

Abstract
Selective activation of the M1 subtype of muscarinic acetylcholine receptor, via positive allosteric modulation (PAM), is an exciting strategy to improve cognition in schizophrenia and Alzheimer’s disease patients. However, highly potent M1 ago-PAMs, such as MK-7622, PF-06764427, and PF-06827443, can engender excessive activation of M1, leading to agonist actions in the prefrontal cortex (PFC) that impair cognitive function, induce behavioral convulsions, and result in other classic cholinergic adverse events (AEs). Here, we report a fundamentally new and highly selective M1 PAM, VU0486846. VU0486846 possesses only weak agonist activity in M1-expressing cell lines with high receptor reserve and is devoid of agonist actions in the PFC, unlike previously reported ago-PAMs MK-7622, PF-06764427, and PF-06827443. Moreover, VU0486846 shows no interaction with antagonist binding at the orthosteric acetylcholine (ACh) site (e.g., neither bitopic nor displaying negative cooperativity with [3H]-NMS binding at the orthosteric site), no seizure liability at high brain exposures, and no cholinergic AEs. However, as opposed to ago-PAMs, VU0486846 produces robust efficacy in the novel object recognition model of cognitive function. Importantly, we show for the first time that an M1 PAM can reverse the cognitive deficits induced by atypical antipsychotics, such as risperidone. These findings further strengthen the argument that compounds with modest in vitro M1 PAM activity (EC50 > 100 nM) and pure-PAM activity in native tissues display robust procognitive efficacy without AEs mediated by excessive activation of M1. Overall, the combination of compound assessment with recombinant in vitro assays (mindful of receptor reserve), native tissue systems (PFC), and phenotypic screens (behavioral convulsions) is essential to fully understand and evaluate lead compounds and enhance success in clinical development.
Keywords: M1, muscarinic acetylcholine receptor, agonist, positive allosteric modulator, ago-PAM, cognition, schizophrenia
Graphical Abstract
INTRODUCTION
Of the five muscarinic acetylcholine receptor subtypes (M1–M5), extensive biochemical, genetic, and patient data have implicated selective activation of M1 as an attractive approach for the treatment of the cognitive deficits associated with both schizophrenia and Alzheimer’s disease (AD).1–15 Early efforts to selectively activate M1 with pan-muscarinic agonists ultimately failed due to a lack of subtype selectivity, resulting in severe gastrointestinal (GI) disturbances and SLUD (salivation, lacrimation, urination, defecation) due to activation of peripheral M2 and M3.16 However, Phase II and Phase III clinical trials with these agonists demonstrated procognitive efficacy.17,18 Thus, the field moved to targeting less conserved allosteric binding sites on the M1 receptor and achieved unprecedented levels of muscarinic subtype selectivity with positive allosteric modulators (PAMs).1,2,12–14 BQCA was the first of these compounds disclosed (Figure 1).19–21 However, BQCA and related analogues, such as the subsequent clinical compound MK-7622,22 are robust ago-PAMs demonstrating direct agonist activity in addition to PAM activity in both in vitro recombinant systems and native tissues.19–23 In addition, they appear to interact with the orthosteric site, which may contribute to their severe seizure liability and other cholinergic adverse effects (AEs), as well as limit their procognitive efficacy.19–23 An early compound derived from our internal high-throughput screening (HTS) campaign,24 VU0453595 (VU595), proved to be a pure-PAM, devoid of direct agonist activity, in both recombinant and native systems. Additionally, VU595 was devoid of AEs and possessed procognitive activity. However, VU0453595 is a weak M1 PAM with a potency at M1 in the low micromolar range.25 Derived from another HTS hit, VU6004256 is a more potent M1 PAM, with agonist activity in high receptor reserve recombinant cell lines, yet is devoid of behavioral convulsions and cholinergic AEs in rodents.26,27 Unlike the structurally related M1 ago-PAM PF-0676442728 and the reported M1 PAM PF-06767832,29 VU6004256 displays procognitive efficacy in rodents.26,27 Furthermore, despite very low agonist activity in a recombinant in vitro cell line, PF-06827443 still elicited robust cholinergic AEs across species and induced seizures in mice.29 Recently, we demonstrated that similar to MK-7622,22 PF-06827443 is a potent agonist in native systems (prefrontal cortex (PFC)), and that agonist activity in native tissues accurately predicts the magnitude of in vivo efficacy/toxicity.30 Thus, the field does not have a robust in vivo M1 PAM with the ideal properties to critically evaluate the role of M1 in CNS biology. Therefore, we are reporting VU0486846, a highly selective, potent M1 PAM devoid of agonist activity in native tissues and cholinergic AEs, with robust procognitive efficacy, for the benefit of the research community to study selective M1 activation in vitro and in vivo.
Figure 1.
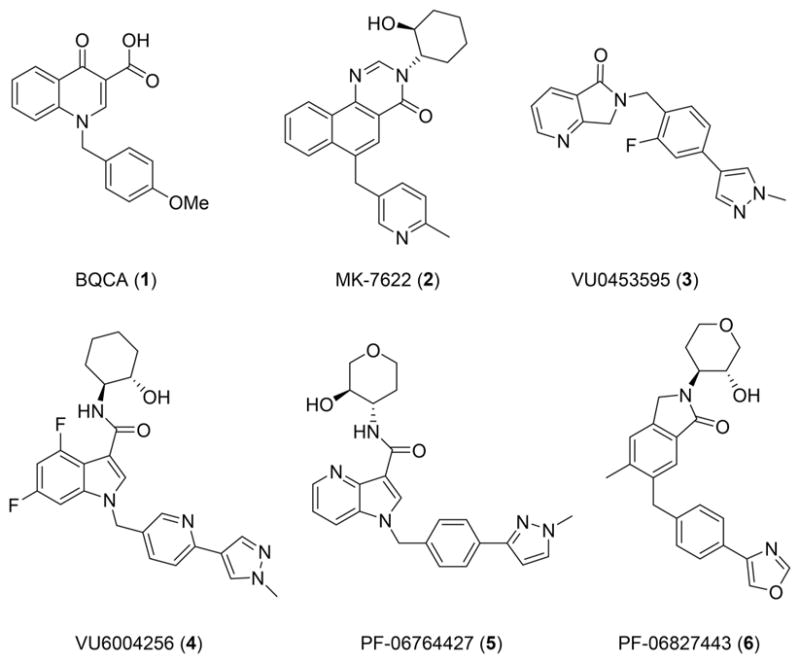
Structures of reported M1 PAMs with varying degrees of M1 PAM potency, varying degrees of M1 ago-PAM activity in recombinant cell lines and native tissues, and a diverse range of cholinergic AEs.
RESULTS AND DISCUSSION
Ligand Design toward the Next Generation of M1 PAMs
Having exhausted our indole-based series of M1 PAMs (e.g., VU6004256),26,27 we revisited where the M1 PAM field began—with BQCA.19–22 The discovery of the (1S,2S)-2-aminocyclohexan-1-ol amide moiety, as replacement for the carboxylic acid functionality of BQCA, as in 7, was a watershed event.31 Even more salient was the recognition that the secondary N–H of (1S,2S)-2-aminocyclohexan-1-ol amide moiety formed an intramolecular hydrogen bond (IMHB) with the carbonyl oxygen of the quinolone core.29–31 It was this recognition that led to the tricyclic core of MK-7622 and the azaindole core of PF-06764427. Utilizing the 6,6-fused ring system of BQCA and 7, we simultaneously increased sp3 character, while bringing the Lewis basic oxygen into the ring by scaffold hopping to a benzomorpholine core 8 (Figure 2). By virtue of this juxtaposition, we also created a new chiral center, and the opportunity for enantioselective M1 PAM activity while still maintaining the key intramolecular hydrogen bonding (IMHB) between the secondary amide N–H and the benzomorpholine oxygen atom.
Figure 2.
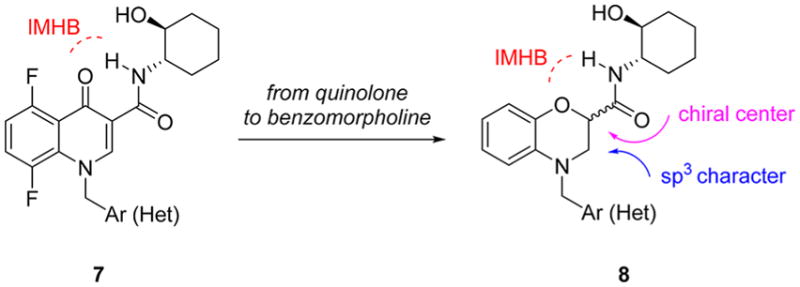
Ligand design concept to scaffold hop from the classical M1 PAM quinolone core of 1 and 7 to a novel, benzomorphoine core 8, bearing a new chiral center and increased sp3 character.
Chemistry and Limited Structure–Activity Relationships (SAR)
The chemistry to access benzomorpholine-based M1 PAMs was straightforward (Scheme 1). Following a standard route to the benzomorpholine core, commercial ethyl 2,3-dibromopropanoate 9 was condensed with 2-aminophenol to provide the key racemic heterocyclic core 10 in 65% yield. Benzylation with 4-bromobenzyl bromide proceeded smoothly delivering 11, suitable for diversification, in 74% yield. A quantitative hydrolysis of the ester gave 12, which then underwent a HATU-mediated amide coupling with (1S,2S)-2-aminocyclohexan-1-ol to provide 13 as a mixture of diastereomers 13. Finally, a copper-mediated coupling with pyrazole produced the putative M1 PAM 14, in 65% yield.
Scheme 1.

