

Abstract
The glomerular basement membrane (GBM) is an important component of the kidney’s glomerular filtration barrier. Like all basement membranes, the GBM contains type IV collagen, laminin, nidogen, and heparan sulfate proteoglycan. It is flanked by the podocytes and glomerular endothelial cells that both synthesize it and adhere to it. Mutations that affect the GBM’s collagen α3α4α5(IV) components cause Alport syndrome (kidney disease with variable ear and eye defects) and its variants, including thin basement membrane nephropathy. Mutations in LAMB2 that impact the synthesis or function of laminin α5β2γ1 (LM-521) cause Pierson syndrome (congenital nephrotic syndrome with eye and neurological defects) and its less severe variants, including isolated congenital nephrotic syndrome. The very different types of kidney diseases that result from mutations in collagen IV vs. laminin are likely due to very different pathogenic mechanisms. A better understanding of these mechanisms should lead to targeted therapeutic approaches that can help people with these rare but important diseases.
Introduction
Glomeruli are spheroid capillary tufts in the kidney cortex that filter the blood to generate the primary urine. This filtrate flows into the tubular segments of the nephron and then into the collecting ducts, which together convert the dilute primary urine into the final concentrated urine. Within the glomerulus is the glomerular basement membrane (GBM), a blanket-like extracellular matrix (ECM) that is situated between two cell types: specialized epithelial cells called podocytes that reside in the urinary space, and specialized fenestrated endothelial cells lining the glomerular capillaries (Fig. 1).
Fig. 1. Transmission electron micrograph showing the structure of a typical glomerular capillary from an adult mouse.
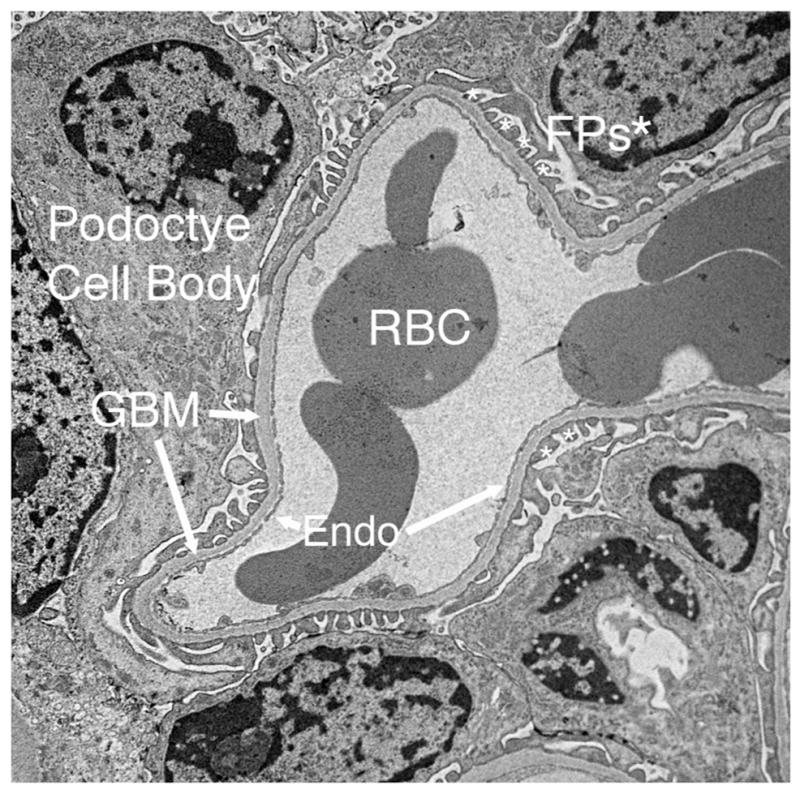
The glomerular basement membrane (GBM) is flanked by podocyte foot processes (FPs, asterisks) and the fenestrated glomerular endothelium (Endo). FPs emanate from podocyte cell bodies. Red blood cells (RBCs) can be seen in the lumen of the capillary.
The GBM together with the podocytes and endothelial cells comprise the glomerular filtration barrier between the blood and the urine [1, 2]. During glomerulogenesis, the GBM arises from fusion of separate basement membranes synthesized by the immature epithelial podocytes and glomerular endothelial cells [3]. Like all basement membranes [4], the GBM contains members of the four major classes of basement membrane proteins: collagen IV, laminin, heparan sulfate proteoglycan (primarily agrin, though perlecan is also present in the immature GBM [5]), and nidogen (isoforms 1 and 2) (Fig. 2) [6–8], though dozens of other matrix proteins are likely present [9, 10], including nephronectin [11, 12].
Fig. 2. Schematic diagrams of the four major GBM components.

Four type IV collagen α3α4α5(IV) triple helical heterotrimers (or protomers) interact via their 7S domains, and two interact via their NC1 domains to form the collagen IV network. LM-521 heterotrimers interact with each other via their laminin N-terminal (LN) domains to form a separate network. Nidogen binds to both collagen IV and laminin networks to help stabilize them and the GBM by linking them to each other. Agrin binds to LM-521 via its N-terminal agrin (NtA) domain. Both agrin and laminin α5 have C-terminal laminin globular (LG) domains that serve as ligands for cell surface receptors. These interactions link the GBM to the cell surface and allows the cell to influence the organization of GBM components.
The GBM is of special interest for both nephrologists and geneticists because there are mutations in type IV collagen and laminin genes that cause human kidney disease [13]. Alport syndrome is caused by mutations that affect the major collagen IV network of the GBM, whereas Pierson syndrome is caused by mutations that affect the laminin network. This makes the GBM also a keen interest of biochemists, since the hundreds of known collagen IV and laminin mutations can provide valuable information about important structural and functional aspects. In addition, recent breakthroughs in super-resolution imaging have provided novel information about how the collagen IV and laminin networks are arranged within the GBM, leading to new insights about how these networks might function in glomerular filtration.
Type IV collagen and Alport syndrome
Collagen IV chains and assembly
The type IV collagen family consists of six α chains, α1–α6, and each is encoded by a separate gene (COL4A1-COL4A6). Like all other collagen chains, collagen IV chains contain a collagenous domain consisting of Gly-X-Y amino acid triplet repeats that allow the intertwining of three α chains into a triple helix. However, unlike fibrillar collagens that form stiff structures, network-forming collagen IV contains multiple interruptions of the Gly-X-Y repeats interspersed throughout the large collagenous domain. These interruptions are thought to confer flexibility to the assembled trimers and to the basement membrane. In addition, each chain has characteristic noncollagenous domains at its NH2- and COOH-termini that are involved in chain recognition and assembly of monomers into trimers, as well as the assembly of trimers into the networks they form [14].
There are three known collagen IV heterotrimers (also called protomers), α1α2α1, α3α4α5, and α5α6α5. These trimers form within the endoplasmic reticulum and then self-assemble to form collagen IV networks after they are secreted into the extracellular space [15]. The NH2-terminal 7S domains interact with 7S domains of three other trimers to form a dodecameric structure, while the COOH-terminal NC1 domains interact with NC1 domains of one other trimer to form a hexameric structure. Through these intertrimer linkages, collagen IV protomers form a chain link fence- or chicken wire-like network [16, 17].
The major collagen IV network of the GBM is composed of α3α4α5 protomers, and there is also a less abundant α1α2α1(IV) network present on the endothelial aspect of the GBM [18]. Super-resolution imaging confirmed that these two networks are distinct and revealed that the collagen α3α4α5(IV) network is near the center of the GBM where it appears too distant from the surface of podocytes and endothelial cells to interact with typical matrix receptors [19]. This is in contrast to the collagen α1α2α1(IV) network, which is directly adjacent to glomerular endothelial cells [19] and should be capable of binding cell surface receptors.
