g for 15 min. Protein pellets were collected and washed twice with acetone and once with 80% acetone. Then, the pellets were suspended in 50 μL 50 mM NH4HCO3 pH 8.0 solution. Finally, 50 μL trypsin buffer (2.5 μg trypsin in 50 mM NH4HCO3) was added and digested at 37°C for 16–18 h. The digested peptides were dried in Speed Vacuum.
2.3 Liquid chromatography-tandem mass spectrometry analysis
Dried tryptic peptides were dissolved with 2% ACN and 0.5% formic acid for nano LC-MS/MS analysis, which was carried out using an Orbitrap Fusion™ Tribrid™ (Thermo-Fisher Scientific, San Jose, CA) mass spectrometer with a nanospray Flex Ion Source, and coupled with a Dionex UltiMate3000RSLCnano system (Thermo, Sunnyvale, CA) as reported previously [22]. For each group, three biological and duplicate technical repeats were performed. The peptide samples (5 μL) were injected onto a PepMap C-18 RP nano trap column (100 μm × 20 mm, 5 μm, Dionex) with nanoViper Fittings at 20 μL/min flow rate for on-line desalting and then separated on a PepMap C-18 RP nano column (75 μm × 25 cm, 2 μm) at 35°C, and eluted in a 90 min gradient of 5–35% ACN in 0.1% formic acid at 300 nL/min, followed by a 7 min ramping to 90% ACN/0.1% formic acid and an 8 min hold at 90% ACN/0.1% formic acid. The column was re-equilibrated with 0.1% formic acid for 25 min before next injection. The Orbitrap Fusion is operated in positive ion mode with spray voltage set at 1.6 kV and source temperature at 275°C. External calibration for Fourier transform (FT), ion trap and quadrupole mass analyzers was performed. In data-dependent acquisition (DDA) analysis, the instrument was operated using FT mass analyzer in MS scan to select precursor ions followed by 3 s “Top Speed” data-dependent CID ion trap MS/MS scans at 1.6 m/z quadrupole isolation for precursor peptides with multiple charged ions above a threshold ion count of 10 000 and normalized collision energy of 30%. MS survey scans at a resolving power of 120 000 (fwhm at m/z 200), for the mass range of m/z 375–1575. Dynamic exclusion parameters were set at repeat count 1 with a 20 s repeat duration, an exclusion list size of 500, 40 s of exclusion duration with ± 10 ppm exclusion mass width. The activation time was 10 ms for CID analysis. All data were acquired under Xcalibur 3.0 operation software (Thermo-Fisher Scientific).
2.4 Protein identification and quantitation
The DDA raw files of duplicate technical repeats from each biological sample were combined and subjected to database searches using Proteome Discoverer 2.2 software (Thermo Fisher Scientific, Bremen, Germany) with the Sequest HT algorithm. The database search was conducted against a Bos taurus (41 062 entries downloaded on 4/12/2016 from National Center for Biotechnology Information non-redundant protein database) with two-missed trypsin cleavage sites allowed. The peptide precursor tolerance was set to 10 ppm and fragment ion tolerance was set to 0.6 Da. Variable modification of cysteine carbamidomethylation, methionine oxidation, protein N-terminal acetylation and deamidation of asparagines/glutamine were set for the database search. Only high confidence peptides defined by Sequest HT with a 1% false discovery rate by Percolator were considered for the peptide identification. Based on label free quantitiation (LFQ) workflow in Proteome Discoverer 2.2, the protein abundance values are the sums of the abundances of all the peptides. Proteins grouped was handled according the parsimony principle and no normalization was applied. The final protein identification numbers represent protein groups that were filtered with at least two peptides per protein. Precursor-based LFQ was used in Proteome Discoverer 2.2 for relative quantitation analysis of six different sample preparation methods.
2.5 Data analysis
Analyses of the identified MFGM proteins associated with annotated functions were performed according to the DAVID Functional Annotation Tools (david.ncifcrf.gov). The identified proteins were analyzed transmembrane domain and grand average of hydropathy (GRAVY) using TMHMM 2.0 (cbs.dtu.dk/services/TMHMM/) and Protparam (web.expasy.org/protparam/) software, respectively [23, 24]. Differences in identified peptides and proteins from each group were analyzed by one-way ANOVA using SPSS software (v16.0, IBM). Tukey’s test was used to evaluate differences between sample preparation groups and p-values of less than 0.05 were defined as significant.