Synthesis of Benzomorpholine-Based M1 PAM 14a
aReagents and conditions: (a) 2-aminophenol, CH3CN, K2CO3, 80 °C, 65%; (b) 4-bromobenzyl bromide, K2CO3, CH3CN, 80 °C, 74%; (c) KOH, THF/H2O (2:1), rt, 98%; (d) (1S,2S)-2-aminocyclohexan-1-ol, HATU, DIEA, DMF, rt, 80%; (e) pyrazole, (1S,2S)-N1,N2-dimethylcyclohexane-1,2- diamine, CuI, K3PO4, dioxane, 100 °C, 65%.
Upon testing, 14 proved to be a human M1 PAM (EC50 = 0.92 μM, pEC50 = 6.1 ± 0.13, 77 ± 7% acetylcholine (ACh) Max) with weak M1 agonist activity (EC50 > 10 μM, 22% ACh Max). The diastereomers at the benzomorpholine chiral center were readily separable by column chromatography, affording isomer-1, 15 (VU0486834), and isomer-2, 16 (VU0486846). A single crystal X-ray structure determined the absolute stereochemistry of 16 (Figure 3) as the (R)-enantiomer and highlighted the IMHB.32 Moreover, enantioselective M1 PAM activity was noted, with 15 (EC50 = 0.63 μM, pEC50 = 6.21 ± 0.05 89 ± 2% ACh Max and weak agonist activity EC50 > 10 μM, 16% ACh Max) being less potent than VU0486846 (EC50 = 0.31 μM, pEC50 = 6.53 ± 0.05, 85 ± 2% ACh Max and weak agonist activity EC50 = 4.0 μM, pEC50 = 5.39 ± 0.07, 39 ± 3% ACh in our high expression line). These positive data necessitated a more in depth pharmacological evaluation of novel M1 PAM VU0486846, which also possessed a favorable central nervous system multiparameter optimization (CNS MPO) score (>5).33
Figure 3.

Structures and human M1 PAM activities of 14–16, as well as a single X-ray crystal structure of the more active (R)-enantiomer 16 (VU0486846).
Molecular Pharmacology Studies and Adverse Effect Liability
As shown previously,26,27 in our high expression human M1 cell line, VU0486846 was a moderately potent M1 PAM (EC50 = 0.31 μM, pEC50 = 6.53 ± 0.05, 85 ± 2% ACh Max) with weak, partial agonist activity (EC50 = 4.5 μM, pEC50 = 5.37 ± 0.07, 29 ± 6% ACh). In our rat M1 cell line (also a high expression system), VU0486846 was an equipotent PAM (EC50 = 0.25 μM, pEC50 = 6.63 ± 0.06, 83 ± 1% ACh Max) with weak, partial agonist activity (EC50 = 5.6 μM, pEC50 = 5.27 ± 0.05, 26 ± 6% ACh). In contrast, in our mouse M1 cell line with low expression, VU0486846 showed little agonism (9 ± 1% ACh Max), and good M1 PAM potency (EC50 = 0.6 μM, pEC50 = 6.25 ± 0.10, 77 ± 4% ACh Max). Similarly, in our low expression M1 dog line, VU0486846 was a PAM (EC50 = 0.38 μM, pEC50 = 6.41 ± 0.03, 78 ± 1% ACh Max) with minimal agonist activity (EC50 > 10 μM, 18 ± 0.2% ACh Max). Agonism (full/partial) is a receptor reserve/expression-dependent driven pharmacology, whereas PAM activity is conserved across varying degrees of expression/reserve, potentiating the EC20 orthosteric agonist effect.23 Therefore, the lower expression cell lines are more indicative of the native system. Demonstrating a lack of agonist activity in these in vitro systems or native tissues, such as the PFC, more accurately predicts a lack of CNS AEs.23,27,30 The lack of agonist activity in the lower expressing cell line by VU0486846, suggests that this compound will be devoid of seizure liability in vivo. A point worth mentioning is the potential for overstimulation of M1 with either a highly potent M1 PAM or a potent M1 ago-PAM exists, and would thereby lose the mechanistic advantage of potentiation by these allosteric modulators. As the in vitro cellular functional assays employ a subthreshold concentration of ACh (typically an EC20 concentration), in vivo PAM potency is arguably underestimated, as cholinergic tone in many brain regions may approach an EC100 levels and leftward shifts in PAM potency are observed with increasing ACh concentration. For example (Supporting Figure 1),32 with a very low level of ACh (an ~EC9), VU0486846 exhibited an M1 PAM potency of 430 ± 120 nM (84 ± 2% ACh Max), but as ACh concentration was increased to an EC70, M1 PAM potency improved to 68 ± 11 nM (90 ± 1% ACh Max).32 Similarly, PF-06827443 also showed an increase in M1 PAM potency from 40 ± 6 nM to 7 ± 1 nM at the same ACh concentration range.32 Thus, a very potent PAM or ago-PAM in vitro may lead to overactivation of M1 in vivo leading to M1-mediated seizure liability. After in vitro and in vivo evaluation of M1 PAMs across multiple chemotypes, M1 functional potencies in the 100–400 nM potency range in vitro in our cell-based systems have proven to be a first step toward avoiding seizure liability in vivo. As shown in Figure 4, M1 PAM VU0486846 was highly selective for M1 against both human M2–M5 (Figure 4A) and rat M2-M5 (Figure 4B). While M1 VU0486846 had no interaction with the orthosteric site (Figure 4C), i.e., no displacement of [3H]-NMS binding, other M1 PAMs, such as 1, 2, 5, and 6, with reported adverse effect liability, show weak to significant displacement of [3H]-NMS, and in some cases found to possess negative cooperativity.19,20,23,25,27–30 With regard to this parameter, we feel M1 PAMs should not interact with the orthosteric site, and that PAMs with negative cooperativity can present as procognitive efficacy (reversal of scopolamine-induced deficits) when, in reality, they are inducing a dose-dependent reversal of scopolamine binding. This is further supported by ligands such as 2, which failed to show efficacy in a rat novel object recognition (NOR) model, yet the same structural class of compounds were efficacious in scopolamine challenge models.23,34,35
Figure 4.

Molecular pharmacology profile of 16. (A) Human M1 CRC (EC50 = 308 nM) versus M2–M5 (inactive). (B) Rat M1 CRC (EC50 = 253 nM) versus M2–M5 (inactive). (C) Inhibition of orthosteric radioligand binding with [3H]-NMS by VU0486846 and atropine control. VU0486846 does not substantially inhibit binding of [3H]-NMS up to 30 μM.
Finally, prior to any further characterization, a high dose (100 mg/kg intraperitoneal (i.p.)) phenotypic mouse screen was performed to assess seizure liability, with a clean profile allowing a PAM to advance in development. Whereas seizure activity in mouse indicates over-stimulation of M1 (either too potent of an M1 PAM or robust ago-PAM), the latter are compounds not suitable for further advancement down the lead optimization work flow.22,26 VU0486846 was devoid of seizure liability in mice up to 3 h at a 100 mg/kg dose; by comparison, both 222 and 526 displayed Racine scale 4/5 behavioral convulsions that develop rapidly and lasted for the 3 h duration of the study.
Electrophysiology Studies in Layer V Medial Prefrontal Cortex (mPFC) Pyramidal Neurons Reveal That VU0486846, in Contrast to Other Ago-PAMs, Maintains the Activity-Dependence of Muscarinic Agonists
A critical feature of an M1 PAM that appears to differentiate procognitive efficacy from cognitive disruption is agonist effects in a native system such as the rodent PFC.20,23,25,27–30 As agonist activity in cell lines can be accentuated or disguised by the level of receptor expression, assessment in native systems is the ultimate arbiter.30 As we recently reported, 2 (MK-7622), 5 (PF-06764427), and 6 (PF-06827443) have agonist activity in the mouse PFC, which overactivates M1 in the absence of endogenous ACh release and disrupts PFC function potentially leading to the observed lack of procognitive efficacy in rodent models. In contrast, pure M1 PAMs do not.23,30 Evaluation of VU0486846 in this same assay was shown to induce no significant change in field excitatory post synaptic potentials (fEPSPs) electrically evoked in layer II/III and recorded in layer V, and therefore maintain activity dependence of PFC function (Figure 5A). A submaximal concentration of the prototypical acetylcholine receptor (AChR) agonist carbachol (10 μM) does not induce a significant long-term depression (LTD) of layer V fEPSPs (Figure 5B) as previously shown,23,25,30 but can be potentiated to a robust LTD by the M1 PAM VU0486846 (Figure 5C), similar to other previously reported M1 pure-PAMs such as VU045359525 and VU055164.23 Furthermore, at the ventral hippocampal-mPFC synapse, a pathway thought to be dysregulated in schizophrenia, 36,37 a submaximal concentration (3 μM) of the muscarinic acetylcholine receptor (mAChR) agonist oxotremorine-M (OxoM), does not induce a significant LTD of layer V fEPSPs evoked by optical stimulation of ventral hippocampal afferents (Figure 5D). However, VU0486846 can robustly potentiate the effects of 3 μM OxoM to induce a substantial LTD at the ventral hippocampal-PFC synapse. A significant increase in the magnitude of LTD was observed with coapplication of 3 μM VU0486846 + 10 μM CCh compared to 10 μM CCh alone (p < 0.05) (Figure 5F). In addition, the magnitude of LTD observed with coapplication of 10 μM VU0486846 + 3 μM OxoM compared to 3 μM OxoM alone (p < 0.05) was also significantly enhanced (Figure 5G). Therefore, in this native system, VU0486846 displays no agonist activity on its own but can potentiate the effects of cholinergic agonists and thus holds promise for utility as an in vivo tool to assess the procognitive efficacy of M1 PAMs in rodent models.
Figure 5.