Work that took advantage of state-of-the-art mass spectrometry has documented the unique nature of the inter-trimeric NC1 domain bonds that are present in collagen IV hexamers and are thought to provide increased stability to the collagen IV network in the context of significant tissue stress. These sulfilimine bonds occur between a methionine from one NC1 domain and a hydroxylysine within an adjoining NC1 domain [20]. Subsequent studies showed that formation of the bond requires a bromide ion [21] and is catalyzed by an enzyme called peroxidasin [22]. The importance of peroxidasin for the stability and function of the GBM may be called into questions by the existence of PXDN mutations that cause anterior segment dysgenesis of the eye in humans and in mouse without reported renal involvement [23–25]. However, only a few mutations have been discovered, and their impact on peroxidasin’s enzymatic activity or other functions has not yet been determined.
Diagnosis and genetics of Alport syndrome
Early signs of Alport syndrome, which usually appear in children, include hematuria (red blood cells in the urine) and evidence of a high tone sensorineural hearing deficit. By light microscopy, renal biopsies taken at early stages can appear normal or show signs of podocyte hypertrophy and tubular red blood cell casts [26], while at later stages, the GBM becomes focally and segmentally thickened, with eventual sclerosis and glomerular obsolescence. Light microscopy is not sufficient for an unequivocal diagnosis of Alport syndrome, whereas ultrastructural analysis of the glomerular capillary wall can usually provide a specific diagnosis. By standard transmission electron microscopy, the Alport GBM is initially thinner than normal. But as patients age the GBM usually becomes irregularly thickened, with outpocketing towards the podocyte aspect and a typical “basket weave” appearance rather than the normal homogenous lamina densa (Fig. 3A). Although this is the presentation in a majority of patients, some present with a uniformly thin GBM without obvious irregularly thickened areas [26].
Fig. 3. Transmission electron microscopy demonstrates splitting and thickening of the GBM in the Col4a3−/− mouse model of Alport syndrome.

A. Early stage of pathology shows mild GBM splitting and thickening (asterisk) with preserved podocyte foot processes (arrows) and endothelial cell fenestrations (arrowheads). B. Later stage of pathology shows extensive GBM splitting and thickening (black asterisk) and effacement of podocyte foot processes (arrows). Endothelial cell fenestrations appear to be maintained (arrowheads), and there is evidence of podocyte invasion into the GBM towards the endothelium (white asterisks). Some of these processes appear embedded in the GBM due to the plane of section.
The genetic basis for Alport syndrome was described in the early 1990s [27–29]. Mutations in any one of the three genes encoding components of the collagen α3α4α5(IV) network (COL4A3, COL4A4, and COL4A5) can cause the disease. Most mutations prevent assembly and/or secretion of collagen α3α4α5(IV) heterotrimers such that all 3 proteins are absent from the GBM. There is a compensatory expansion of the normally endothelial collagen α1α2α1(IV) network towards the podocyte aspect of the GBM [18, 19]; this apparently maintains GBM architecture temporarily, resulting in the delayed onset disease. A minority of patients carry a missense mutation that allows mutant collagen α3α4α5(IV) trimer assembly, secretion, and network formation, but there is functional impairment that can in some cases result in milder disease severity. Moreover, missense mutations could lead to collagen IV chain or trimer misfolding which could secondarily cause podocyte endoplasmic reticulum (ER) stress [30].
Overall, the genetics of Alport syndrome are quite complex [31], and the relationship between genotype and phenotype/disease status is currently undergoing a close reexamination by experts in the field. Complexity arises in part because the COL4A5 gene is on the X chromosome, whereas the COL4A3 and COL4A4 genes are tightly linked in a head-to-head orientation on chromosome 2. As with other diseases with an X-linked component, there are many more males affected than females because males have only a single X chromosome, so only one mutation is required in males for the disease to occur. But unlike many other X-linked diseases, females with a heterozygous mutation (COL4A5+/−) usually do show some symptoms, and some can even progress to kidney failure [32]. This can be explained by the concept that only podocytes secrete collagen α3α4α5(IV) protomers, and these protomers tend to assemble into the GBM where they are secreted. Due to random X chromosome inactivation, about half of all podocytes will secrete normal collagen α3α4α5(IV) protomers because their mutant X chromosome was inactivated, but the other half will have the normal X chromosome inactivated and will not be able to secrete normal protomers. Any clustering of podocytes unable to secrete collagen α3α4α5(IV) will lead to focal areas of abnormal GBM that can develop typical Alport GBM lesions and lead to the Alport phenotype. Preferential inactivation of the normal X chromosome will exacerbate disease presentation and speed progression to kidney failure. On the other hand, preferential inactivation of the mutant X chromosome should result in milder disease.
An additional layer of complexity arises for the autosomal COL4A3 and COL4A4 genes. Alport patients without an X-linked mutation carry mutations in either COL4A3 or COL4A4, usually homozygous or compound heterozygous, that prevent secretion of normal collagen α3α4α5(IV) trimers. These patients are diagnosed with autosomal recessive Alport syndrome. Interestingly, some COL4A3 and COL4A4 heterozygotes have hematuria and thin GBMs but not the typical split, thickened GBM observed in Alport patients. This condition has historically been called benign familial hematuria and/or thin basement membrane nephropathy. However, it is now clear that a significant percentage of these individuals will eventually progress to kidney failure, and many with high proteinuria will be biopsied and diagnosed as having focal segmental glomerulosclerosis (FSGS), a pathological diagnosis that had not until relatively recently been associated with collagen IV mutations [33–36]. Because ~1% of the population is thought to carry a heterozygous mutation in COL4A3 or COL4A4 and is therefore at increased risk for developing kidney disease, it is important to identify and monitor these individuals. As patients with classic Alport syndrome have been shown to benefit greatly from angiotensin converting enzyme inhibition (ACEi) therapy [37] such that it has become the standard of care, these heterozygotes could also benefit from ACEi if they are found to have kidney problems early enough.
Further genetic complexity and variation in disease penetrance can arise in patients with heterozygous mutations in more than one COL4 gene [38, 39]. In addition, evidence in mice shows that a LAMB2 variant can hasten progression to ESRD in Col4a3−/− Alport mice [40], suggesting that variants in non-collagenous components of the GBM may be able to impact progression of kidney disease in Alport patients. There are also reports of patients who carry both a COL4 gene mutation and a mutation or variant in NPHS2 that can exacerbate manifestation of disease [41, 42]. NPHS2 is a gene expressed primarily in podocytes that encodes podocin, a regulator of the integrity of the glomerular slit diaphragm, a critical podocyte cell-cell junction that bridges the gap between foot processes and is involved in maintaining the glomerular filtration barrier. This genetic interaction between a COL4 gene and NPHS2 demonstrates a pathophysiological linkage between the GBM and the slit diaphragm.
Pathogenesis of Alport syndrome
There has been much progress in investigating the pathophysiology of Alport syndrome using mouse and dog genetic models, but there are still many questions about exactly why a change in the collagen IV component of the GBM from collagen α3α4α5(IV) to collagen α1α2α1(IV) causes kidney disease. An important clue must lie in the typical ultrastructural defects observed in the Alport GBM and later in the podocytes in humans and animal models (Fig. 3). Despite the inevitable appearance of tubular and interstitial histopathology (inflammation and fibrosis) as kidney disease ensues, any hypotheses about the pathogenic mechanisms of Alport syndrome must begin with the impact of the defective GBM in the glomerulus.