3 Results and discussion
MFGM proteins were extracted from whole cow milk with SDS solution in triplicate and used to assess six different sample preparation methods in this study. Raw data are uploaded to PRIDE repository with the dataset identifier PXD009288. MFGM proteins are complexed with various lipids [2, 25], and this interaction may reduce their solubility. SDS is believed to be a superior reagent for protein solubilization and denaturation, and is widely used to solubilize membrane proteins. Due to its incompatibility with the subsequent LC-MS/MS analysis, it is required to completely remove the SDS during the sample preparation steps. Protein precipitation by Chl/MeOH is one of the common methods to remove SDS. Then precipitated proteins require reduction, alkylation and enzymatic digestion, and therefore the desalting of tryptic peptides by this Chl/MeOH method was needed prior to MS/MS analysis. Compared to the classical in-solution digestion procedure, the omission of peptide clean-up methods were implemented. That is, protein samples were precipitated after the proteins alkylated with iodoacetamide solution and then protein pellets were dissolved with volatile buffer: NH4HCO3 solution for tryptic digestion and direct LC-MS/MS analysis. This modified method was slightly different compared to a developed in-solution digestion of protein mixtures approach, in which protein pellets were first digested with trypsin, and followed by reduction and alkylation of tryptic peptides and then cleaned with ZipTips before MS/MS analysis [26]. Thus, this modified method for in-solution digestion without peptide cleanup is called either Acetone-M or Chl/MeOH-M depending on which solvent used for protein precipitation. Obviously, the modified method was more time efficient and economical.
3.1 Properties of identified peptides
The results of identified peptides and proteins from six different procedures are summarized in Table 1 and Supporting Information Table 1. The number of acquired peptide spectrum matches (PSMs) was found to be the highest in S-Trap and Chl/MeOH methods and the lowest in in-gel and FASP methods. Similarly, the number of peptides identified from S-Trap and Chl/MeOH methods turned out to be the highest, whereas in-gel and FASP methods were the lowest. Based on per protein with at least two identified peptides [14], we found the highest number of the identified proteins were obtained in S-Trap and FASP methods, followed by Acetone-M method, whereas the lowest number of identified proteins by in-gel digestion. According to the number of identified peptides and proteins, we found the highest average protein sequence coverage was achieved by Chl/MeOH, in-gel and S-Trap methods with 28.3, 26.0, and 25.3%, respectively, the lowest by FASP method down to 14.9%. Our results were different with previous study, where more identified peptides and a higher mean sequence coverage in E. coli were observed in FASP method [27]. This result may associate with the difference in sample types. A previous study found that in-gel digestion with multiple gel slices was the most advantageous approach for analysis of plasma membrane proteins [13], which makes sense given the separation capability at protein level in gel approach. However, to maintain the equal number of fraction for downstream MS/MS analysis, only one fraction of digests by six different sample preparation methods was performed in our study. We found the number of the identified proteins from one fraction of in-gel digestion was the lowest, compared to those of the other in-solution digestion methods in this study. This may reflect the biased proteins/peptides loss occurred in the in-gel digestion/extraction process more serious than the other in-solution digestion methods in MFGM sample.
Table 1.
