

Summary
AT-rich interactive domain-containing protein 1A and 1B (ARID1A, ARID1B) are mutually exclusive subunits of the chromatin remodeler SWI/SNF. ARID1A is the most frequently mutated chromatin regulator across all cancers, and ovarian clear cell carcinoma (OCCC) carries the highest prevalence of ARID1A mutations (~57%). Despite evidence implicating ARID1A in tumorigenesis, the mechanism remains elusive. Here, we demonstrate that in OCCC ARID1A binds active regulatory elements. Depletion of ARID1A represses RNA Polymerase II (RNAPII) transcription, but results in modest changes to accessibility. Specifically, pausing of RNAPII is severely impaired after loss of ARID1A. Compromised pausing results in transcriptional dysregulation of active genes, which is compensated by upregulation of ARID1B. However, a subset of ARID1A-dependent genes is not rescued by ARID1B, including many p53 and estrogen receptor (ESR1) targets. Our results provide new insight on ARID1A-mediated tumorigenesis and unveil new functions of SWI/SNF in modulating RNAPII dynamics.
Introduction
AT-rich interactive domain-containing protein 1A and 1B (ARID1A, ARID1B) are mutually exclusive subunits of the chromatin remodeler complex SWI/SNF (Raab et al., 2015; Ryme et al., 2009). As part of the core SWI/SNF complex, ARID1A and ARID1B are conserved throughout metazoans and expressed across all human tissues. While the AT-rich interactive domain (ARID) has been shown to bind DNA in vitro (Kortschak et al., 2000; Wilsker et al., 2004), the function of ARID1A and ARID1B in vivo is poorly understood. ARID1A and ARID1B are components of the BAF sub-family of SWI/SNF complexes, in association with the BRG1/BRM ATPases that maintain accessibility at most promoters and enhancers (Bao et al., 2015; Tolstorukov et al., 2013). There are limited genome-wide studies on ARID1A and ARID1B, which suggest substantial overlap of their target loci but functionally distinct roles (Raab et al., 2015). Importantly, ARID1A is the most frequently mutated chromatin regulator across all human cancers (Kadoch et al., 2013). In particular, Ovarian Clear Cell Carcinoma (OCCC) carries the highest prevalence of ARID1A mutations [~57%] (Bitler et al., 2015; Bitler et al., 2017; Jones et al., 2010). Conversely, ARID1B is rarely found mutated. Importantly, the large majority of ARID1A mutations are frameshift indels or nonsense point mutations resulting in functional loss of the entire protein (Ayhan et al., 2012; Jones et al., 2010). Recent studies have proposed ARID1A as regulator of H3 acetylation (H3K27ac) (Alver et al., 2017; Mathur et al., 2017) and chromatin accessibility (Kelso et al. 2017) at distal enhancer regions. Using a model of OCCC, we examine the immediate consequences of ARID1A loss on transcription and nucleosome positioning. We find only a marginal role for ARID1A in maintaining accessibility at regulatory elements. Instead, we describe an unanticipated role for this tumor suppressor in regulating widespread RNAPII pausing. The mechanism of polymerase pausing is largely conserved across higher metazoans and represents a pivotal instrument to allow fast, coordinated, and robust transcription at hundreds of loci during cell homeostasis and development (Adelman and Lis, 2012; Levine, 2011). Paused RNA polymerase is centrally regulated by the negative elongation factor, NELF, which prevents productive elongation and determines arrest of the RNAPII holoenzyme within the first 100bp from the Transcription Start Site (TSS). Depletion of NELF and loss of paused RNAPII result in overall loss of polymerase from the gene locus and transcriptional downregulation (Gilchrist et al., 2010; Williams et al., 2015). While several complexes that allow for coordinated release of paused polymerase have been identified (Gardini et al., 2014; Patel et al., 2013; Rahl et al., 2010), little is known about additional mechanisms and proteins that actively enforce pausing genome-wide (Chen et al., 2015). Here, we identify a link between the chromatin remodeler SWI/SNF and RNAPII pausing in ovarian epithelial cells, mediated by the ARID1 subunits. We propose that dysregulated pausing mediates the oncogenic effects of ARID1A loss.