The splitting and lamellation of the Alport GBM is thought to reflect an intrinsic structural defect due to the compensating collagen α1α2α1(IV) network being less crosslinked and/or more susceptible to proteases [17]. Alternatively, the GBM may simply have too little collagen IV, regardless of isoform. Although there is no good evidence for this, if podocytes are unable to secrete collagen α3α4α5(IV) and they do not secrete α1α2α1(IV) trimers instead, there could easily be less than normal levels of collagen IV in the GBM. (This could also be the case, though to a lesser extent, for COL4A3 and COL4A4 heterozygotes who have thin GBMs.) Imaging shows an expansion of the collagen α1α2α1(IV) network towards the podocyte aspect of the GBM in both human and mouse Alport syndrome [18, 19], but there are neither definitive quantitative nor qualitative data for understanding this compensating network; most researchers would likely agree that the GBM would disintegrate without sufficient collagen IV. Because knockouts in collagen receptors discoidin-like domain receptor 1, integrin α1, and integrin α2 provide some protection from Alport disease progression in mice [43–45], an attractive hypothesis is that the expanded domain of collagen α1α2α1(IV) in the Alport GBM leads to aberrant activation of these receptors on podocytes and results in loss of homeostasis [46].
Aside from the change in collagen IV composition, there are also changes to the GBM’s laminin composition. Human, dog, and mouse Alport GBM show ectopic accumulation of laminin α2 [43, 47], which is normally confined to the mesangial matrix. This ectopic laminin has been shown to provide aberrant signals to adjacent receptors [48], but it might also inhibit normal polymerization or signaling by LM-521, even though levels of laminin α5 are increased in Alport mouse GBM [49]. These changes in GBM composition and their effects on signaling, coupled with the characteristic changes to the architecture and shape of the Alport GBM, would be expected to impact the behavior of the adjacent cells: podocytes, mesangial cells, and endothelial cells. In fact, changes have been documented for all three cell types.
Podocytes experience changes in gene expression due to biomechanical strain associated with the abnormal GBM [50, 51], elevated focal adhesion kinase activation [48], and accelerated detachment from the GBM [52, 53], which typically causes glomerulosclerosis. Due to their critical role in establishing and maintaining the glomerular filtration barrier, protection of podocytes from injury should be paramount in therapeutic approaches in Alport syndrome [54]. As determined by 3-D electron microscopic reconstruction, and sometimes apparent in standard images (Fig. 3B), podocytes acquire an invasive phenotype evidenced by basal protrusions into the GBM, accompanied by foot process effacement [55]. In contrast, at least some of the observed processes in the GBM are thought to be derived from mesangial cells; this conclusion is based on positive staining in peripheral capillary loops for integrin α8 [56, 57], which is normally a marker of mesangial cells [12]. In support of a role for mesangial cells in the pathogenesis of Alport syndrome, activation of the endothelin A receptor on cultured mesangial cells initiates the formation of actin spikes, and pharmacological inhibition of the endothelin A receptor in Alport mice ameliorates some GBM abnormalities [58]. The increased activation of the endothelin A receptor on mesangial cells is thought to be due to increased endothelin A expression by glomerular endothelial cells, perhaps secondary to altered podocyte secretion of cytokines. Finally, using state-of-the-art helium ion scanning microscopy of Alport glomeruli, defects in both endothelial cells and podocytes were revealed [59].
Therapeutic approaches for Alport syndrome
Based on the routes of pathogenesis discussed above, various approaches to therapy have been attempted in Alport models. The most successful has been angiotensin converting enzyme (ACE) inhibition, which reduces blood pressure and proteinuria. Successful proof of concept studies in Alport mice using Ramipril [60] have been translated to humans such that ACE inhibition is now the standard of care for Alport patients [37, 61]. Although ACE inhibition extends the time to the appearance of proteinuria and to the need for dialysis and/or transplantation, it is not a cure for Alport syndrome. The notion that ACE inhibition might be effective at prolonging kidney function by reducing proteinuria is supported by the finding in Alport mice that knockout of the Alb gene that encodes albumin extends life span by 64%, presumably by reducing injury to the renal parenchyma due to albumin overload [62]. Although it is not feasible to remove albumin from Alport patients, targeting the signaling pathways activated by uptake of filtered albumin could be a successful therapeutic approach.
The podocyte-driven glomerulosclerosis and the likely indirect tubulointerstitial fibrosis that eventually destroys the function of the kidney have been major targets of therapeutic approaches. Transforming growth factor β promotes fibrosis in Alport syndrome, as it does in many types of organ fibroses. Anti-TGFβ therapy reduces fibrosis in Alport mice, but only in combination with integrin α1 knockout was there a dramatic impact on kidney function [43]. In contrast, targeting microRNA-21 (miR-21), which can be induced by TGFβ, with specific anti-miR-21 oligonucleotides was effective at reducing the level of albuminuria and increasing the 50% survival time point for Alport mice on the 129X1/SvJ background from 80 days to 110 days, a 38% increase [63]. There was a corresponding reduction in histopathology throughout the renal parenchyma. The mechanism involves enhanced PPARα/retinoid X receptor activation and enhanced mitochondrial function that leads to stimulation of protective metabolic pathways [63]. A therapeutic approach targeting the mesangial cell activation observed in Alport syndrome used sitaxentan, an endothelin A antagonist that is known to be toxic. In mice, treatment with sitaxentan nevertheless delayed proteinuria, improved GBM architecture, inhibited mesangial cell invasion of the GBM, increased lifespan by 20%, and prevented glomerulosclerosis and interstitial fibrosis [58]. It will be interesting to determine whether combined ACE inhibition with either anti-miR-21 therapy or endothelin A receptor antagonism can improve outcomes.
The finding that podocyte ER stress is associated with Alport syndrome [30], likely due to misfolded mutant collagen chains or to accumulation of chains that are unable to properly fold due to lack of one of the three required, has led to studies in vitro aimed at using chemical chaperones to reduce ER stress in cells from Alport patients [64]. In addition, amniotic fluid stem cells have shown promise in Alport mice for delaying progression of renal fibrosis via paracrine/endocrine modulation of profibrotic cytokine expression and recruitment of macrophages [65]. These innovative approaches could turn out to be effective tools for slowing progression to renal failure.
Laminin and Pierson syndrome
Laminin-521
The GBM’s major laminin isoform is laminin-521 (LM-521), a cross-shaped heterotrimeric glycoprotein composed of the laminin α5, β2, and γ1 chains. LM-521 trimers are secreted by both podocytes and endothelial cells [66, 67] and polymerize in the ECM to form separate lattice-like networks at each edge of the GBM. Polymerization of trimers into a network is directed by ternary interactions among the α5, β2, and γ1 chain laminin NH2-terminal (LN) domains [68, 69]. This leaves the large laminin globular (LG) domain at the COOH-terminus of the α5 chain to bind receptors (integrin and perhaps also non-integrin) [70] on the surfaces of the podocytes, endothelial cells, and mesangial cells so that they can adhere to and perhaps modify and influence the organization of the GBM. Nidogens and heparan sulfate proteoglycans are thought to link the independently formed collagen IV and laminin networks in the extracellular matrix to each other and in some cases to cellular receptors to establish the basement membrane and its linkage to cell surfaces.
Pierson syndrome and related diseases
Mutations affecting the laminin β2 gene (LAMB2) cause either Pierson syndrome or congenital nephrotic syndrome with or without ocular abnormalities, which is designated nephrotic syndrome type 5 (NPHS5) by Online Mendelian Inheritance in Man (OMIM #614199). Although pathogenic LAMB2 mutations are relatively rare compared to mutations in other genes that cause nephrotic syndrome [71], LAMB2 is considered to be an important candidate for genetic analysis in cases of nephrotic syndrome in the first year of life [72].