M1 PAM VU0486846 can robustly potentiate a submaximal cholinergic agonist-induced long-term depression (LTD) in layer V of the prelimbic mPFC evoked by either electrical or optical stimulation. (A) Time course graph showing that bath application of 3 μM VU0486846 for 20 min led to no significant change in fEPSP slope. (B) Time course graph showing that bath application of 10 μM carbachol (CCh), a cholinergic agonist, induces a negligible LTD of fEPSPs in layer V electrically evoked by stimulation of layer II/III in the mPFC. (C) 10 min pretreatment of the M1 PAM VU0486846 (3 μM) followed by a 10 min coapplication of PAM and 10 μM CCh led to a robust LTD of electrically evoked fEPSP slope. Inset shows representative fEPSP traces for each condition for baseline (black trace) and 50 min after CCh washout (gray trace). (D) Time course graph showing that bath application of 3 μM OxoM, a selective muscarinic agonist, for 10 min led to an acute depression followed by a minimal LTD of optically evoked fEPSPs (ofEPSP) measured 46–50 min following drug washout. (E) Under similar conditions, bath application 10 min pretreatment of the M1 PAM VU0486846 (10 μM) followed by a 10 min coapplication of PAM with 3 μM OxoM led to a robust LTD of ofEPSP slope. Inset shows representative ofEPSP traces for each condition for baseline (black trace) and 50 min after OxoM washout (blue trace). (F) Quantification of fEPSP slope 46–50 min following drug washout (shaded area) indicates a significant depression of fEPSP slope with 3 μM VU0486846 + 10 μM CCh compared to 10 μM CCh alone (n = 9 brain slice experiments per group). (G) Quantification of ofEPSP slope 46–50 min following drug washout (shaded area) indicates a significant depression of ofEPSP with 10 μM VU0486846 + 3 μM OxoM compared to 3 μM OxoM alone (n = 6–8 brain slice experiments per group). Scale bars denote 0.5 mV and 5 ms. Data are expressed as mean ± SEM; *p < 0.05, Student’s t test.
Drug Metabolism Studies
Due to the differentiated pharmacological profile of VU0486846 (16) from the other M1 PAMs (Figure 1),19,20,23,25,27–30 we evaluated VU0486846 in a battery of in vitro and in vivo drug metabolism and pharmacokinetic (DMPK) assays. VU0486846 was predicted to be a low to moderately cleared compound in hepatic microsomes (human CLhep = 11.1 mL/min/kg; rat CLhep = 23 mL/min/kg; cyno CLhep = 43 mL/min/kg) with attractive fraction unbound in plasma (human f u = 0.12, rat f u = 0.11, cyno f u = 0.016) and rat brain homogenate (f u = 0.03). The CYP450 profile was acceptable for a tool compound (3A4 IC50 = 5.0 μM; 1A2 IC50 > 30 μM; 2C9 IC50 = 18.8 μM; 2D6 IC50 = 29.8 μM), but precluded further advancement as a putative clinical candidate due to the inhibition of 3A4. Like other M1 PAMs harboring the (1S,2S)-2-aminocyclohexan-1-ol amide moiety, CNS exposure varied across rodent species (rat Kp = 0.18, Kp,uu = 0.05; mouse Kp = 0.67; Kp,uu = 0.17), but the compound was not a human P-gp substrate (MDCK ER = 0.9, Papp = 30 × 10−6 cm/s). In vivo PK for VU0486846 was evaluated in both rats and nonhuman primates. From a standard rat PK IV/PO crossover study, VU0486846 showed high clearance (CLp = 89 mL/min/kg) and a lack of an in vitro:in vivo correlation (IVIVC) from predicted in vitro parameters. 16 also displayed a 1.2 h half-life, a reasonable volume (Vss = 1.8 L/kg), and excellent oral bioavailability (%F = 95.9) in rat. A similarly designed study in male cynomolgus monkey was more favorable, with a 4.2 h half-life, moderate clearance (CLp = 18 mL/min/kg), good volume (Vss = 1 L/kg), and good oral bioavailability (%F= 37) from the amorphous solid. No adverse events or cholinergic side effects were noted in these PK studies. While VU0486846 was highly selective versus M2-M5, we wanted a broader assessment of ancillary pharmacology prior to initiating extensive in vivo studies. Evaluation of VU0486846 in a Eurofins Lead Profiling panel of 68 GPCRs, ion channels, and transporters revealed no significant activity at any target in the panel (no inhibition > 50% @10 μM).32 Thus, we were ready to assess the behavioral pharmacology of a structurally novel M1 PAM with good CNS penetration, an attractive DMPK profile, devoid of agonist activity in the PFC, and free from seizure/cholinergic liability in mouse, the most sensitive species.
Behavioral Pharmacology Assessment
Prior to behavioral pharmacology assessment for efficacy of the novel M1 PAM VU0486846, we evaluated the potential of VU0486846 to produce cholinergic or CNS adverse events in both mice and rats. Thus, we performed a Modified Irwin Toxicology Battery (Supporting Figure 2)32 test in mice dosed at 100 mg/kg i.p., which mirrored the mouse seizure study. Over the 3 h evaluation period, VU0486846 did not produce any cholinergic side effects or pronounced adverse effects in mice, as opposed to nonselective muscarinic agonists and M1 ago-PAMs.23,27 Moreover, exposure from this study was more than sufficient to elicit adverse pharmacology. Here, total plasma levels reached 18.2 μM (30x above the mouse EC50), free plasma levels were 2 μM, with correlating high brain levels (total brain reached 12.2 μM, free brain levels were 342 nM). A similar Modified Irwin Toxicology Battery (Supporting Figure 3)32 test in rats was also performed at 56.6 mg/kg i.p, and, as with mouse, VU0486846 did not produce any cholinergic side effects or pronounced adverse effects in rats, as opposed to nonselective muscarinic agonists/M1 ago-PAMs, over the 3 h evaluation period. Here, total terminal plasma exposure reached 4.4 μM (489 nM free) with acceptable brain levels (1.15 μM total, 32 nM free). We attempted a related nonhuman primate (NHP) assessment, but the physiochemical properties and exposure limitations of VU0486846 precluded definitive data; however, we achieved total plasma levels in NHP of ~2 μM, without any adverse effects. The physiochemical properties of VU0486846 did not allow for higher dose formulations, and is another limitation, beyond 3A4 inhibition, that precluded further development toward a clinical candidate.
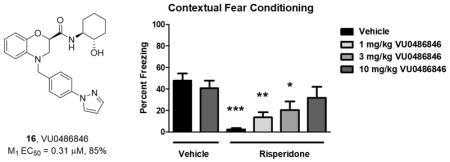
Previously, we demonstrated that M1 PAMs with no agonist activity in native systems (PFC), afforded robust efficacy in the novel object recognition (NOR) paradigm, whereas M1 PAMs and ago-PAMs with agonist activity in the PFC showed little efficacy in NOR.23,27,30 As shown in Figure 6A, PAM VU0486846 dose-dependently enhanced recognition memory in rats, with a minimum effective dose of 3 mg/kg i.p (n = 12; p = 0.0133; F = 4; R2 = 0.2143). Thus, suggesting that VU0486846 could serve as an M1 PAM to investigate efficacy in other models of cognition. The cognitive and negative symptom clusters of schizophrenia are a major focus of our lab. However, a major unmet need in the schizophrenia patient population is the added cognitive dysfunction induced by standard atypical antipsychotics.,38–40 Indeed, first line agents, such as risperidone, are known to engender cognitive deficits in humans (and in rodent models), where cognitive dysfunction is already a major symptom of the disease.38–41 In Figure 6B, we show for the first time that an M1 PAM can reverse the cognitive deficits induced by risperidone (n = 11–13; p < 0.0001; F = 6.682; R2 = 0.3430), and suggest coadministration of an M1 PAM with an atypical antipsychotic might attenuate cognitive impairments in schizophrenia patients. In these studies, VU0486846 dose-dependently reversed risperidone-induced deficits in acquisition of contextual fear conditioning. When coadministered with 3 mg/kg risperidone, 10 mg/kg VU0486846 restored conditioned freezing to vehicle-treated control levels, with a minimum effective dose (MED) of 1 mg/kg. Future efforts will evaluate the cognitive dysfunction induced by other typical/atypical antipsychotics and the ability of VU0486846 to reverse these deficits.
Figure 6.

Novel object recognition and contextual fear conditioning. (A) VU0486846 dose-dependently enhanced recognition memory in rats. Pretreatment with 3 and 10 mg/kg VU0486846 (i.p., 10% Tween 80 in water, 30 min) prior to exposure to identical objects significantly enhanced recognition memory assessed 24 h later. (B) VU0486846 dose-dependently reversed risperidone-induced deficits in acquisition of contextual fear conditioning. When coadministered with 3 mg/kg risperidone, 10 mg/kg VU0486846 restored conditioned freezing to vehicle-treated control levels. Data are expressed as mean ± SEM (n = 11–13). Statistical analysis was conducted using a one-way analysis of variance. If significant (p < 0.05), comparison of group effects relative to the vehicle group was completed using a Dunnett’s test, *p < 0.05, **p < 0.01, ***p < 0.001.
CONCLUSIONS
Here we report the discovery of a novel M1 PAM with enhanced potency, physiochemical properties, and DMPK profile to assess M1 function in CNS disorders. VU0486846 is a highly selective, potent M1 PAM, devoid of agonist activity in the PFC, as well as cholinergic or other adverse effects in mice, rats and NHP, which results in robust procognitive activity in rodent models. These data afford the community a new in vivo tool compound to study M1 PAM activity in mouse and rat models, as well as supporting the pursuit of M1 PAMs as either a monotherapy to enhance cognitive function or has an adjunct therapy with current standard of care therapies for individuals suffering from cognitive deficits, such as schizophrenia patients. Further studies with VU0486846 (16) are underway and will be reported in due course.