Proteins identified with at least two peptides from cow MFGM samples using six experimental procedures
| Items | Chl/MeOH | Chl/MeOH-M | Acetone-M | FASP | S-Trap | In-gel |
|---|---|---|---|---|---|---|
| Proteins | 356 ± 29 c | 340 ± 6 c | 382 ± 8 b | 387 ± 19 ab | 412 ± 3 a | 184 ± 21 d |
| Peptides | 3069 ± 256 a | 2311 ± 10 c | 2583 ± 41 b | 1823 ± 122 d | 3127 ± 53 a | 1856 ± 166 d |
| PSMs | 19266 ± 1629 a | 16146 ± 278 b | 15495 ± 125 c | 11447 ± 216 d | 19012 ± 475 a | 9797 ± 826 e |
| Coverage | 28.3 ± 0.3 | 21.0 ± 0.1 | 23.2 ± 0.1 | 14.9 ± 0.2 | 25.3 ± 0.5 | 26.0 ± 0.5 |
| Proteins (3 runs) | 286 | 280 | 321 | 314 | 346 | 142 |
Peptides with missed tryptic cleavages were analyzed and the results are listed in Fig. 1. We found the lowest proportion of peptides with zero missed tryptic cleavages was in-gel digestion, followed by Chl/MeOH, whereas the highest proportion of peptides with zero missed cleavages were S-Trap, Acetone-M and Chl/MeOH-M methods. Due to the lack of tryptic cleavage sites across transmembrane chain fragments, we found proportions of peptides with one and two missed tryptic cleavages from in-gel digestion were significantly higher than Chl/MeOH, Chl/MeOH-M, S-Trap, Acetone-M and FASP methods. In addition, S-Trap, Acetone-M and Chl/MeOH-M methods shared similar proportions of peptides with zero, one and two missed tryptic cleavages, and their missed cleavages were not significant difference, respectively. Similarly, in-solution digestion method produced more missed cleavage sites than FASP were reported in previous study [28]. These results suggested that reduction/alkylation at protein level shown in our Acetone-M and Chl/MeOH-M enables to improve the accessibility of cleavage sites to trypsin. This is most likely due to the fact that protein level alkylation to chemically block cysteine residues to prevent Cys-containing proteins from forming disulphide bonds again in the protein renaturation processes. Effects of trypsin digestion on the MFGM proteins from each protocol were observed in our study, because protein digestion is usually performed by trypsin. Besides trypsin, other proteases such as, chymotrypsin, glu-C, lys-C, lys-N, and arg-C were also used in proteomics that may help to evaluate considerable bias in proteolytic digestion. Moreover, multi-enzyme digestion protocol had been implicated in improving proteome coverage [29]. These methods would be considered in the next experiment.
Figure 1.
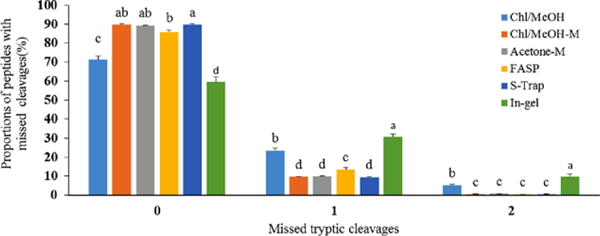
Distribution of peptides with missed tryptic cleavages in six sample preparation procedures.
3.2 Physicochemical properties of the identified proteins
The identified proteins from all studied protocols were evaluated on the basis of the hydrophobicity, transmembrane helices, molecular weight (MW) and isoelectric point (pI). The hydrophobicity of the identified MFGM proteins is listed in Fig. 2A. Besides in-gel digestion, distributions of GRAVY scores in the other experiments were very similar. All sample preparation methods were maximally represented between GRAVY scores -0.6 and 0. Proteins with GRAVY scores 0 and 0.2 from Chl/MeOH were higher than those of Chl/MeOH-M method. The distribution of transmembrane domains in the identified MFGM proteins from six methods were comparable from each other (Fig. 2B). Distributions of proteins containing one or more transmembrane helices in all methods were similar. The number of transmembrane proteins identified in FASP and S-Trap methods was the highest, and these identified by in-gel method was the lowest, compared to those by three in-solution methods, in which the number of the identified transmembrane proteins was similar.
Figure 2.