Pierson syndrome refers to autosomal recessive congenital nephrotic syndrome with diffuse mesangial sclerosis, distinct ocular abnormalities, and neurological deficits [73–75]. This “full blown” version of Pierson syndrome is usually associated with nonsense and splice site mutations in LAMB2 that are predicted to prevent synthesis of a full-length laminin β2 protein. In contrast, the less severe versions of the disease, often isolated congenital nephrotic syndrome with only minimal extrarenal involvement or NPHS5 [76], are associated with single amino acid changes/missense LAMB2 mutations. Based on these phenotypes, it seems that the GBM is more sensitive to LAMB2 defects than other sites where LAMB2 is found. Many of these missense mutations are clustered in the laminin β2 LN domain, suggesting that they could impair laminin polymerization. Based on studies in mice and in vitro, these missense mutations are thought to impair folding, secretion, and/or polymerization in the extracellular matrix.
Mechanisms of pathogenesis
The Lamb2−/− mouse model of Pierson syndrome was published in 1995, a decade before the corresponding human disease was identified genetically. The mouse develops nephrotic syndrome with foot process effacement (Fig. 4) and has been instrumental in helping to investigate the role of the GBM in establishing the filtration barrier to plasma albumin and the mechanisms by which LAMB2 missense mutations cause albuminuria and nephrotic syndrome. Lamb2−/− mice have a GBM that is more readily penetrated by the large tracer molecule ferritin than is the normal GBM, suggesting that the absence of LM-521 results in a GBM with larger pores [77]. Interestingly, forced compensatory expression of laminin β1 in podocytes, which results in overproduction of LM-511, restores the normal filtration barrier [78]. This indicates that LM-511 functions in the GBM similarly to LM-521 and that nephrotic syndrome might develop due to there being too little laminin in the GBM rather than to the absence of LM-521 per se.
Fig. 4. Ultrastructural analysis of glomerular capillary walls in control and Lamb2 mutant kidneys that model Pierson syndrome.

A. Lamb2+/− control mouse shows typical podocyte foot processes and endothelial fenestrations adjacent to the GBM. B. A Lamb2−/− mouse shows effaced or broadened podocyte foot processes but retained endothelial cell fenestrations and a GBM of normal appearance. C. A Lamb2−/− mouse was injected intravenously on consecutive days with LM-521. This treatment attenuated podocyte foot process effacement and delayed the onset of high level proteinuria. Arrows, podocyte foot processes; arrowheads, endothelial cell fenestrations; asterisks, GBM.
One of the main functions of laminin as a basement membrane component is to serve as a ligand for cellular receptors so that cells can adhere and receive signals. Podocytes express integrin α3β1, a receptor for LM-521’s α5 LG domain that is critical for podocyte homeostasis [79, 80]. Lamb2 null mice have reduced levels of laminin α5 in the GBM [78]. The pathogenesis of Pierson syndrome could therefore include impaired podocyte-GBM interactions in addition to intrinsic defects in the GBM’s architecture.
In an effort to investigate the pathogenic mechanisms of three different human LAMB2 missense mutations that cause nephrotic syndrome, we introduced them into mice either as transgenes specifically expressed in podocytes or via CRISPR/Cas9-mediated gene editing of Lamb2. These studies revealed different mechanisms leading to proteinuria. The LAMB2-R246Q and -C321R mutant proteins showed impaired secretion, likely due to protein misfolding, which reduces the total level of LM-521 in the GBM [81, 82]. Moreover, podocyte ER stress occurs due to LAMB2-C321R expression [82], likely impairing podocyte homeostasis and hastening their detachment from the GBM.
Although the mechanism for pathogenesis has not been rigorously investigated, a LAMB2-C185Y mutation, which is present in the LN domain and could impact folding, secretion, or polymerization, was identified in mice with early nephrotic syndrome, termed Nephertiti mice [83]. That these mice could live for at least 6 months suggests an impact on LAMB2 expression and/or function that is less severe than we observed in LAMB2-R246Q or -C321R transgenic mice. It is interesting to speculate that the C185Y mutation leaves an unpaired cysteine in the LN domain, causing either impaired LN domain folding or impaired polymerization, or both.
In contrast to the clear nephrotic phenotypes in these missense mutant mice, our CRISPR/Cas9-induced Lamb2S83R mouse model of the human LAMB2-S80R mutation found in a patient with delayed onset proteinuria did not recapitulate the human phenotype. This is despite the fact that our in vitro studies revealed a polymerization defect of LM-121 harboring the LAMB2-S83R mutation [84]. Furthermore, the analogous S68R mutation in LAMB1 also shows a polymerization defect [85]. To address the hypothesis that additional glomerular stress was necessary to reveal the pathogenicity of the mouse LAMB2-S83R mutation, we bred the mutation onto the Col4a3−/− Alport syndrome mouse background. Just one copy of the mutant Lamb2S83R allele exacerbated kidney disease progression, and two copies caused kidney failure by 2 weeks of age. We concluded that even a mild defect in the GBM’s laminin network in the context of the abnormal collagen IV network can hasten progression of Alport syndrome [84]. By extension, this suggests that variants in non-collagen IV components of the GBM can impact progression of Alport syndrome even if the variant is innocuous on its own.
Therapeutic approaches for Pierson syndrome
Nonspecific treatments that reduce blood pressure and albuminuria, such as angiotensin converting enzyme inhibitors, can be somewhat effective at mitigating the symptoms of nephrotic syndrome, but they are not a long-term cure. We reasoned that replacing at least some of the missing LM-521 in Pierson syndrome should decrease proteinuria by both reducing the average size of the pores in the GBM and providing missing signals to the adjacent cells. Because the GBM is accessible to proteins within the bloodstream, we simply injected full-sized LM-521 trimers into Lamb2 mutant mice beginning at 10–12 days of age for 5 or more days [86]. We found that the injected protein accumulated in the GBM and was stable there for at least twenty days, suggesting that it had properly integrated. Although the injected protein attenuated foot process effacement (Fig. 4) and delayed the onset of high level proteinuria, it did not prevent progression to nephrotic range proteinuria. Super-resolution imaging revealed that the injected LM-521 integrated on the endothelial side of the GBM but did not reach the podocyte aspect, likely because at 800 kDa it is simply too large to cross the dense collagen IV layer at the center of the GBM. Nevertheless, these data show that the GBM’s composition can be therapeutically manipulated [86]. This agrees with our proof-of-principle studies showing that inducible postnatal expression of collagen α3α4α5(IV) in the podocytes of Alport mice allows them to deposit a functional collagen α3α4α5(IV) network--into an existing and already functioning abnormal GBM--that slows disease progression [87]. Further work is necessary to find approaches that will be effective over the long term, and it is likely that important lessons will be learned from experimental therapeutic approaches in a model of congenital muscular dystrophy due to Lama2 mutation [88, 89].
Concluding thoughts
Both Alport syndrome and Pierson syndrome are caused by mutations that impact the structure and function of the GBM, yet the kidney aspects of these diseases are very different. Alport syndrome causes a gradual decline in kidney function, beginning with hematuria, but proteinuria begins later in the disease course. In stark contrast, Pierson syndrome and the related NPHS5 are congenital nephrotic syndromes with very high levels of proteinuria at or shortly after birth. Deciphering the mechanisms by which the different changes to GBM collagen IV and laminin composition causes these very different manifestations of kidney dysfunction will lead to novel, targeted therapeutic approaches that can improve the lives of patients.
Highlights.
The kidney glomerular basement membrane (GBM) is an extracellular matrix that helps filter the blood to make urine.
The GBM contains specialized isoforms of type IV collagen (collagen α3α4α5(IV)) and laminin (laminin α5β2γ1, or LM-521).
Mutations that impact the type IV collagen components cause Alport syndrome and thin basement membrane nephropathy.
Mutations in LAMB2 cause Pierson syndrome and isolated congenital nephrotic syndrome.