METHODS
Chemical Synthesis and Purification
All 1H NMR and 13C NMR spectra were recorded on a Bruker AV-400 (400 MHz) or Bruker AV-NMR (600 MHz) instrument. Chemical shifts are reported in ppm relative to residual solvent peaks as an internal standard set to δH 7.26 or δC 77.0 (CDCl3) and δH 3.31 or δC 49.0 (CD3OD). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, m = multiplet), integration, and coupling constant (Hz). IR spectra were recorded as thin films and are reported in wavenumbers (cm−1). Low resolution mass spectra were obtained on an Agilent 1200 LCMS with electrospray ionization. High resolution mass spectra were recorded on a Waters Qtof-API-US plus Acquity system. The value Δ is the error in the measurement (in ppm) given by the equation Δ = [(ME − MT)/MT] × 106, where ME is the experimental mass and MT is the theoretical mass. The HRMS results were obtained with ES as the ion source and leucine enkephalin as the reference. Optical rotations were measured on a PerkinElmer-341 polarimeter. Analytical thin layer chromatography was performed on 250 μM silica gel 60 F254 plates. Visualization was accomplished with UV light, and/or the use of ninhydrin, anisaldehyde and ceric ammonium molybdate solutions followed by charring on a hot-plate. Chromatography on silica gel was performed using Silica Gel 60 (230–400 mesh) from Sorbent Technologies. Analytical HPLC was performed on an Agilent 1200 analytical LCMS with UV detection at 214 and 254 nm along with ELSD detection. Solvents for extraction, washing, and chromatography were HPLC grade. All reagents were purchased from Aldrich Chemical Co. and were used without purification. All polymer-supported reagents were purchased from Biotage, Inc. Flame-dried (under vacuum) glassware was used for all reactions. All reagents and solvents were commercial grade and purified prior to use when necessary. High-resolution mass spectrometry (HRMS) data were obtained using a Micromass Q-Tof API-US mass spectrometer.
Ethyl 3,4-Dihydro-2H-1,4-benzoxazine-2-carboxylate (10)
A solution of 2-aminophenol (15740 mg, 144.28 mmol), potassium carbonate (33717 mg, 240.46 mmol), ethyl 2,3-dibromopropionate (25000 mg, 96.18 mmol), and acetonitrile (300 mL) was heated to 80 °C and was allowed to stir for 16 h. The reaction was diluted with water and extracted with EtOAc (3×). The layers were separated, and the organic phases were combined, washed with brine (1×), dried with MgSO4, filtered, and concentrated in vacuo. Crude product was purified by flash chromatography (Teledyne ISCO Combi-Flash system, 0–30% EtOAc in hexanes) to provide a brown oil (14595 mg, 70.43 mmol, 73% yield). 1H NMR (400 MHz, CDCl3) δ 6.93 (dd, J = 7.92 Hz, 1.4 Hz, 1H), 6.79 (td, J = 7.5 Hz, 1.4 Hz, 1H), 6.72 (td, J = 7.6 Hz, 1.6 Hz, 1H), 6.61 (dd, J = 7.7 Hz, 1.6 Hz, 1H), 4.81–4.79 (m, 1H), 4.26 (m, 2H), 3.63–3.55 (m, 2H), 1.28 (t, J = 7.2, 3H). 13C NMR (100 MHz, CDCl3) δ 169.4, 142.9, 132.6, 121.6, 119.5, 116.9, 115.7, 72.7, 61.5, 42.6, 14.1 ppm. HRMS (TOF, ES+) calcd for C11H13NO3, 207.0895; found, 207.0892.
Ethyl 4-[(4-Bromophenyl)methyl]-2,3-dihydro-1,4-benzoxazine-2-carboxylate (11)
A solution of ethyl 3,4-dihydro-2H-benzo[b]-[1,4]oxazine-2-carboxylate (2165 mg, 10.45 mmol), potassium carbonate (1831 mg, 13.06 mmol), 4-bromobenzyl bromide (3263.88 mg, 13.06 mmol), and acetonitrile (18 mL) was heated to 80 °C and allowed to stir for 16 h. The reaction was diluted with water and extracted with EtOAc (3×). The layers were separated and the organic phases were combined, washed with brine (1×), dried over MgSO4, filtered, and concentrated in vacuo. Crude product was purified using flash chromatography (Teledyne ISCO Combi-Flash system 0–40% EtOAc in hexanes) to provide an off white solid (2935 mg, 7.80 mmol, 74% yield). 1H NMR (400 MHz, CDCl3) δ 7.45 (d, J = 8.4 Hz, 2H), 7.14 (d, J = 8.3 HZ, 2H), 6.97 (dd, J = 7.9 Hz, 1.5 Hz, 1H), 6.80 (td, J = 7.7 Hz, 1.5 Hz, 1H), 6.72 (td, J = 7.6 Hz, 1.4 Hz, 1H), 6.61 (dd, J = 8.0 Hz, 1.3 Hz, 1H), 4.83 (t, J = 4.0 Hz, 1H), 4.45–4.17 (m, 4H), 3.51 (d, J = 4.1 Hz, 2H), 1.25 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 169.2, 142.9, 136.7, 134.4, 131.7, 128.7, 121.8, 121.0, 119.0, 116.6, 112.8, 72.5, 61.6, 54.4, 48.6, 14.1 ppm. HRMS (TOF, ES+) calcd for C18H18BrNO3, 375.0470; found, 375.0467.
4-[(4-Bromophenyl)methyl]-2,3-dihydro-1,4-benzoxazine-2-carboxylic acid (12)
A solution of ethyl 4-[(4-bromophenyl)methyl]-2,3-dihydro-1,4-benzoxazine-2-carboxylate (2935 mg, 7.8 mmol), potassium hydroxide (437.7 mg, 7.8 mmol), THF (16 mL), and Water (8 mL) was heated to 60 °C and allowed to stir for 3 h. The reaction was diluted in water, acidified to pH = 3, extracted with EtOAc (3x), and concentrated in vacuo to afford the desired material, which was carried forward to the next step without any further purification (2715 mg, quantitative yield). 1H NMR (400 MHz, CDCl3) δ 7.40 (d, J = 8.3 Hz, 2H), 7.13 (d, J = 8.2 Hz, 2H), 6.96 (dd, J = 7.8 Hz, 1.4 Hz, 1H), 6.82 (td, J = 8.0 Hz, 1.4 Hz, 1H), 6.73 (td, J = 7.4 Hz, 1.4 Hz, 1H), 6.63 (dd, J = 8.0 Hz, 1.1 Hz, 1H), 4.90 (t, J = 3.8, 1H), 4.44–4.28 (m, 2H), 3.52 (d, J = 3.80 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 174.4, 142.7, 136.5, 134.5, 131.9, 129.0, 122.3, 121.3, 119.4, 116.8, 113.2, 72.4, 54.6, 48.4 ppm. HRMS (TOF, ES+) calc’d for C16H14BrNO3, 347.0157; found, 347.0150.
4-[(4-Bromophenyl)methyl]-N-[(1S,2S)-2-hydroxycyclohexyl]-2,3-dihydro-1,4-benzoxazine-2 carboxamide (13)
A solution of 4-[(4-bromophenyl)methyl]-2,3-dihydro-1,4-benzoxazine-2-carboxylic acid (2700 mg, 7.75 mmol), N,N-diisopropylethylamine (6.08 mL, 34.8 9 mmol), HATU (3243.3 mg, 8.53 mmol), and DMF (18 mL) was prestirred for 10 min, to which was then added (1S,2S)-2-aminocyclohexanol (1071.69 mg, 9.31 mmol) and the reaction was allowed to stir at room temperature for 12 h. The reaction was diluted with water and extracted with EtOAc (3×). The layers were separated and the combined organics were washed with brine (1×), dried over MgSO4, filtered, and concentrated in vacuo. Crude product was purified using flash chromatography (Teledyne ISCO Combi-Flash system, 0–5% MeOH in DCM) to provide an off white solid (2785 mg, 6.2535 mmol, 80% yield, 8:1 dr)). 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 8.3 Hz, 2H), 7.16 (d, J = 8.3 Hz, 2H), 6.92 (dd, J = 7.9 Hz, 1.3 Hz, 1H), 6.83 (td, J = 7.7 Hz, 1.3 Hz, 1H), 6.70 (td, J = 7.7 Hz, 1.2 Hz, 1H), 6.63 (dd, J = 8.1 Hz, 1.1 Hz, 1H), 6.50 (d, J = 7.4 Hz, 1H), 4.72 (dd, J = 6.7 Hz, 2.9 Hz, 1H), 4.43–4.42 (m, 2H), 3.72–3.63 (m, 1H), 3.59 (dd, J = 11.9 Hz, 3.0 Hz, 1H), 3.51–3.46 (m, 1H), 3.28– 3.23 (m, 1H), 2.06–1.97 (m, 2H), 1.77–1.69 (m, 2H), 1.38–1.17 (m, 5H). 13C NMR (100 MHz, CDCl3) δ 170.2, 141.9, 136.5, 134.8, 131.8, 128.8, 122.6, 121.0, 118.6, 116.3, 113.2, 74.9, 73.8, 55.5, 54.7, 49.0, 34.1, 31.2, 24.4, 23.8 ppm. HRMS (TOF, ES+) calcd for C22H25BrN2O3, 444.1049; found, 444.1048.