Results
ARID1A binds active enhancers and promoters in ovarian epithelial cells
While ARID1A is a constitutive component of the evolutionarily conserved SWI/SNF complex, its genome-wide localization has remained elusive. To profile ARID1A binding in ovarian epithelial cells, we performed ChIP-seq in the OCCC derived RMG-1 cell line (Nozawa et al., 1988) using two different antibodies (Fig. 1; Supplemental Fig. S1A). We assessed specificity of ARID1A antibodies and its association with SWI/SNF by co-immunoprecipitation (Supplemental Fig. S1B). Alongside ARID1A, we profiled H3K27ac, a chromatin mark that encompasses transcriptionally active regulatory elements. We attributed H3K27ac/ARID1A peaks to promoters when located in the vicinity (<1kb) of an annotated transcription start site (TSS), whereas distal peaks (>1kb from TSS) were classified as enhancers. ChIP-seq revealed that ARID1A is bound to nearly all active regulatory elements in OCCC cells (37,773 enhancers, 13,167 promoters). ARID1A and H3K27ac profiles were highly correlated across all human genes (Fig. 1B-C), suggesting that ARID1A is broadly distributed across active genes and that its occupancy increases with transcriptional activity (e.g. we found robust ARID1A binding across the top 200 genes expressed in RMG-1, Supplemental Fig. S1C). Altogether, our results are consistent with recent reports suggesting association of SWI/SNF with enhancers (Alver et al., 2017; Mathur et al., 2017), supporting the notion that ARID1A also targets a wide spectrum of active genes in mammalian cells (Mathur et al., 2017; Raab et al., 2015; Tolstorukov et al., 2013).
Figure 1 – ARID1A binds active enhancers and promoters in OCCC cells.

(A) Genomic distribution of ARID1A ChIP-seq peaks. The pie chart shows that ARID1A is broadly distributed across TSS, intronic regions and intergenic regulatory elements. (B) Heatmap of H3K27ac and ARID1A ChIP-seq signal across the ARID1A peaks suggests that ARID1A occupancy is enriched across all active regulatory elements. (C) Correlation plot of H3K27ac and ARID1A ChIP-seq experiments reveals a strong overlap (Pearson correlation=0.86) between ARID1A and active transcription genome-wide. (D) Average profile of ATAC-seq and ARID1A ChIP-seq in OCCC suggests that ARID1A binds the +1 and −1 nucleosomes flanking the TSS. (E) Average profile of ATAC-seq and ARID1A ChIP-seq in OCCC reveals that ARID1A binds the nucleosome depleted region of enhancers. (F, G) Genome Browser screenshot of ATAC-seq and ChIP-seq (ARID1A, H3K27ac) at a representative promoter and enhancer, respectively.
To further characterize the binding pattern of ARID1A in OCCC, we employed ATAC-seq to chart DNA accessibility and found, as expected, that all ARID1A target regions are highly accessible (Supplemental Fig. S1D). However, we observed unanticipated differences in the occupancy profiles of ARID1A at enhancers and promoters. Specifically, at most promoters ARID1A presents a bimodal distribution overlapping the “−1” and “+1” nucleosomes, while the nucleosome depleted region (NDR) surrounding the transcription start site (TSS) is devoid of ARID1A (Fig. 1D, F). Conversely, ARID1A binding profile at enhancers mirrors that of the NDR defined by ATAC-seq (Fig. 1E, G).
Loss of ARID1A determines widespread transcriptional decrease in absence of major accessibility changes
To recapitulate early events in ovarian carcinogenesis, we depleted ARID1A in OCCC derived cells. We used the OCCC cell line RMG-1, bearing two wild-type alleles of ARID1A, to generate stable clones with an inducible shRNA against ARID1A under the control of a tetracycline-responsive promoter. In this system, we nearly eliminate chromatin-bound ARID1A within 72h (Fig. 2A, Supplemental Fig. S1E). Next, we measured changes in accessibility and transcription upon depletion of ARID1A. Previous reports postulate that ARID1A may specifically modulate a subset of enhancers (Kelso et al., 2017), however our ChIP-seq analysis (Fig. 1) suggests a widespread role of ARID1A at most protein coding genes. Notably, ATAC-seq analyses indicated that accessibility at promoters is largely unchanged upon ARID1A-KD (Fig. 2B). We next examined enhancer regions before and after ARID1A-KD (Fig. 2C-E). We clustered enhancers into two categories based on H3K27ac level: i) weak enhancers (quartiles 1 and 2 of H3K27ac intensity) and ii) strong enhancers (H3K27ac quartiles 3 and 4). While we observed loss of accessibility at cis-regulatory elements (Kelso et al., 2017), this effect was largely confined to weak enhancers (Fig. 2C) that are poorly bound by ARID1A (Fig. 2E). Conversely, accessibility at strong enhancers, highly acetylated and enriched in ARID1A, was slightly affected (Fig. 2D). Statistical analysis of differential accessibility (edgeR; Table S1) confirmed that significant changes (FDR<10%) in accessibility were detectable in only ~2% of the tested regions, all of which represented weak enhancers. To further examine the enhancer landscape of OCCC, we performed unbiased motif search and retrieved the same transcription factor (TF) signature (E2F, ELF3, SOX8; Supplemental Data S1) at both categories of enhancers, with the sole exception of AP-1 that was only found at strong enhancers, suggesting that FOS/JUN TFs drive enhancer activity in RMG-1 regardless of the presence of ARID1A.