Acknowledgments
Our research into Alport syndrome and Pierson syndrome has been supported by NIH grants R01DK078314, R56DK100593, R01DK058366, and T32DK007126 and by grants from the Alport Syndrome Foundation-Pedersen Family-Kidney Foundation of Canada.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Miner JH. Building the glomerulus: a matricentric view. J Am Soc Nephrol. 2005;16(4):857–861. doi: 10.1681/ASN.2004121139. [DOI] [PubMed] [Google Scholar]
- 2.Miner JH. Organogenesis of the kidney glomerulus: Focus on the glomerular basement membrane. Organogenesis. 2011;7(2) doi: 10.4161/org.7.2.15275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abrahamson DR. Origin of the glomerular basement membrane visualized after in vivo labeling of laminin in newborn rat kidneys. J Cell Biol. 1985 Jun;100:1988–2000. doi: 10.1083/jcb.100.6.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pozzi A, Yurchenco PD, Iozzo RV. The nature and biology of basement membranes. Matrix Biol. 2017;57–58:1–11. doi: 10.1016/j.matbio.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harvey SJ, Jarad G, Cunningham J, Rops AL, van der Vlag J, Berden JH, Moeller MJ, Holzman LB, Burgess RW, Miner JH. Disruption of glomerular basement membrane charge through podocyte-specific mutation of agrin does not alter glomerular permselectivity. Am J Pathol. 2007;171(1):139–152. doi: 10.2353/ajpath.2007.061116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miner JH. Glomerular basement membrane composition and the filtration barrier. Pediatr Nephrol. 2011;26(9):1413–1417. doi: 10.1007/s00467-011-1785-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miner JH. The glomerular basement membrane. Exp Cell Res. 2012;318(9):973–978. doi: 10.1016/j.yexcr.2012.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suh JH, Miner JH. The glomerular basement membrane as a barrier to albumin. Nat Rev Nephrol. 2013;9(8):470–477. doi: 10.1038/nrneph.2013.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lennon R, Byron A, Humphries JD, Randles MJ, Carisey A, Murphy S, Knight D, Brenchley PE, Zent R, Humphries MJ. Global analysis reveals the complexity of the human glomerular extracellular matrix. J Am Soc Nephrol. 2014;25(5):939–951. doi: 10.1681/ASN.2013030233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Randles MJ, Humphries MJ, Lennon R. Proteomic definitions of basement membrane composition in health and disease. Matrix Biol. 2017;57–58:12–28. doi: 10.1016/j.matbio.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 11.Muller-Deile J, Dannenberg J, Schroder P, Lin MH, Miner JH, Chen R, Brasen JH, Thum T, Nystrom J, Staggs LB, Haller H, Fiedler J, Lorenzen JM, Schiffer M. Podocytes regulate the glomerular basement membrane protein nephronectin by means of miR-378a-3p in glomerular diseases. Kidney Int. 2017;92(4):836–849. doi: 10.1016/j.kint.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zimmerman SE, Hiremath C, Tsunezumi J, Yang Z, Finney B, Marciano DK. Nephronectin Regulates Mesangial Cell Adhesion and Behavior in Glomeruli. J Am Soc Nephrol. 2018;29 doi: 10.1681/ASN.2017070752. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chew C, Lennon R. Basement Membrane Defects in Genetic Kidney Diseases. Front Pediatr. 2018;6:11. doi: 10.3389/fped.2018.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khoshnoodi J, Sigmundsson K, Cartailler JP, Bondar O, Sundaramoorthy M, Hudson BG. Mechanism of chain selection in the assembly of collagen IV: a prominent role for the alpha2 chain. J Biol Chem. 2006;281(9):6058–6069. doi: 10.1074/jbc.M506555200. [DOI] [PubMed] [Google Scholar]
- 15.Brown KL, Cummings CF, Vanacore RM, Hudson BG. Building collagen IV smart scaffolds on the outside of cells. Protein Sci. 2017;26(11):2151–2161. doi: 10.1002/pro.3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hudson BG, Reeders ST, Tryggvason K. Type IV collagen: Structure, gene organization, and role in human diseases. J Biol Chem. 1993 Dec 15;268:26033–26036. [PubMed] [Google Scholar]
- 17.Hudson BG, Tryggvason K, Sundaramoorthy M, Neilson EG. Alport’s syndrome, Goodpasture’s syndrome, and type IV collagen. N Engl J Med. 2003;348(25):2543–2556. doi: 10.1056/NEJMra022296. [DOI] [PubMed] [Google Scholar]
- 18.Kashtan CE, Kim Y. Distribution of the alpha 1 and alpha 2 chains of collagen IV and of collagens V and VI in Alport syndrome. Kidney Int. 1992;42(1):115–126. doi: 10.1038/ki.1992.269. [DOI] [PubMed] [Google Scholar]
- 19.Suleiman H, Zhang L, Roth R, Heuser JE, Miner JH, Shaw AS, Dani A. Nanoscale protein architecture of the kidney glomerular basement membrane. Elife. 2013;2:e01149. doi: 10.7554/eLife.01149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vanacore R, Ham AJ, Voehler M, Sanders CR, Conrads TP, Veenstra TD, Sharpless KB, Dawson PE, Hudson BG. A sulfilimine bond identified in collagen IV. Science. 2009;325(5945):1230–1234. doi: 10.1126/science.1176811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCall AS, Cummings CF, Bhave G, Vanacore R, Page-McCaw A, Hudson BG. Bromine Is an Essential Trace Element for Assembly of Collagen IV Scaffolds in Tissue Development and Architecture. Cell. 2014;157(6):1380–1392. doi: 10.1016/j.cell.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhave G, Cummings CF, Vanacore RM, Kumagai-Cresse C, Ero-Tolliver IA, Rafi M, Kang JS, Pedchenko V, Fessler LI, Fessler JH, Hudson BG. Peroxidasin forms sulfilimine chemical bonds using hypohalous acids in tissue genesis. Nat Chem Biol. 2012;8(9):784–790. doi: 10.1038/nchembio.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi A, Lao R, Ling-Fung Tang P, Wan E, Mayer W, Bardakjian T, Shaw GM, Kwok PY, Schneider A, Slavotinek A. Novel mutations in PXDN cause microphthalmia and anterior segment dysgenesis. Eur J Hum Genet. 2015;23(3):337–341. doi: 10.1038/ejhg.2014.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khan K, Rudkin A, Parry DA, Burdon KP, McKibbin M, Logan CV, Abdelhamed ZI, Muecke JS, Fernandez-Fuentes N, Laurie KJ, Shires M, Fogarty R, Carr IM, Poulter JA, Morgan JE, Mohamed MD, Jafri H, Raashid Y, Meng N, Piseth H, Toomes C, Casson RJ, Taylor GR, Hammerton M, Sheridan E, Johnson CA, Inglehearn CF, Craig JE, Ali M. Homozygous mutations in PXDN cause congenital cataract, corneal opacity, and developmental glaucoma. Am J Hum Genet. 2011;89(3):464–473. doi: 10.1016/j.ajhg.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan X, Sabrautzki S, Horsch M, Fuchs H, Gailus-Durner V, Beckers J, Hrabe de Angelis M, Graw J. Peroxidasin is essential for eye development in the mouse. Hum Mol Genet. 2014;23(21):5597–614. doi: 10.1093/hmg/ddu274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heidet L, Gubler MC. The renal lesions of Alport syndrome. J Amer Soc Nephrol. 2009;20(6):1210–1215. doi: 10.1681/ASN.2008090984. [DOI] [PubMed] [Google Scholar]