(2R)-N-[(1S,2S)-2-Hydroxycyclohexyl]-4-[(4-pyrazol-1-ylphenyl)-methyl]-2,3-dihydro-1,4-benzoxazine-2-carboxamide (VU0486846, 16)
A degassed solution of 4-[(4-bromophenyl)methyl]-N-[(1S,2S)-2-hydroxycyclohexyl]-2,3-dihydro-1,4-benzoxazine-2-carboxamide (900 mg, 2.02 mmol), pyrazole (192.61 mg, 2.83 mmol), potassium phosphate (943.74 mg, 4.45 mmol), copper(I) iodide (115.46 mg, 0.61 mmol), trans-N,N′-dimethylcyclohexane-2-diamine (0.09 mL, 0.61 mmol), and 1,4-dioxane (6.7 mL) was heated to 100 °C and was allowed to stir for 24 h. The reaction was diluted in water and extracted with EtOAc (3×). The layers were separated, and the organic phases were combined, washed with saturated NH4Cl (1×) and saturated brine (1×), dried over MgSO4, filtered, and concentrated in vacuo. Crude product was purified using flash chromatography (Teledyne ISCO Combi-Flash system, 0–60% EtOAc in hexanes) to provide a white solid (219 mg, 0.506 mmol, 25% yield). 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 2.3 Hz, 1H), 7.70 (d, J = 1.2 Hz, 1H), 7.64 (d, J = 8.5 Hz, 2H), 7.36 (d, J = 8.4 Hz, 2H), 6.91 (dd, J = 8.2 Hz, 1.0 Hz, 1H), 6.84 (td, J = 8.2 Hz, 1.2 Hz, 1H), 6.73–6.68 (m, 2H), 6.52 (d, J = 7.5 Hz, 1H), 6.44 (t, J = 2.0 Hz, 1H), 4.73 (dd, J = 6.7 Hz, 2.9 Hz, 1H), 4.51–4.40 (m, 2H), 3.73–3.61 (m, 2H), 3.53– 3.39 (m, 1H), 3.30–3.24 (m, 1H), 2.01 (d, J = 10.9 Hz, 2H), 1.76– 1.68 (m, 2H), 1.38–1.20 (m, 5H). 13C NMR (100 MHz, CDCl3) δ 170.3, 142.0, 140.9, 139.4, 135.7, 134.9, 128.1, 126.7, 122.6, 119.5, 118.5, 116.2, 113.3, 107.5, 74.8, 73.8, 55.5, 54.8, 49.0, 34.1, 31.2, 24.4, 23.8 ppm. HRMS (TOF, ES+) calcd for C25H28N4O3, 432.2161; found, 432.2161; [α]D21 + 58.571 (c = 0.91, MeOH).
Cell Lines
Chinese hamster ovary (CHO) cells stably expressing muscarinic receptor isoforms were maintained in Ham’s F-12 growth medium containing 10% FBS, 20 mM HEPES, antibiotic/antimycotic, 500 μg/mL G418 in the presence of 5% CO2 at 37 °C. For Gi-coupled M2 and M4 receptors, chimeric Gqi5 was stably coexpressed to elicit Ca response. To determine the functional activity at dog M1, the dog M1 full-length open reading frame (ORF) was amplified from the dog hippocampus cDNAs (Zyagen, San Diego, CA). The ORF was then subcloned into the EcoR I and Xho I sites of pcDNA3.1 (+) vector (Life Technologies, Carlsbad, CA). Sequencing of the plasmid confirmed the presence of dog M1 ORF (XM_540897). CHO cells were transfected with dog M1 expression plasmid using Fugene 6 (Promega, Madison, WI), the transfected cells were incubated with the selection medium containing 1 mg/mL G418 for 2 weeks, and the resulting polyclones were used for the calcium mobilization assay described below.
Calcium Mobilization Assay
To determine the potency and efficacy of M1 ago-PAMs, calcium flux was measured using the Functional Drug Screening System (FDSS7000, Hamamatsu, Japan) as previously described (Rook et al. 2016). Briefly, All muscarinic receptor-CHO cells including multi species M1 and M2–M5 cells were plated in black-walled, clear-bottomed 384 well plates (Greiner Bio-One, Monroe, NC) at 20,000 cells/well in 20 μL of growth medium without G418 the day before assay. The following day, cells were washed with assay buffer (Hank’s balanced salt solution, 20 mM HEPES, and 2.5 mM probenecid) and immediately incubated with 20 μL of 1.15 μM fluo-4-acetomethoxyester (Fluo-4 AM) dye solution prepared in assay buffer for 45 min at 37 °C. During the incubation time, all compounds were serial diluted (1:3) in DMSO for 10 point concentration–response curves (CRC), and further diluted in assay buffer at starting final concentration 30 μM using Echo liquid handler (Labcyte, Sunnyvale CA). Dye was removed and replaced with assay buffer. Immediately, calcium flux was measured using the FDSS7000. The CRC of compounds or vehicle was added to cells for 2.5 min and then an EC20 concentration of acetylcholine (ACh) was added and incubated for 1 min. ECmax concentration was also added to cells that were incubated with DMSO vehicle to calculate the EC20 calcium response. Using a four point logistical equation in GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA), the concentration response curves were generated for determination of the potency and efficacy of the agonist and PAM.
Radioligand Binding Assay
Competition binding assays were performed using [3H]-N-methylscopolamine ([3H-]NMS, PerkinElmer. Boston, MA) as previously described. Briefly, compounds were serial diluted 1:3 in DMSO for an 11 point CRC, then further diluted for a final top concentration of 30 μM in binding buffer (20 mM HEPES, 10 mM MgCl2, and 100 mM NaCl, pH 7.4). Membranes from rat M1-CHO cells (10 μg) were incubated with the serial diluted compounds in the presence of a Kd concentration of [3H-]NMS, 0.088 nM, at room temperature for 1 h with constant shaking. Nonspecific binding was determined in the presence of 10 μM atropine. Binding was terminated by rapid filtration through GF/B Unifilter plates (PerkinElmer) using a Brandel 96-well plate Harvester (Brandel Inc., Gaithersburg, MD), followed by three washes with ice-cold harvesting buffer (25 mM Tris-HCl, pH 7.4, 150 mM NaCl). Plates were air-dried overnight, 50 μL of Microscint20 added to the plate, and radioactivity was counted using a TopCount Scintillation Counter (PerkinElmer Life and Analytical Sciences).
Drug Metabolism Methods
In Vitro
Protein binding of M1 PAMs was determined in plasma via equilibrium dialysis employing Single-Use RED Plates with inserts (ThermoFisher Scientific, Rochester, NY). Briefly plasma (220 μL) was added to the 96-well plate containing test article (5 μL) and mixed thoroughly. Subsequently, 200 μL of the plasma–test article mixture was transferred to the cis chamber (red) of the RED plate, with an accompanying 350 μL of phosphate buffer (25 mM, pH 7.4) in the trans chamber. The RED plate was sealed and incubated 4 h at 37 °C with shaking. At completion, 50 μL aliquots from each chamber were diluted 1:1 (50 μL) with either plasma (cis) or buffer (trans) and transferred to a new 96-well plate, at which time ice-cold acetonitrile (2 volumes) was added to extract the matrices. The plate was centrifuged (3000 rpm, 10 min), and supernatants were transferred to a new 96-well plate. The sealed plate was stored at −20 °C until LC/MS/MS analysis.
A cocktail of substrates for cytochrome P450 enzymes (1A2: phenacetin, 10 μM; 2C9: diclofenac, 5 μM; 2D6: dextromethorphan, 5 μM; 3A4: midazolam, 2 μM) were mixed to assess the ability of compounds to inhibit the major cytochrome P450 enzymes. A reaction mixture of 100 mM Kpi, pH 7.4, 0.1 mg/mL human liver microsomes (HLM) and Substrate Mix was prepared and aliquoted into a 96-deep-well block. Test compound and positive control (in duplicate) were then added such that the final concentration of test compound ranged from 0.1–30 μM. The plate was vortexed briefly and then preincubated at 37 °C while shaking for 15 min. The reaction was initiated with the addition of NADPH (1 mM final concentration). The incubation continued for 8 min and the reaction quenched by 2x volume of cold acetonitrile containing internal standard (50 nM carbamazepine). The plate was centrifuged for 10 min (4000 rcf, 4 °C) and the resulting supernatant diluted 1:1 with water for LC/MS/MS analysis. A 12 point standard curve of substrate metabolites over the range of 0.98 nM to 2000 nM. The IC50 values for each compound were obtained for the individual CYP enzymes by quantitating the inhibition of metabolite formation for each probe substrate. A 0 μM compound condition (or control) was set to 100% enzymatic activity and the effect of increasing test compound concentrations on enzymatic activity could then be calculated from the % of control activity. Curves were fitted using XLfit 5.2.2 (four-parameter logistic model, eq 201) to determine the concentration that produces halfmaximal inhibition (IC50).
The in vitro biotransformation of compounds was investigated using hepatic microsomes incubated with or without NADPH (1 mM). Reactions were terminated by adding 1 volume of acetonitrile, and proteins were removed by centrifugation. The supernatants were saved for HPLC/UV/MS analysis. Samples were analyzed on a Waters Acquity UPLC system with PDA detector with a flow rate of 0.5 mL/min (A: Water/Acetonitrile 95/5 with 0.1% formic acid; B: Acetonitrile) and a Waters Xevo QTOF mass spectrometer with positive ESI source.
The metabolic stability of M1 PAMs was investigated in multispecies hepatic microsomes (BD Biosciences, Billerica, MA) using substrate depletion methodology (% test article remaining). A potassium phosphate-buffered reaction mixture (0.1 M, pH 7.4) of test article (1 μM) and microsomes (0.5 mg/mL) was preincubated (5 min) at 37 °C prior to the addition of NADPH (1 mM). The incubations, performed in 96-well plates, were continued at 37 °C under ambient oxygenation and aliquots (80 μL) were removed at selected time intervals (0, 3, 7, 15, 25, and 45 min). Protein was precipitated by the addition of chilled acetonitrile (160 μL), containing glyburide as an internal standard (50 ng/mL), and centrifuged at 3000 rpm (4 °C) for 10 min. Resulting supernatants were transferred to new 96-well plates in preparation for LC/MS/MS analysis. The in vitro half-life (t1/2, min, eq 1), intrinsic clearance (CLint, mL/min/kg, eq 2) and subsequent predicted hepatic clearance (CLhep, mL/min/kg, eq 3) was determined employing the following equations:
| (1) |
where k represents the slope from linear regression analysis (% test article remaining)
| (2) |
where the superscript a indicates scale-up factors of 20 (human) and 45 (rat) and Q = heptatic blood flow
| (3) |
In Vivo
All animal studies were approved by the Vanderbilt University Medical Center Institutional Animal Care and Use Committee. The animal care and use program is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International.