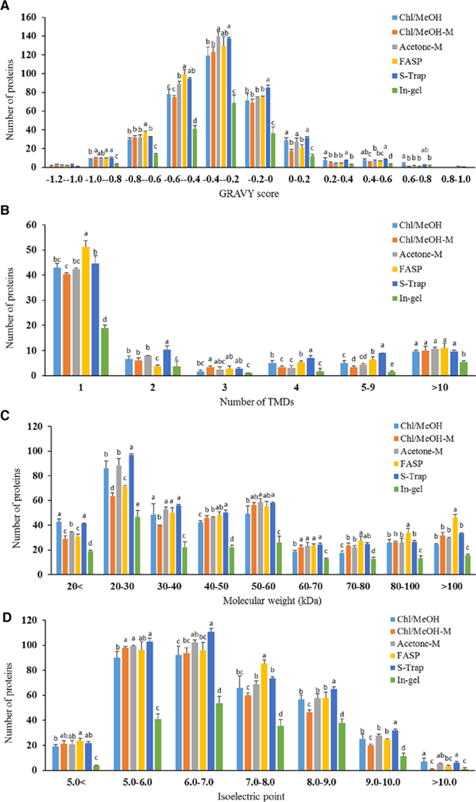
Distribution of the hydrophobicity (A), transmembrane helices (B), molecular weight (C) and isoelectric point (D) of the identified proteins from six methods.
Comparative analysis of MW of the identified MFGM proteins, the highest number of proteins with MW more than 100 kDa was identified in FASP method, and the highest number of proteins with MW less than 30 kDa was identified in S-Trap and Chl/MeOH methods (Fig. 2C). Our results indicated that FASP method produced a slight selective enrichment in high MW proteins, whereas S-Trap and Chl/MeOH methods tended in favor for low MW proteins. Distributions of proteins with pI in all experiments were similar (Fig. 2D), in which the number of proteins identified by in-gel method was the lowest, compared to those of the other methods. All sample preparation methods were maximally represented between pI 5.0 and 7.0.
3.3 Components of the identified proteins
To obtain the robust results, proteins identified with at least two peptides per protein in all three biological runs from each method were listed in Table 1 and used to do the following analysis. Effect of different sample preparation procedures on the components of the identified proteins were carried out by Venn diagrams (bioinformatics.psb.ugent.be/webtools/Venn/). We found only 101 overlap proteins that were presented all six different procedures. Some distinct proteins were identified in one sample preparation procedure alone. The highest number of distinct proteins was obtained by FASP (83 proteins), followed by S-Trap (26 proteins), and then three in-solution digestion (Chl/MeOH, Acetone-M and Chl/MeOH-M), whereas no distinct proteins were observed by in-gel digestion (Supporting Information Table 2). These results indicated that MFGM proteome identified strongly depends on the sample preparation procedure, due apparently to the biased protein loss occurred in each protocol. The S-Trap method appears the best procedure for membrane samples in terms of missed cleavage sites, average protein sequence coverage, and number of the identified proteins and their components. Considerable differences in components of the identified proteins were observed among FASP, in-gel and in-solution digestion methods in previous studies [9, 13]. As discussed previously, there is no single protocol that can be used to detect the entire proteome of MFGM fraction. Our results suggested combination of multiple methods with specific properties of sample preparation (FASP, S-Trap and one of in-solution digestion methods either Acetone-M or Chl/MeOH-M) would be the best complementary for better characterization of the proteome in membrane samples. In addition, we found that the number of the identified protein between Chl/MeOH and Chl/MeOH-M methods were very similar. However, protein components between Chl/MeOH and Chl/MeOH-M methods were obviously different. These results indicated that slight changes in the sample preparation steps could result in differences in the identified proteins. This phenomenon need further investigation on more different membrane samples.
3.4 Location of the identified proteins
Cellular components of the identified proteins were predicted using DAVID Functional Annotation Tools (Supporting Information Fig. 1). Most of the identified proteins were subcategorized as membrane and extracellular exosome as expected, and a small number of proteins were annotated as mitochondrion and endosome. This phenomena was related to fat droplets released into the milk, milk fat globules containing components of the cytoplasm are retained between the membrane layers [30]. Distributions of cellular components of the identified proteins in all experiments were similar.