- 27.Barker DF, Hostikka SL, Zhou J, Chow LT, Oliphant AR, Gerken SC, Gregory MC, Skolnick MH, Atkin CL, Tryggvason K. Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science. 1990 Jun 8;248:1224–1227. doi: 10.1126/science.2349482. [DOI] [PubMed] [Google Scholar]
- 28.Mochizuki T, Lemmink HH, Mariyama M, Antignac C, Gubler MC, Pirson Y, Verellen-Dumoulin C, Chan B, Schroder CH, Smeets HJ, Reeders ST. Identification of mutations in the a3(IV) and a4(IV) collagen genes in autosomal recessive Alport syndrome. Nature Genet. 1994 Sep;8:77–82. doi: 10.1038/ng0994-77. [DOI] [PubMed] [Google Scholar]
- 29.Lemmink HH, Mochizuki T, van den Heuvel PWJ, Schroder CH, Barrientos A, Monnens LAH, van Oost BA, Brunner HG, Reeders ST, Smeets HJM. Mutations in the type IV collagen alpha3 (COL4A3) gene in autosomal recessive Alport syndrome. Hum Mol Genet. 1994;3(8):1269–1273. doi: 10.1093/hmg/3.8.1269. [DOI] [PubMed] [Google Scholar]
- 30.Pieri M, Stefanou C, Zaravinos A, Erguler K, Stylianou K, Lapathitis G, Karaiskos C, Savva I, Paraskeva R, Dweep H, Sticht C, Anastasiadou N, Zouvani I, Goumenos D, Felekkis K, Saleem M, Voskarides K, Gretz N, Deltas C. Evidence for Activation of the Unfolded Protein Response in Collagen IV Nephropathies. J Amer Soc Nephrol. 2014;25(2):260–275. doi: 10.1681/ASN.2012121217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miner JH. Pathology vs. molecular genetics: (re)defining the spectrum of Alport syndrome. Kidney Int. 2014;86(6):1081–1083. doi: 10.1038/ki.2014.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Savige J, Colville D, Rheault M, Gear S, Lennon R, Lagas S, Finlay M, Flinter F. Alport Syndrome in Women and Girls. Clin J Am Soc Nephrol. 2016;11(9):1713–1720. doi: 10.2215/CJN.00580116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pierides A, Voskarides K, Athanasiou Y, Ioannou K, Damianou L, Arsali M, Zavros M, Pierides M, Vargemezis V, Patsias C, Zouvani I, Elia A, Kyriacou K, Deltas C. Clinico-pathological correlations in 127 patients in 11 large pedigrees, segregating one of three heterozygous mutations in the COL4A3/COL4A4 genes associated with familial haematuria and significant late progression to proteinuria and chronic kidney disease from focal segmental glomerulosclerosis. Nephrol Dial Transplant. 2009;24(9):2721–2729. doi: 10.1093/ndt/gfp158. [DOI] [PubMed] [Google Scholar]
- 34.Voskarides K, Damianou L, Neocleous V, Zouvani I, Christodoulidou S, Hadjiconstantinou V, Ioannou K, Athanasiou Y, Patsias C, Alexopoulos E, Pierides A, Kyriacou K, Deltas C. COL4A3/COL4A4 mutations producing focal segmental glomerulosclerosis and renal failure in thin basement membrane nephropathy. J Amer Soc Nephrol. 2007;18(11):3004–3016. doi: 10.1681/ASN.2007040444. [DOI] [PubMed] [Google Scholar]
- 35.Deltas C, Savva I, Voskarides K, Papazachariou L, Pierides A. Carriers of Autosomal Recessive Alport Syndrome with Thin Basement Membrane Nephropathy Presenting as Focal Segmental Glomerulosclerosis in Later Life. Nephron. 2015;130(4):271–280. doi: 10.1159/000435789. [DOI] [PubMed] [Google Scholar]
- 36.Malone AF, Phelan PJ, Hall G, Cetincelik U, Homstad A, Alonso A, Jiang R, Lindsey T, Wu G, Sparks MA, Smith SR, Webb NJA, Kalra P, Adeyemo A, Shaw AS, Conlon PJ, Jennette JC, Howell DN, Winn MP, Gbadegesin RA. A high frequency of hereditary nephritis with rare COL4A3/COL4A4 variants erroneously included in a familial FSGS cohort. Kidney Int. 2014 doi: 10.1038/ki.2014.305. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gross O, Licht C, Anders HJ, Hoppe B, Beck B, Tonshoff B, Hocker B, Wygoda S, Ehrich JH, Pape L, Konrad M, Rascher W, Dotsch J, Muller-Wiefel DE, Hoyer P, Knebelmann B, Pirson Y, Grunfeld JP, Niaudet P, Cochat P, Heidet L, Lebbah S, Torra R, Friede T, Lange K, Muller GA, Weber M. Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int. 2012;81(5):494–501. doi: 10.1038/ki.2011.407. [DOI] [PubMed] [Google Scholar]
- 38.Fallerini C, Baldassarri M, Trevisson E, Morbidoni V, La Manna A, Lazzarin R, Pasini A, Barbano G, Pinciaroli AR, Garosi G, Frullanti E, Pinto AM, Mencarelli MA, Mari F, Renieri A, Ariani F. Alport syndrome: impact of digenic inheritance in patients management. Clin Genet. 2017;92(1):34–44. doi: 10.1111/cge.12919. [DOI] [PubMed] [Google Scholar]
- 39.Mencarelli MA, Heidet L, Storey H, van Geel M, Knebelmann B, Fallerini C, Miglietti N, Antonucci MF, Cetta F, Sayer JA, van den Wijngaard A, Yau S, Mari F, Bruttini M, Ariani F, Dahan K, Smeets B, Antignac C, Flinter F, Renieri A. Evidence of digenic inheritance in Alport syndrome. J Med Genet. 2015;52(3):163–174. doi: 10.1136/jmedgenet-2014-102822. [DOI] [PubMed] [Google Scholar]
- 40.Funk SD, Bayer RH, Malone AF, McKee KK, Yurchenco PD, Miner JH. Pathogenicity of a Human Laminin beta2 Mutation Revealed in Models of Alport Syndrome. J Am Soc Nephrol. 2018;29 doi: 10.1681/ASN.2017090997. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tonna S, Wang YY, Wilson D, Rigby L, Tabone T, Cotton R, Savige J. The R229Q mutation in NPHS2 may predispose to proteinuria in thin-basement-membrane nephropathy. Pediatr Nephrol. 2008;23(12):2201–2207. doi: 10.1007/s00467-008-0934-7. [DOI] [PubMed] [Google Scholar]
- 42.Voskarides K, Arsali M, Athanasiou Y, Elia A, Pierides A, Deltas C. Evidence that NPHS2-R229Q predisposes to proteinuria and renal failure in familial hematuria. Pediatr Nephrol. 2012;27(4):675–679. doi: 10.1007/s00467-011-2084-6. [DOI] [PubMed] [Google Scholar]
- 43.Cosgrove D, Rodgers K, Meehan D, Miller C, Bovard K, Gilroy A, Gardner H, Kotelianski V, Gotwals P, Amatucci A, Kalluri R. Integrin alpha1beta1 and transforming growth factor-beta1 play distinct roles in Alport glomerular pathogenesis and serve as dual targets for metabolic therapy. Am J Pathol. 2000;157(5):1649–1659. doi: 10.1016/s0002-9440(10)64802-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gross O, Girgert R, Beirowski B, Kretzler M, Kang HG, Kruegel J, Miosge N, Busse AC, Segerer S, Vogel WF, Muller GA, Weber M. Loss of collagen-receptor DDR1 delays renal fibrosis in hereditary type IV collagen disease. Matrix Biol. 2010;29(5):346–356. doi: 10.1016/j.matbio.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 45.Rubel D, Kruegel J, Martin M, Leibnitz A, Girgert R, Miosge N, Eckes B, Muller GA, Gross O. Collagen receptors integrin alpha2beta1 and discoidin domain receptor 1 regulate maturation of the glomerular basement membrane and loss of integrin alpha2beta1 delays kidney fibrosis in COL4A3 knockout mice. Matrix Biol. 2014;34:13–21. doi: 10.1016/j.matbio.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 46.Cosgrove D, Liu S. Collagen IV diseases: A focus on the glomerular basement membrane in Alport syndrome. Matrix Biol. 2017;57–58:45–54. doi: 10.1016/j.matbio.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kashtan CE, Kim Y, Lees GE, Thorner PS, Virtanen I, Miner JH. Abnormal glomerular basement membrane laminins in murine, canine and human Alport syndrome: aberrant laminin alpha2 deposition is species-independent. J Amer Soc Nephrol. 2001;12(2):252–260. doi: 10.1681/ASN.V122252. [DOI] [PubMed] [Google Scholar]