Male Sprague–Dawley rats (n = 2) weighing around 300 g were purchased from Harlon laboratories (Indianapolis, IN) and implanted with catheters in the carotid artery and jugular vein. The cannulated animals were acclimated to their surroundings for approximately 1 week before dosing and provided food and water ad libitum. IV cassette PK experiments in rats were carried out according to methods described previously (Bridges et al. Pharmacol. Res. Perspect. 2014; reference 49). Briefly, A cassette of compounds (n = 4–5/cassette) were formulated from 10 mM solutions of compounds in DMSO. In order to reduce the absolute volume of DMSO that was administered, the compounds were combined and diluted with ethanol and PEG400 to achieve a final concentration of 0.4–0.5 mg/mL for each compound (2 mg/mL total) administered in each cassette. The final dosing solutions consisted of approximately 10% ethanol, 40% PEG400, and 50% DMSO (v/v). Each cassette dose was administered IV via the jugular vein to two dual-cannulated (carotid artery and jugular vein) adult male Sprague–Dawley rats, each weighing between 250 and 350 g (Harlan, Indianapolis, IN) for a final dose of 0.2–0.25 mg/kg per compound. Whole blood collections via the carotid artery were performed at 0.033, 0.117, 0.25, 0.5, 1, 2, 4, 7, and 24 h post dose and plasma samples prepared for bioanalysis. For tissue distribution studies in cassette format, brain dissection and blood collections via the carotid artery were performed at 0.25 h post dose. Blood samples were collected into chilled, EDTA-fortified tubes, centrifuged for 10 min at 3000 rpm (4 °C), and resulting plasma aliquoted into 96-well plates for LC/MS/MS analysis. The brain samples were rinsed in PBS, snap frozen and stored at −80 °C. Prior to LC/MS/MS analysis, brain samples were thawed to room temperature and subjected to mechanical homogenation employing a Mini-Beadbeater and 1.0 mm Zirconia/Silica Beads (BioSpec Products). Discrete IV PK experiments in rats (n = 2) were carried out analogously at a dose of 1.0 mg/kg in 10% EtOH, 50% PEG 400, 40% saline, while discrete PO PK experiments in rats (n = 2) were carried out using a 3 mg/kg dose of compounds in a fine microsuspension in 30% Captisol in H2O via oral gavage to fasted animals. Whole blood collections via the carotid artery were performed at 0.117, 0.25, 0.5, 1, 2, 4, 7, and 24 h post dose. Plasma samples were prepared for bioanalysis as described above.
Determination of the pharmacokinetic profile of VU0486846 following a single IV bolus dose of 1 mg/kg (Ethanol: Peg400: Saline, 10:60:30, v/v/v) and a single PO dose of 3 mg/kg (30% aqueous Captisol) to male Cynomolgus monkeys (n = 3) was carried out at Frontage Laboratories (Exton, Pennsylvania). Blood was collected at standard PK time points and plasma prepared for bioanalysis as per internal protocols at Frontage. All bioanalysis and calculation of PK parameters was carried out by Frontage and a final report issued upon study completion.
Liquid Chromatography/Mass Spectrometry Analysis
M1 PAMs were analyzed via electrospray ionization (ESI) on an AB Sciex API-4000 (Foster City, CA) triple-quadrupole instrument that was coupled with Shimadzu LC-10AD pumps (Columbia, MD) and a Leap Technologies CTC PAL autosampler (Carrboro, NC). Analytes were separated by gradient elution using a Fortis C18 2.1 × 50 mm, 3.5 μm column (Fortis Technologies Ltd., Cheshire, UK) thermostated at 40 °C. HPLC mobile phase A was 0.1% formic acid in water (pH unadjusted), mobile phase B was 0.1% formic acid in acetonitrile (pH unadjusted). The gradient started at 30% B after a 0.2 min hold and was linearly increased to 90% B over 0.8 min; held at 90% B for 0.5 min and returned to 30% B in 0.1 min followed by a re-equilibration (0.9 min). The total run time was 2.5 min and the HPLC flow rate was 0.5 mL/min. The source temperature was set at 500 °C and mass spectral analyses were performed using multiple reaction monitoring (MRM) utilizing a Turbo-Ionspray source in positive ionization mode (5.0 kV spray voltage). All data were analyzed using AB Sciex Analyst 1.4.2 software. For in vivo studies, the final PK parameters were calculated by noncompartmental analysis using Phoenix (version 6.2) (Pharsight Inc., Mountain View, CA).
Mouse Plasma-Brain Exposure
M1 PAMs were dissolved in 10% Tween 80 at the concentration of 1–10 mg/mL (base form) and administered intraperitoneally to male C57BL/6J (Jackson Laboratory, Sacramento, CA) mice aged 7–9 weeks at a volume of 10 mL/kg. The blood and brain were collected at 0.25 h. Animals were euthanized and decapitated, and the brains were removed, thoroughly washed with cold saline and immediately frozen on dry ice. Trunk blood was collected in EDTA coated Eppendorf tubes, and plasma was separated by centrifugation and stored at −80 °C until processed for LC/MS/MS analysis as previously described. Three animals were used for each time point.
Electrophysiology Methods
Animals
All animal studies were approved by the Vanderbilt University Medical Center Institutional Animal Care and Use Committee and were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Male C57BL6/J mice (Jackson laboratories) were used in electrophysiology and behavioral studies (6–10 weeks old). Animals were group housed 4–5 per cage, maintained on a 12 h light/dark cycle, and provided food and water ad libitum.
Extracellular Field Electrophysiology
Briefly, 6–10 week old male C57BL6/J mice were anesthetized using a mixture of ketamine and xylazine (100 mg/kg and 10 mg/kg, respectively, intraperitoneal injection) then transcardially perfused with ice-cold cutting solution (in mM: 230 sucrose, 2.5 KCl, 8 MgSO4, 0.5 CaCl2, 1.25 NaH2PO4, 10 D-glucose, 26 NaHCO3), and then the brains were removed and submerged in ice-cold cutting solution. Coronal slices containing the prelimbic prefrontal cortex were cut at 400 μm using Leica VT1200 vibratome and were transferred to a holding chamber containing NMDG-HEPES recovery solution (in mM: 93 NMDG, 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 D-glucose, 5 sodium ascorbate, 2 thiourea, 3 sodium pyruvate, 10 MgSO4, 0.5 CaCl2, 12 N-acetyl-L-cysteine, pH 7.3, <310 mOsm) for 8–10 min at 32 °C. Slices were then transferred to a room temperature holding chamber for at least 1.5 h containing ACSF (in mM: 126 NaCl, 1.25 NaH2PO4, 2.5 KCl, 10 D-glucose, 26 NaHCO3, 2 CaCl2, 1 MgSO4) supplemented with 600-μM sodium ascorbate for slice viability. All buffers were continuously bubbled with 95% O2/5% CO2. Subsequently, slices were transferred to a 30–32 °C submersion recording chamber (Warner Instruments) where they were perfused with ACSF at a rate of 2 mL/min. Field excitatory postsynaptic potentials (fEPSPs) were recorded from layer V of the prelimbic cortex and evoked electrically by a concentric bipolar stimulating electrode (200 μs duration, 0.05 Hz; interpulse interval of 50 ms) in the superficial layers II–III as described previously (Ghoshal et al., 2017). VU0486846 was diluted to the appropriate concentrations in DMSO (<0.1% final) in ACSF and applied to the bath for 20 min using a peristaltic pump perfusion system. Carbachol and OxoM (Tocris Bioscience, Bristol, UK) were diluted in H2O. See the Supporting Information for additional information.
Stereotaxic Viral Injections
Mice underwent stereotaxic injections for viral-mediated gene transfer of channelrhodopsin-2 (ChR2) at 4–5 weeks of age. Mice were anaesthetized with 1–2% isoflurane for the duration of the surgery and were administered carprofen (10 mg/kg, s.c.) before the surgery commenced. Following a craniotomy, one injection per hemisphere (800 nL at a rate of 100 nL/min) of AAV5-CaMKII-ChR2-EYFP (UNC Viral Vector Core) was delivered into the target regions through a 28G needle attached to a 10 μL Hamilton syringe. Injection site coordinates were as follows (relative to bregma): vHipp [ML: ±3.4, AP: −3.4, DV: −4.0]. Carprofen (10 mg/kg, s.c.) was administered for at least 72 h post procedure. Recordings were made 4–5 weeks following surgery to allow sufficient expression of ChR2 in axon terminals within the PFC.
Optical Extracellular Field Electrophysiology
Brain slices from viral injected mice were prepared in a similar manner described above. For studies involving optical stimulation, blue light (470 nm) was delivered using a High-Power LED (Thorlabs Inc. Newton, NJ), which was mounted to the epi-illumination port of an Olympus BX50WI upright microscope (Olympus). Blue light was shined onto slices through the 40× objective lens at 0.05 Hz (Maximum light intensity at the site of illumination is 2.6 mW).
Behavioral Pharmacology Studies
Animals
In vivo studies utilized either 7–8 week old male C57Bl/6 mice (Jackson Laboratory, Sacramento, CA) or male Sprague–Dawley rats weighing 275–300 g (Envigo, Indianapolis, IN). The animals were cared for in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All experimental procedures were approved by the Vanderbilt University Animal Care and Use Committee.