3.5 Quantitative comparison of the identified MFGM proteins
To reveal the quantitative differences at the protein level, relative abundance of the identified proteins was evaluated among six methods (Supporting Information Table 3). Based on Perseus software (www.perseus-framework.org), Pearson correlation coefficients of three replicates for each procedure were more than 0.97 (Supporting Information Fig. 2) that was represented highly reproducible. A total of 771 proteins with at least two peptides were identified and quantified and selected on the basis of all three runs of each procedure. Of them, the highest number of the identified proteins was also quantified in S-Trap and FASP methods, followed by Acetone-M method. Out of the 771 proteins, 85 proteins including mammaglobin-A, olfactomedin-like protein 3, retinol dehydrogenases 10 and 14, were first identified in cow MFGM fraction (as shown Supporting Information Table 4), compared to the previously identified proteins of the pasteurized milk, in which 74 cow MFGM proteins were identified by shotgun proteomic method [31], and of raw milk in recent studies [5,6,32]. In raw milk, 554 proteins were identified and quantified by LFQ approach, and 520 proteins quantified by iTRAQ proteomics approach in bovine MFGM fraction [5,6]. Our data increase the total number of the identified proteins in MFGM fraction and facilitate the in-depth MFGM proteome coverage. Interestingly, these proteins have been implicated in the multiple potential physiological functions. For example, retinoid dehydrogenases families were involved in metabolism of various retinoid isomers and contributed to regulating vitamin A function [33]. Of them, retinol dehydrogenase 10 mediated the first step in retinoic acid synthesis from retinol to retinal and considered as a feedback regulator of retinoic acid signaling [34]. Thus, a broad in-depth proteins profile of cow MFGM fraction in pasteurized milk was achieved in this study that would contribute to better understanding the structural characteristic and physiological function of MFGM fraction. As shown in Supporting Information Table 4, GO analysis shows that majority of the 85 newly identified proteins in this study belong to the membrane proteins functioning as binding and catalytic activities for various biomoelcules.
The principal components analysis (PCA) was performed and score plot is shown in Fig. 3A. Protein profiles of FASP and in-gel methods were distinguished from the other studied groups in the direction of PC1, whereas protein components of these groups were distinguished in the direction of PC2. An obviously separated clustering of FASP, and a less effective but also clear discernment of S-Trap, Chl/MeOH, Acetone-M and Chl/MeOH-M were also observed. Hierarchical clustering analysis of the identified proteins yielded a pattern consisting of four major sample clusters. S-Trap, Acetone-M and Chl/MeOH-M shared similar proteomic patterns and comprised one subcluster. Chl/MeOH affiliated with the S-Trap, Acetone-M and Chl/MeOH-M to form a larger cluster. Subsequently, FASP joined this group to constitute another cluster, and then in-gel incorporated into this cluster (Fig. 3B). These results may be related to the protein precipitation implemented with acetone or methanol, resulted in the similar proteins loss among S-Trap, Acetone-M, Chl/MeOH and Chl/MeOH-M methods, compared to FASP method, in which most proteins retained on the filter unit (cut-off ≤ 30 kDa) [14, 35]. However, little information is now available that proteome profiles of Acetone-M and Chl/MeOH-M methods were more similar to these of S-Trap. In addition, differences in the quantitative proteins were observed between Chl/MeOH and Chl/MeOH-M methods that were similar to the results of Venn diagrams analysis. This phenomenon may be associated with the biased proteins/peptides loss occurred in protein level of reduction/alkylation prior to or after Chl/MeOH precipitation followed by in-solution digestion. Results of PCA and hierarchical clustering analysis highlight relationships of the quantitative cow MFGM proteins associated with different sample preparation procedures.
Figure 3.