- 48.Delimont D, Dufek BM, Meehan DT, Zallocchi M, Gratton MA, Phillips G, Cosgrove D. Laminin alpha2-mediated focal adhesion kinase activation triggers Alport glomerular pathogenesis. PLoS ONE. 2014;9(6):e99083. doi: 10.1371/journal.pone.0099083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abrahamson DR, Isom K, Roach E, Stroganova L, Zelenchuk A, Miner JH, St John PL. Laminin compensation in collagen alpha3(IV) knockout (Alport) glomeruli contributes to permeability defects. J Am Soc Nephrol. 2007;18(9):2465–2472. doi: 10.1681/ASN.2007030328. [DOI] [PubMed] [Google Scholar]
- 50.Meehan DT, Delimont D, Cheung L, Zallocchi M, Sansom SC, Holzclaw JD, Rao V, Cosgrove D. Biomechanical strain causes maladaptive gene regulation, contributing to Alport glomerular disease. Kidney Int. 2009;76(9):968–976. doi: 10.1038/ki.2009.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller RT. Mechanical properties of basement membrane in health and disease. Matrix Biol. 2017;57–58:366–373. doi: 10.1016/j.matbio.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 52.Ding F, Wickman L, Wang SQ, Zhang Y, Wang F, Afshinnia F, Hodgin J, Ding J, Wiggins RC. Accelerated podocyte detachment and progressive podocyte loss from glomeruli with age in Alport Syndrome. Kidney Int. 2017;92(6):1515–1525. doi: 10.1016/j.kint.2017.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wickman L, Hodgin JB, Wang SQ, Afshinnia F, Kershaw D, Wiggins RC. Podocyte Depletion in Thin GBM and Alport Syndrome. PLoS ONE. 2016;11(5):e0155255. doi: 10.1371/journal.pone.0155255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gross O, Kashtan CE, Rheault MN, Flinter F, Savige J, Miner JH, Torra R, Ars E, Deltas C, Savva I, Perin L, Renieri A, Ariani F, Mari F, Baigent C, Judge P, Knebelman B, Heidet L, Lagas S, Blatt D, Ding J, Zhang Y, Gale DP, Prunotto M, Xue Y, Schachter AD, Morton LCG, Blem J, Huang M, Liu S, Vallee S, Renault D, Schifter J, Skelding J, Gear S, Friede T, Turner AN, Lennon R. Advances and unmet needs in genetic, basic and clinical science in Alport syndrome: report from the 2015 International Workshop on Alport Syndrome. Nephrol Dial Transplant. 2017;32(6):916–924. doi: 10.1093/ndt/gfw095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Randles MJ, Collinson S, Starborg T, Mironov A, Krendel M, Konigshausen E, Sellin L, Roberts IS, Kadler KE, Miner JH, Lennon R. Three-dimensional electron microscopy reveals the evolution of glomerular barrier injury. Sci Rep. 2016;6:35068. doi: 10.1038/srep35068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zallocchi M, Johnson BM, Meehan DT, Delimont D, Cosgrove D. alpha1beta1 integrin/Rac1-dependent mesangial invasion of glomerular capillaries in Alport syndrome. Am J Pathol. 2013;183(4):1269–1280. doi: 10.1016/j.ajpath.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clark SD, Nabity MB, Cianciolo RE, Dufek B, Cosgrove D. X-Linked Alport Dogs Demonstrate Mesangial Filopodial Invasion of the Capillary Tuft as an Early Event in Glomerular Damage. PLoS ONE. 2016;11(12):e0168343. doi: 10.1371/journal.pone.0168343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dufek B, Meehan DT, Delimont D, Cheung L, Gratton MA, Phillips G, Song W, Liu S, Cosgrove D. Endothelin A receptor activation on mesangial cells initiates Alport glomerular disease. Kidney Int. 2016;90(2):300–310. doi: 10.1016/j.kint.2016.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsuji K, Suleiman H, Miner JH, Daley JM, Capen DE, Paunescu TG, Lu HAJ. Ultrastructural Characterization of the Glomerulopathy in Alport Mice by Helium Ion Scanning Microscopy (HIM) Sci Rep. 2017;7(1):11696. doi: 10.1038/s41598-017-12064-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gross O, Beirowski B, Koepke ML, Kuck J, Reiner M, Addicks K, Smyth N, Schulze-Lohoff E, Weber M. Preemptive ramipril therapy delays renal failure and reduces renal fibrosis in COL4A3-knockout mice with Alport syndrome. Kidney Int. 2003;63(2):438–446. doi: 10.1046/j.1523-1755.2003.00779.x. [DOI] [PubMed] [Google Scholar]
- 61.Kashtan CE, Ding J, Gregory M, Gross O, Heidet L, Knebelmann B, Rheault M, Licht C. Clinical practice recommendations for the treatment of Alport syndrome: a statement of the Alport Syndrome Research Collaborative. Pediatr Nephrol. 2013;28(1):5–11. doi: 10.1007/s00467-012-2138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jarad G, Knutsen RH, Mecham RP, Miner JH. Albumin contributes to kidney disease progression in Alport syndrome. Am J Physiol Renal Physiol. 2016;311(1):F120–130. doi: 10.1152/ajprenal.00456.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gomez IG, MacKenna DA, Johnson BG, Kaimal V, Roach AM, Ren S, Nakagawa N, Xin C, Newitt R, Pandya S, Xia TH, Liu X, Borza DB, Grafals M, Shankland SJ, Himmelfarb J, Portilla D, Liu S, Chau BN, Duffield JS. Anti-microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J Clin Invest. 2015;125(1):141–156. doi: 10.1172/JCI75852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang D, Mohammad M, Wang Y, Tan R, Murray LS, Ricardo S, Dagher H, van Agtmael T, Savige J. The Chemical Chaperone PBA, Reduces ER Stress and Autophagy and Increases Collagen IV alpha5 Expression in Cultured Fibroblasts From Men With X-Linked Alport Syndrome and Missense Mutations. Kidney Int Rep. 2017;2(4):739–748. doi: 10.1016/j.ekir.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sedrakyan S, Da Sacco S, Milanesi A, Shiri L, Petrosyan A, Varimezova R, Warburton D, Lemley KV, De Filippo RE, Perin L. Injection of amniotic fluid stem cells delays progression of renal fibrosis. J Amer Soc Nephrol. 2012;23(4):661–673. doi: 10.1681/ASN.2011030243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.St John PL, Abrahamson DR. Glomerular endothelial cells and podocytes jointly synthesize laminin-1 and -11 chains. Kidney Int. 2001;60(3):1037–1046. doi: 10.1046/j.1523-1755.2001.0600031037.x. [DOI] [PubMed] [Google Scholar]
- 67.Di Russo J, Hannocks MJ, Luik AL, Song J, Zhang X, Yousif L, Aspite G, Hallmann R, Sorokin L. Vascular laminins in physiology and pathology. Matrix Biol. 2017;57–58:140–148. doi: 10.1016/j.matbio.2016.06.008. [DOI] [PubMed] [Google Scholar]