Novel Object Recognition Task
Novel object recognition memory was assessed as previously described.26 Briefly, rats were habituated to an empty novel object recognition (NOR) arena. On test day, rats were administered vehicle (10% tween 80) or VU0486846 (1–10 mg/kg, intraperitoneally (i.p.), 1 mL/kg, n = 12) and returned to their home cage for 30 min. Rats were then placed in the NOR arena containing 2 identical objects for 10 min. Following the exposure period, rats were placed back into their home cages for 24 h. The rats were then returned to the arena in which one of the previously exposed (familiar) objects was replaced by a novel object and exploration behavior was assessed for 3 min. Time spent exploring each object was scored by an observer blinded to the experimental conditions and the recognition index was calculated as [(time spent exploring novel object) − (time spent exploring familiar object)]/total time exploring objects.
Contextual Fear Conditioning
The effects of VU0486846 on acquisition of contextual fear conditioning were evaluated in rats as previously described.42 Rats were given a 30 min pretreatment of vehicle (10% Tween 80), risperidone alone (3 mg/kg, i.p., 1 mL/kg), or risperidone coadministered with VU0486846 (1–10 mg/kg) and placed in a sound-attenuating conditioning chamber (Med Associates). Following a 2 min habituation period, rats received three shock-tone pairing trials (30 s 3000 Hz 80 dB tone coterminated with a 1 s, 0.5 mA shock) in the presence of a 10% vanilla solution scent and then returned to their home cages. Twenty four hours later, acquisition of fear conditioning was assessed in the same conditioning environment for 4 min by measuring freezing behavior in the absence of any shock stimuli or drug. Testing sessions were recorded and time spent freezing was automatically scored using video freeze software (MED-VFC-RS, MedAssociates).
Modified Irwin Toxicology Battery
The potential CNS adverse effects of VU0486846 were evaluated using the Modified Irwin Toxicology Battery. Baseline assessments were conducted prior to administration of compounds. Mice were administered vehicle or VU0486846 (100 mg/kg, 10% Tween 80, 10 mL/kg, i.p., n = 3) while rats were administered vehicle or VU0486846 (56.6 mg/kg, 10% Tween 80, 2 mL/kg, i.p., n = 3). Animals were then placed back into their home cages and evaluated by an observer blinded to dosing conditions at 15, 30, 60, 180, and 360 min for autonomic nervous and somatomotor system parameters. Animals were assigned a score for each parameter of 0 (normal), 1 (slight/light), 2 (moderate), or 3 (marked) relative to vehicle-treated controls.
Supplementary Material
Acknowledgments
Funding
This work was funded by the NIH and NIMH (MH082867 and MH106839).
We thank William K. Warren, Jr. and the William K. Warren Foundation who funded the William K. Warren, Jr. Chair in Medicine (to C.W.L.).
ABBREVIATIONS
- ACh
acetylcholine
- i.p
intraperioneal
- p.o
oral dosing
- LTS
GPCR, G protein-coupled receptor
- M1
muscarinic acetylcholine receptor subtype 1
- NOR
novel object recognition
- CFC
contextual fear conditioning
Footnotes
Author Contributions
J.M.R., J.L.B., H.P.C., and P.M.G.-B. contributed equally to this work. C.W.L., S.R.S., P.J.C., and C.M.N., and J.M.R. and C.K.J., and A.L.B. oversaw and designed the chemistry, molecular pharmacology, behavioral pharmacology, and DMPK, respectively. J.L.B. and P.M.G. performed synthetic/medicinal chemistry and scaled up key compounds for in vivo studies. H.P.C. designed and executed advanced molecular pharmacology assays. K.D.N. and P.M.G.-B. performed molecular pharmacology assays. S.C. and A.L.B. performed DMPK and bioanalysis. J.W.D. and D.H.R. performed in vivo behavioral assays. S.P.M. and J.T.M. performed electrophysiology. J.M.H. obtained the single crystal X-ray structure. C.W.L. and J.M.R. wrote the manuscript.
Notes
The authors are developing M1 PAMs for the treatment of schizophrenia and AD, and have an open-IND/Phase I trial for the same as well as a patent portfolio of M1 PAMs.
The authors declare the following competing financial interest(s): We have an M1 PAM in clinical development and possess IP with respect to M1 PAMs.
The Supporting Information is available free of charge on the ACS Publications Web site at DOI: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro.8b00131.
Pharmacology and drug metabolism data (PDF)
References
- 1.Melancon BJ, Tarr JC, Panarese JD, Wood MR, Lindsley CW. Allosteric modulation of the M1 muscarinic acetylcholine receptor: improving cognition and a potential treatment for schizophrenia and Alzheimer’s disease. Drug Discovery Today. 2013;18:1185–1199. doi: 10.1016/j.drudis.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bridges TM, LeBois EP, Hopkins CR, Wood MR, Jones JK, Conn PJ, Lindsley CW. Antipsychotic potential of muscarinic allosteric modulation. Drug News Perspect. 2010;23:229–240. doi: 10.1358/dnp.2010.23.4.1416977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levey AI, Kitt CA, Simonds WF, Price DL, Brann MR. Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J Neurosci. 1991;11:3218–3226. doi: 10.1523/JNEUROSCI.11-10-03218.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levey AI, Edmunds SM, Koliatsos V, Wiley RG, Heilman CJ. Expression of M1-M4 muscarinic acetylcholine receptor proteins in rat hippocampus and regulation by cholinergic innervations. J Neurosci. 1995;15:4077–4092. doi: 10.1523/JNEUROSCI.15-05-04077.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levey AI. Muscarinic acetylcholine receptor expression in memory circuits: implications for treatment of Alzheimer disease. Proc Natl Acad Sci U S A. 1996;93:13541–13546. doi: 10.1073/pnas.93.24.13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Felder CC, Porter AC, Skillman TL, Zhang L, Bymaster FP, Nathanson NM, Hamilton SE, Gomeza J, Wess J, McKinzie DL. Elucidating the role of muscarinic receptors in psychosis. Life Sci. 2001;68:2605–2613. doi: 10.1016/s0024-3205(01)01059-1. [DOI] [PubMed] [Google Scholar]
- 7.Anagnostaras SG, Murphy GG, Hamilton SE, Mitchell SL, Rahnama NP, Nathanson NM, Silva AJ. Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor in mutant mice. Nat Neurosci. 2003;6:51–58. doi: 10.1038/nn992. [DOI] [PubMed] [Google Scholar]
- 8.Caccamo A, Oddo S, Billings LM, Green KN, MartinezCoria H, Fisher A, LaFerla FM. M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron. 2006;49:671–682. doi: 10.1016/j.neuron.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 9.Caccamo A, Fisher A, LaFerla FM. M1agonists as a potential disease-modifying therapy for Alzheimer’s disease. Curr Alzheimer Res. 2009;6:112–117. doi: 10.2174/156720509787602915. [DOI] [PubMed] [Google Scholar]
- 10.Dean B, Hopper S, Conn PJ, Scarr E. Changes in BQCA allosteric modulation of [3H]-NMS binding to human cortex within schizophrenia and by divalent cations. Neuropsychopharmacology. 2016;41:1620–1628. doi: 10.1038/npp.2015.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scarr E, Udawela M, Thomas EA, Dean B. Changed gene expression in subjects with schizophrenia and low cortical muscarinic M1 receptors predict disrupted upstream pathways interacting with that receptor. Mol Psychiatry. 2018;23:295–303. doi: 10.1038/mp.2016.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conn PJ, Lindsley CW, Meiler J, Niswender CM. Opportunities and challenges in the discovery of allosteric modulators of GPCRs for the treatment of CNS disorders. Nat Rev Drug Discovery. 2014;13:692–708. doi: 10.1038/nrd4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A, Conn PJ, Lindsley CW. Allosteric Modulation of 7 Transmembrane Spanning Receptors: Theory, Practice and Opportunities for CNS Drug Discovery. J Med Chem. 2012;55:1445–1464. doi: 10.1021/jm201139r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Menniti FS, Lindsley CW, Conn PJ, Pandit J, Zagouras P, Volkmann RA. Allosteric modulation for the treatment of schizophrenia: Targeting glutamatergic networks. Curr Top Med Chem. 2013;13:26–54. doi: 10.2174/1568026611313010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raedler TJ, Bymaster FP, Tandon R, Copolov D, Dean B. Towards a Muscarinic Hypothesis of Schizophrenia. Mol Psychiatry. 2007;12:232–246. doi: 10.1038/sj.mp.4001924. [DOI] [PubMed] [Google Scholar]
- 16.Bender AM, Jones CK, Lindsley CW. Classics in Chemical Neuroscience: Xanomeline. ACS Chem Neurosci. 2017;8:435–443. doi: 10.1021/acschemneuro.7b00001. [DOI] [PubMed] [Google Scholar]