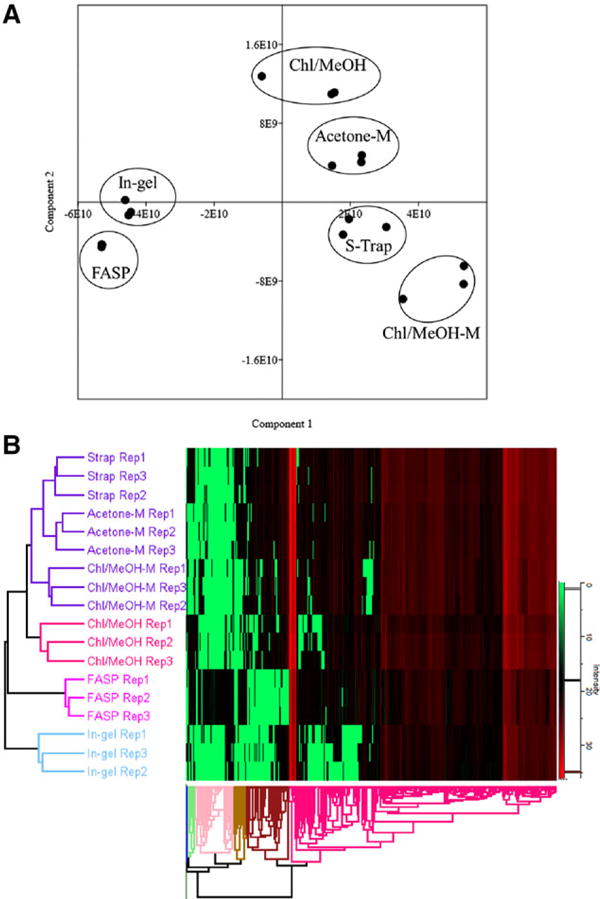
PCA scores plot (A) and hierarchical clustering (B) of the components of cow milk fat globule membrane proteins from the studied groups.
Based on the LFQ results, the abundance ranking of the proteins present in FASP is obviously different from that observed in the other methods. Top six of most abundance proteins in FASP are alpha-S2-casein, butyrophilin subfamily 1 member A1 (BTN1A1), beta-lactoglobulin, lactadherin, polymeric immunoglobulin receptor and lactotransferrin. While in S-Trap the equivalents are alpha-S1-casein, glycosylation-dependent cell adhesion molecule 1 (Glycam1), alpha-S2-casein, lactadherin, BTN1A1 and beta-lactoglobulin, whereas in three other methods the top abundance proteins are alpha-S1-casein, Glycam1, alpha-S2-casein, lactadherin, BTN1A1 and kappa-casein. Our results indicated that major MFGM proteins in pasteurized whole cow milk were from skim milk proteins incorporated into MFGM fraction, and some peptides of MFGM proteins affected by biochemical modification were difficult to detect [36], compared to proteins identified in raw milk, in which most abundant proteins included lactadherin, BTN1A1, xanthine dehydrogenase/oxidase and Glycam1 [2, 37]. Similar to the qualitative changes, relative abundance of the identified proteins shows obvious difference apparently due to biased proteins/peptides loss occurred in each protocol. Thus, quantitative protein expression profiling provided by a single protocol is most likely not to reflect the whole proteome picture in samples, which in turn indicates the undiscovered challenge for expression proteomics. Based on these results, we found S-Trap in combination with FASP methods yielded the most proteins (760) being quantified, followed by FASP coupled with Acetone-M methods (750 proteins). Thus, we believe that multiple procedures with orthogonal properties of sample preparation methods are one of the solutions to provide more confident and relatively accurate protein expression profiles. Further investigation using spiked proteins/peptides standard in samples is needed that may help to assess the recovery of each procedure and to determine their advantages and disadvantages for complementary roles.
4 Concluding remarks
In our study, six different sample preparation procedures for SDS removal and identification of MFGM proteins were compared and assessed. Of them, in-solution digestion methods with alkylation at protein level with omission of peptides cleanup (Acetone-M and Chl/MeOH-M) were demonstrated as a favorable method that saved cost and time. We found the highest number of MFGM proteins identified in S-Trap and FASP methods, followed by Acetone-M method. PCA, hierarchical clustering and the abundance ranking of quantitative proteins highlight differences in the MFGM fraction among the sample preparation procedures. Our results showed that the biased protein loss occurred in each sample preparation protocol by qualitative and quantitative proteomics analysis. As a result, an in-depth proteins profile of MFGM fraction in pasteurized cow milk was achieved and reported on the basis of six sample preparation procedures in this study. We conclude that a combination of multiple procedures with orthogonal properties of sample preparation that provide complementary coverage is required for global identification and quantitative characterization of the membrane proteome.