- 68.Yurchenco PD. Basement membranes: cell scaffoldings and signaling platforms. Cold Spring Harb Perspect Biol. 2011;3(2) doi: 10.1101/cshperspect.a004911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yurchenco PD, Cheng YS. Self-assembly and calcium-binding sites in laminin. A three-arm interaction model. J Biol Chem. 1993;268(23):17286–17299. [PubMed] [Google Scholar]
- 70.Colognato H, Winkelmann DA, Yurchenco PD. Laminin polymerization induces a receptor-cytoskeleton network. J Cell Biol. 1999;145(3):619–631. doi: 10.1083/jcb.145.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Matejas V, Hinkes B, Alkandari F, Al-Gazali L, Annexstad E, Aytac MB, Barrow M, Blahova K, Bockenhauer D, Cheong HI, Maruniak-Chudek I, Cochat P, Dotsch J, Gajjar P, Hennekam RC, Janssen F, Kagan M, Kariminejad A, Kemper MJ, Koenig J, Kogan J, Kroes HY, Kuwertz-Broking E, Lewanda AF, Medeira A, Muscheites J, Niaudet P, Pierson M, Saggar A, Seaver L, Suri M, Tsygin A, Wuhl E, Zurowska A, Uebe S, Hildebrandt F, Antignac C, Zenker M. Mutations in the human laminin beta2 (LAMB2) gene and the associated phenotypic spectrum. Hum Mutat. 2010;31(9):992–1002. doi: 10.1002/humu.21304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hinkes BG, Mucha B, Vlangos CN, Gbadegesin R, Liu J, Hasselbacher K, Hangan D, Ozaltin F, Zenker M, Hildebrandt F. Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2) Pediatrics. 2007;119(4):e907–919. doi: 10.1542/peds.2006-2164. [DOI] [PubMed] [Google Scholar]
- 73.Zenker M, Aigner T, Wendler O, Tralau T, Muntefering H, Fenski R, Pitz S, Schumacher V, Royer-Pokora B, Wuhl E, Cochat P, Bouvier R, Kraus C, Mark K, Madlon H, Dotsch J, Rascher W, Maruniak-Chudek I, Lennert T, Neumann LM, Reis A. Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum Mol Genet. 2004;13(21):2625–2632. doi: 10.1093/hmg/ddh284. [DOI] [PubMed] [Google Scholar]
- 74.Zenker M, Pierson M, Jonveaux P, Reis A. Demonstration of two novel LAMB2 mutations in the original Pierson syndrome family reported 42 years ago. Am J Med Genet A. 2005;138:73–74. doi: 10.1002/ajmg.a.30894. [DOI] [PubMed] [Google Scholar]
- 75.Zenker M, Tralau T, Lennert T, Pitz S, Mark K, Madlon H, Dotsch J, Reis A, Muntefering H, Neumann LM. Congenital nephrosis, mesangial sclerosis, and distinct eye abnormalities with microcoria: an autosomal recessive syndrome. Am J Med Genet A. 2004;130(2):138–145. doi: 10.1002/ajmg.a.30310. [DOI] [PubMed] [Google Scholar]
- 76.Hasselbacher K, Wiggins RC, Matejas V, Hinkes BG, Mucha B, Hoskins BE, Ozaltin F, Nurnberg G, Becker C, Hangan D, Pohl M, Kuwertz-Broking E, Griebel M, Schumacher V, Royer-Pokora B, Bakkaloglu A, Nurnberg P, Zenker M, Hildebrandt F. Recessive missense mutations in LAMB2 expand the clinical spectrum of LAMB2-associated disorders. Kidney Int. 2006;70(6):1008–1012. doi: 10.1038/sj.ki.5001679. [DOI] [PubMed] [Google Scholar]
- 77.Jarad G, Cunningham J, Shaw AS, Miner JH. Proteinuria precedes podocyte abnormalities in Lamb2−/− mice, implicating the glomerular basement membrane as an albumin barrier. J Clin Invest. 2006;116(8):2272–2279. doi: 10.1172/JCI28414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Suh JH, Jarad G, VanDeVoorde RG, Miner JH. Forced expression of laminin beta1 in podocytes prevents nephrotic syndrome in mice lacking laminin beta2, a model for Pierson syndrome. Proc Natl Acad Sci USA. 2011;108(37):15348–15353. doi: 10.1073/pnas.1108269108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kreidberg JA, Donovan MJ, Goldstein SL, Rennke H, Shepherd K, Jones RC, Jaenisch R. Alpha3 beta1 integrin has a crucial role in kidney and lung organogenesis. Development. 1996;122:3537–3547. doi: 10.1242/dev.122.11.3537. [DOI] [PubMed] [Google Scholar]
- 80.Pozzi A, Jarad G, Moeckel GW, Coffa S, Zhang X, Gewin L, Eremina V, Hudson BG, Borza DB, Harris RC, Holzman LB, Phillips CL, Fassler R, Quaggin SE, Miner JH, Zent R. Beta1 integrin expression by podocytes is required to maintain glomerular structural integrity. Dev Biol. 2008;316(2):288–301. doi: 10.1016/j.ydbio.2008.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen YM, Kikkawa Y, Miner JH. A missense LAMB2 mutation causes congenital nephrotic syndrome by impairing laminin secretion. J Amer Soc Nephrol. 2011;22:849–858. doi: 10.1681/ASN.2010060632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen YM, Zhou Y, Go G, Marmerstein JT, Kikkawa Y, Miner JH. Laminin beta2 gene missense mutation produces endoplasmic reticulum stress in podocytes. J Am Soc Nephrol. 2013;24(8):1223–33. doi: 10.1681/ASN.2012121149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bull KR, Mason T, Rimmer AJ, Crockford TL, Silver KL, Bouriez-Jones T, Hough TA, Chaudhry S, Roberts IS, Goodnow CC, Cornall RJ. Next-generation sequencing to dissect hereditary nephrotic syndrome in mice identifies a hypomorphic mutation in Lamb2 and models Pierson’s syndrome. J Pathol. 2014;233(1):18–26. doi: 10.1002/path.4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Funk SD, Bayer RH, Malone AF, McKee KK, Yurchenco PD, Miner JH. Pathogenecity of a human laminin beta2 mutation revealed in models of Alport syndrome. J Amer Soc Nephrol. 2018;29 doi: 10.1681/ASN.2017090997. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Purvis A, Hohenester E. Laminin network formation studied by reconstitution of ternary nodes in solution. J Biol Chem. 2012;287(53):44270–44277. doi: 10.1074/jbc.M112.418426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lin MH, Miller JB, Kikkawa Y, Suleiman HY, Tryggvason K, Hodges BL, Miner JH. Laminin-521 Protein Therapy for Glomerular Basement Membrane and Podocyte Abnormalities in a Model of Pierson Syndrome. J Am Soc Nephrol. 2018;29 doi: 10.1681/ASN.2017060690. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lin X, Suh JH, Go G, Miner JH. Feasibility of repairing glomerular basement membrane defects in Alport syndrome. J Amer Soc Nephrol. 2014;25(4):687–692. doi: 10.1681/ASN.2013070798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McKee KK, Crosson SC, Meinen S, Reinhard JR, Ruegg MA, Yurchenco PD. Chimeric protein repair of laminin polymerization ameliorates muscular dystrophy phenotype. J Clin Invest. 2017;127(3):1075–1089. doi: 10.1172/JCI90854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Reinhard JR, Lin S, McKee KK, Meinen S, Crosson SC, Sury M, Hobbs S, Maier G, Yurchenco PD, Ruegg MA. Linker proteins restore basement membrane and correct LAMA2-related muscular dystrophy in mice. Sci Transl Med. 2017;9(396) doi: 10.1126/scitranslmed.aal4649. [DOI] [PMC free article] [PubMed] [Google Scholar]