- 17.Dencker D, Thomsen M, Wörtwein G, Weikop P, Cui Y, Jeon J, Wess J, Fink-Jensen A. Muscarinic Acetylcholine Receptor Subtypes as Potential Drug Targets for the Treatment of Schizophrenia, Drug Abuse, and Parkinson’s Disease. ACS Chem Neurosci. 2012;3:80–89. doi: 10.1021/cn200110q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kruse AC, Kobilka BK, Gautam D, Sexton PM, Christopoulos A, Wess J. Muscarinic Acetylcholine Receptors: Novel Opportunities for Drug Development. Nat Rev Drug Discovery. 2014;13:549–560. doi: 10.1038/nrd4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma L, Seager M, Wittman M, Bickel N, Burno M, Jones K, Graufelds VK, Xu G, Pearson M, McCampbell A, Gaspar R, Shughrue P, Danzinger A, Regan C, Garson S, Doran S, Kreatsoulas C, Veng L, Lindsley CW, Shipe W, Kuduk S, Jacobson M, Sur C, Kinney G, Seabrook GR, Ray WJ. Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc Natl Acad Sci U S A. 2009;106:15950–15955. doi: 10.1073/pnas.0900903106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shirey JK, Brady AE, Jones PJ, Davis AA, Bridges TM, Jadhav SB, Menon U, Christain EP, Doherty JJ, Quirk MC, Snyder DH, Levey AI, Watson ML, Nicolle MM, Lindsley CW, Conn PJ. A selective allosteric potentiator of the M1 muscarinic acetylcholine receptor increases activity of medial prefrontal cortical neurons and can restore impairments of reversal learning. J Neurosci. 2009;29:14271–14286. doi: 10.1523/JNEUROSCI.3930-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang FV, Shipe WD, Bunda JL, Nolt MB, Wisnoski DD, Zhao Z, Barrow JC, Ray WJ, Ma L, Wittman M, Seager M, Koeplinger K, Hartman GD, Lindsley CW. Parallel synthesis of N-Birayl Quinolone Carboxylic Acids as selective M1 positive allosteric modulators. Bioorg Med Chem Lett. 2010;20:531–536. doi: 10.1016/j.bmcl.2009.11.100. [DOI] [PubMed] [Google Scholar]
- 22.Uslaner JM, Kuduk SD, Wittmann M, Lange HS, Fox SV, Min C, Pajkovic N, Harris D, Cilissen C, Mahon C, Mostoller K, Warrington S, Beshore DC. Preclinical to Human Translational Pharmacology of the Novel M1 Positive Allosteric Modulator MK-7622. J Pharmacol Exp Ther. 2018 doi: 10.1124/jpet.117.245894. jpet.117.245894. [DOI] [PubMed] [Google Scholar]
- 23.Moran SP, Dickerson JW, Plumley HC, Xiang Z, Maksymetz J, Remke DH, Doyle CA, Niswender CM, Engers DW, Lindsley CW, Rook JM, Conn PJ. M1 positive allosteric modulators lacking agonist activity provide the optimal profile for enhancing cognition. Neuropsychopharmacology. 2018 doi: 10.1038/s41386-018-0033-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bridges TM, Kennedy JP, Cho HP, Conn PJ, Lindsley CW. Chemical optimization of an M1, M3, M5 positive allosteric modulator (PAM) lead. Part II Development of a highly selective M1 PAM. Bioorg Med Chem Lett. 2010;20:1972–1975. doi: 10.1016/j.bmcl.2010.01.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghoshal A, Rook J, Dickerson J, Roop G, Morrison R, Jalan-Sakrikar N, Lamsal A, Noetzel M, Poslusney M, Stauffer SR, Xiang Z, Daniels JS, Niswender CM, Jones CK, Lindsley CW, Conn PJ. Selective potentiation of M1 muscarinic receptors reverses deficits in plasticity and negative and cognitive symptoms in repeated phencyclidine treated mouse model of schizophrenia. Neuropsychopharmacology. 2016;41:598–610. doi: 10.1038/npp.2015.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grannan MD, Mielnik CA, Moran SP, Gould RW, Ball J, Bubser M, Ramsey AJ, Abe M, Cho HP, Nance KD, Blobaum AL, Niswender CM, Conn PJ, Lindsley CW, Jones CK. Prefrontal cortex-mediated impairments in a genetic model of NMDA receptor hypofunction are reversed by the novel M1 PAM VU6004256. ACS Chem Neurosci. 2016;7:1706–1716. doi: 10.1021/acschemneuro.6b00230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rook JM, Abe M, Cho HP, Nance KD, Luscombe VB, Adams JJ, Dickerson JW, Remke DH, Garcia-Barrantes PM, Engers DW, Engers JL, Chang S, Foster JJ, Blobaum AL, Niswender CM, Jones CK, Conn PJ, Lindsley CW. Diverse Effects on M1 Signaling and Adverse Effect Liability within a Series of M1 Ago-PAMs. ACS Chem Neurosci. 2017;8:866–883. doi: 10.1021/acschemneuro.6b00429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davoren JE, O’Neil SV, Anderson DP, Brodeny MA, Chenard L, Dlugolenski K, Edgerton JR, Green M, Garnsey M, Grimwood S, Harris AR, Kauffman GW, LaChapelle E, Lazzaro JT, Lee CW, Lotarski SM, Nason DM, Obach RS, Reinhart V, Salomon-Ferrer R, Steyn SJ, Webb D, Yan J, Zhang L. Design and optimization of selective azaindole amides M1 positive allosteric modulators. Bioorg Med Chem Lett. 2016;26:650–655. doi: 10.1016/j.bmcl.2015.11.053. [DOI] [PubMed] [Google Scholar]
- 29.Davoren JE, Lee CW, Garnsey M, Brodney MA, Cordes J, Dlugolenski K, Edgerton JR, Harris AR, Helal CJ, Jenkinson S, Kauffman GW, Kenakin TP, Lazzaro JT, Lotarski SM, Mao Y, Nason DM, Northcott C, Nottebaum L, O’Neil SV, Pettersen B, Popiolek M, Reinhart V, Salomon-Ferrer R, Steyn SJ, Webb D, Zhang L, Grimwood S. Discovery of the Potent and Selective M1 PAM-Agonist N-[(3R,4S)-3-Hydroxytetrahydro-2H-pyran-4-yl]-5-methyl-4-[4-(1,3-thiazol-4-yl)benzyl]pyridine-2-carboxamide (PF-06767832): Evaluation of Efficacy and Cholinergic Side Effects. J Med Chem. 2016;59:6313–6328. doi: 10.1021/acs.jmedchem.6b00544. [DOI] [PubMed] [Google Scholar]
- 30.Moran SP, Cho HP, Maksymetz J, Remke D, Hanson R, Niswender CM, Lindsley CW, Rook JM, Conn PJ. PF-06827443 displays robust allosteric agonist and positive allosteric modulator activity in high receptor reserve and native systems. ACS Chem Neurosci. 2018 doi: 10.1021/acschemneuro.8b00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang ZQ, Shu Y, Ma L, Wittmann M, Ray WJ, Seager MA, Koeplinger KA, Thompson CD, Hartman GD, Bilodeau MT, Kuduk SD. Discovery of naphthyl-fused 5-membered lactams as a new class of M1 positive allosteric modulators. ACS Med Chem Lett. 2014;5:604–608. doi: 10.1021/ml500055h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.See the Supporting Information for full details.
- 33.Wager TT, Hou X, Verhoest PR, Villalobos A. Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery. ACS Chem Neurosci. 2016;7:767–775. doi: 10.1021/acschemneuro.6b00029. [DOI] [PubMed] [Google Scholar]
- 34.Vardigan JD, Cannon CE, Puri V, Dancho M, Koser A, Wittmann M, Kuduk SD, Renger JJ, Uslaner JM. Improved cognition without adverse effects: novel M1 potentiatir compares favorably to donepezil and xanomeline in rhesus monkey. Psychopharmacology. 2015;232:1859–1866. doi: 10.1007/s00213-014-3813-x. [DOI] [PubMed] [Google Scholar]
- 35.Lange HS, Cannon CE, Drott JT, Kuduk SD, Uslaner JM. The M1Muscarinic Positive Allosteric Modulator PQCA Improves Performance on Translatable Tests of Memory and Attention in Rhesus Monkeys. J Pharmacol Exp Ther. 2015;355:442–450. doi: 10.1124/jpet.115.226712. [DOI] [PubMed] [Google Scholar]
- 36.Ghoshal A, Conn PJ. The hippocampal-prefrontal pathway: a possible therapeutic target for the negative and cognitive symptoms of schizophrenia. Future Neurol. 2015;10:115–128. doi: 10.2217/FNL.14.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sigurdsson T, Duvarci S. Hippocampal-prefrontal interactions in cognition, behavior and psychiatric disease. Front Syst Neurosci. 2016;9:190. doi: 10.3389/fnsys.2015.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hill SK, Bishop JR, Palumbo D, Sweeney JA. Effects of second generation antipsychotics on cognition: current issues and future challenges. Expert Rev Neurother. 2010;10:43–57. doi: 10.1586/ern.09.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keefe RS, Bilder RM, Davis SM, Harvey PD, Palmer BW, Gold JM, Meltzer HY, Green MF, Capuano G, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Davis CE, Hsiao JK, Lieberman JA CATIE Investigators, Neurocognitive Working Group. Neurocognitive effects of antipsychotic medications in patients with chronic schizophrenia in the CATIE Trial. Arch Gen Psychiatry. 2007;64:633–647. doi: 10.1001/archpsyc.64.6.633. [DOI] [PubMed] [Google Scholar]
- 40.Nishiyama K, Sugishita M, Kurisaki H, Sakuta M. Reversible memory disturbance and intelligence impairment induced by long-term anticholinergic therapy. Intern Med. 1998;37:514–518. doi: 10.2169/internalmedicine.37.514. [DOI] [PubMed] [Google Scholar]
- 41.Castner SA, Williams GV, Goldman-Rakic PS. Reversal of Antipsychotic Induced Working Memory Deficits by Short-Term Dopamine D1 Receptor Stimulation. Science. 2000;287:2020–2022. doi: 10.1126/science.287.5460.2020. [DOI] [PubMed] [Google Scholar]
- 42.Rook JM, Xiang Z, Lv X, Ghosal A, Dickerson J, Bridges TM, Johnson KA, Bubser M, Gregory KJ, Vinson PN, Byun N, Stauffer SR, Daniels JS, Niswender CM, Lavreysen H, Mackie C, Conde-Ceide S, Alcazar J, Bartolome JM, Macdondald GJ, Steckler T, Jones CK, Lindsley CW, Conn PJ. Biased mGlu5 positive allosteric modulators provide in vivo efficacy without potentiating mGlu5 modulation of NMDAR currents. Neuron. 2015;86:1029–1040. doi: 10.1016/j.neuron.2015.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.