Supplementary Material
Acknowledgments
The work was partially supported by the NIH grant: PPAG14930. We also thank NIH SIG 1S10 PPOD017992-01 grant support for the Orbitrap Fusion mass spectrometer. We thank Mr. Robert Sherwood for his technical support and helpful discussion in sample preparation.
Abbreviations
- Acetone-M
Acetone precipitation without clean-up of tryptic peptides method
- BTN1A1
butyrophilin subfamily 1 member A1
- Chl/MeOH
chloroform/methanol
- Chl/MeOH-M
Chloroform/Methanol precipitation without clean-up of tryptic peptides method
- DDA
data-dependent acquisition
- FASP
filter-aided sample preparation
- FT
Fourier transform
- Glycam1
glycosylation-dependent cell adhesion molecule 1
- GRAVY
grand average of hydropathy
- LFQ
label free quantitiation
- MFGM
milk fat globule membrane
- MW
molecular weight
- PCA
principal components analysis
- PSMs
Peptide spectrum matches
- S-Trap
suspension trapping
- TEAB
triethylammonium bicarbonate
- UT buffer
8 M urea and 100 mM Tris-HCl, pH 8.0
Footnotes
Additional supporting information may be found in the online version of this article at the publisher’s web-site
Author agreement
All authors have reviewed and approved the final version of the manuscript being submitted. The article is the authors’ original work, hasn’t received prior publication and isn’t under consideration for publication elsewhere.
Author declaration
The authors have declared no conflict of interest.
References
- 1.Fong BY, Norris CS. J Agric Food Chem. 2009;57:6021–6028. doi: 10.1021/jf900511t. [DOI] [PubMed] [Google Scholar]
- 2.Fong BY, Norris CS, Akh MG. Int Dairy J. 2007;17:275–288. [Google Scholar]
- 3.Gallier S, Gragson D, Jimenez-Flores R, Everett D. J Agric Food Chem. 2010;58:4250–4257. doi: 10.1021/jf9032409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu J, Argov-Argaman N, Anggrek J, Boeren S, van Hooijdonk T, Vervoort J, Hettinga KA. J Dairy Sci. 2016;99:4726–4738. doi: 10.3168/jds.2015-10375. [DOI] [PubMed] [Google Scholar]
- 5.Lu J, Wang X, Zhang W, Liu L, Pang X, Zhang S, Lv J. Food Chem. 2016;196:665–672. doi: 10.1016/j.foodchem.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 6.Yang Y, Zheng N, Zhao X, Zhang Y, Han R, Ma L, Zhao S, Li S, Guo T, Wang J. J Proteomics. 2015;116:34–43. doi: 10.1016/j.jprot.2014.12.017. [DOI] [PubMed] [Google Scholar]
- 7.Pisanu S, Ghisaura S, Pagnozzi D, Biosa G, Tanca A, Roggio T, Uzzau S, Addis MF. J Proteomics. 2011;74:350–358. doi: 10.1016/j.jprot.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 8.Reinhardt TA, Lippolis JD. J Dairy Res. 2006;73:406–416. doi: 10.1017/S0022029906001889. [DOI] [PubMed] [Google Scholar]
- 9.Pasing Y, Colnoe S, Hansen T. Proteomics. 2017;17:1500462. doi: 10.1002/pmic.201500462. [DOI] [PubMed] [Google Scholar]
- 10.Zhou JY, Dann GP, Shi T, Wang L, Gao X, Su D, Nicora CD, Shukla AK, Moore RJ, Liu T, Camp DG, 2nd, Smith RD, Qian WJ. Anal Chem. 2012;84:2862–2867. doi: 10.1021/ac203394r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wiśniewski JR, Zougman A, Mann M. J Proteome Res. 2009;8:5674–5678. doi: 10.1021/pr900748n. [DOI] [PubMed] [Google Scholar]
- 12.Kuljanin M, Dieters-Castator DZ, Hess DA, Postovit LM, Lajoie GA. Proteomics. 2017;17:1600337. doi: 10.1002/pmic.201600337. [DOI] [PubMed] [Google Scholar]
- 13.Choksawangkarn W, Edwards N, Wang Y, Gutierrez P, Fenselau C. J Proteome Res. 2012;11:3030–3034. doi: 10.1021/pr300188b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wiśniewski JR, Zougman A, Nagaraj N, Mann M. Nat Methods. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 15.Lipecka J, Chhuon C, Bourderioux M, Bessard MA, van Endert P, Edelman A, Guerrera IC. Proteomics. 2016;16:1852–1857. doi: 10.1002/pmic.201600103. [DOI] [PubMed] [Google Scholar]
- 16.Hajba L, Guttman A. Trends Analyt Chem. 2017;90:38–44. [Google Scholar]
- 17.Bodnar J, Hajba L, Guttman A. Electrophoresis. 2016;37:3154–3159. doi: 10.1002/elps.201600405. [DOI] [PubMed] [Google Scholar]
- 18.de Jong N, Visser S, Olieman C. J Chromatogr A. 1993;652:207–213. doi: 10.1016/0021-9673(93)80661-Q. [DOI] [PubMed] [Google Scholar]
- 19.Zougman A, Selby PJ, Banks RE. Proteomics. 2014;14:1006–1000. doi: 10.1002/pmic.201300553. [DOI] [PubMed] [Google Scholar]
- 20.Wessel D, Flügge UI. Anal Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 21.Zhang S, Van Pelt CK, Henion JD. Electrophoresis. 2003;24:3620–3632. doi: 10.1002/elps.200305585. [DOI] [PubMed] [Google Scholar]
- 22.Thomas CJ, Cleland TP, Zhang S, Gundberg CM, Vashishth D. Anal Biochem. 2017;525:46–53. doi: 10.1016/j.ab.2017.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kyte J, Doolittle RF. J Mol Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 24.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 25.Affolter M, Grass L, Vanrobaeys F, Casado B, Kussmann M. J Proteomics. 2010;73:1079–1088. doi: 10.1016/j.jprot.2009.11.008. [DOI] [PubMed] [Google Scholar]
- 26.Kim SC, Chen Y, Mirza S, Xu Y, Lee J, Liu P, Zhao Y. J Proteome Res. 2006;5:3446. doi: 10.1021/pr0603396. [DOI] [PubMed] [Google Scholar]
- 27.Tanca A, Biosa G, Pagnozzi D, Addis MF, Uzzau S. Proteomics. 2013;13:2597–2607. doi: 10.1002/pmic.201200478. [DOI] [PubMed] [Google Scholar]
- 28.Fischer R, Kessler BM. Proteomics. 2015;15:1224–1229. doi: 10.1002/pmic.201400436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo X, Trudgian DC, Lemoff A, Yadavalli S, Mirzaei H. Mol Cell Proteomics. 2014;13:1573–1584. doi: 10.1074/mcp.M113.035170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McManaman JL, Neville MC. Adv Drug Delv Rev. 2003;55:629–641. doi: 10.1016/s0169-409x(03)00033-4. [DOI] [PubMed] [Google Scholar]
- 31.Arena S, Renzone G, Novi G, Scaloni A. J Proteomics. 2011;74:2453–2475. doi: 10.1016/j.jprot.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 32.Tacoma R, Fields J, Ebenstein DB, Lam YW, Greenwood SL. J Proteomics. 2016;130:200–210. doi: 10.1016/j.jprot.2015.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duester G. Eur J Biochem. 2000;267:4315–4324. doi: 10.1046/j.1432-1327.2000.01497.x. [DOI] [PubMed] [Google Scholar]
- 34.Strate I, Min TH, Iliev D, Pera EM. Development. 2009;136:461–472. doi: 10.1242/dev.024901. [DOI] [PubMed] [Google Scholar]
- 35.Wiśniewski JR, Zielinska DF, Mann M. Anal Biochem. 2011;410:307–309. doi: 10.1016/j.ab.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 36.Renzone G, Arena S, Scaloni A. J Proteomics. 2015;117:12–23. doi: 10.1016/j.jprot.2014.12.021. [DOI] [PubMed] [Google Scholar]
- 37.Spertino S, Cipriani V, De Angelis C, Giuffrida MG, Marsano F, Cavaletto M. Mol Biosyst. 2012;8:967–974. doi: 10.1039/c2mb05400k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.