Figure 2 – ARID1A depletion causes transcriptional attenuation in OCCC cells with minor effects on chromatin accessibility.
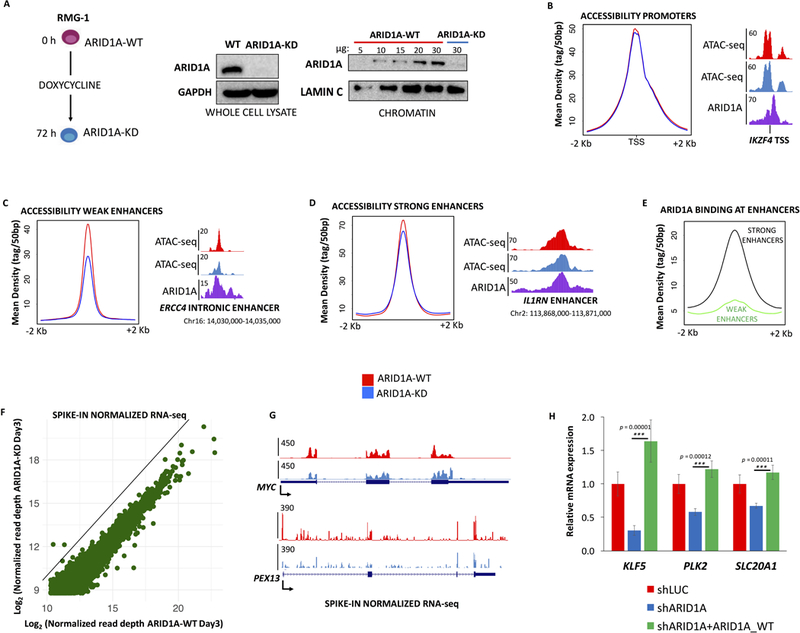
(A) ARID1A is efficiently depleted from RMG-1 cells in 72 hours, using a doxycycline inducible shRNA. Whole cell and chromatin extracts were probed by immunoblot. (B) Average ATAC-seq profiles (ARID1A-WT, KD) show that accessibility at promoters is not affected by ARID1A depletion. (C) Average ATAC-seq profiles (ARID1A-WT, KD) show that accessibility at weak enhancers (3rd and 4th quartile of H3K27ac enrichment) is significantly reduced upon ARID1A depletion. (D) Average ATAC-seq profiles (ARID1A-WT, KD) show that accessibility at strong enhancers (1st and 2nd quartile of H3K27ac enrichment) is marginally affected by ARID1A depletion. (E) Comparison of ARID1A ChIP-seq profiles at weak and strong enhancers. (F) Correlation plot (spike-in normalized RNA-seq) of all expressed coding genes (GENCODE annotations) in RMG-1 cells, ARID1A-KD elicits broad transcriptional downregulation. (G) RNA-seq at select loci displays transcriptional attenuation upon ARID1A-KD. (H) Parental RMG-1 cells were infected with shRNAs against ARID1A and rescued with exogenously expressed full-length ARID1A. Quantitative RT-PCR at three candidate genes shows that downregulation of the mRNA is significantly reversed (n=3) by ARID1A overexpression (fold change compared to shLUC, normalized by 18s rRNA).
Next, we performed RNA-seq analysis (spike-in controlled, two independent replicates) and observed that short-term depletion of ARID1A (72h), in spite of modest or no change in accessibility, elicits widespread transcriptional inhibition (Fig. 2F, G). We tested the possibility that decreased enhancer activity (ARID1A-dependent) may affect global transcription. We identified the active enhancer regions neighboring the 200 most downregulated protein coding genes and analyzed their accessibility and acetylation status. Remarkably, we found no significant changes in DNA accessibility or H3K27ac levels at these enhancers (Supplemental Fig. 2C), suggesting that ARID1A regulates basal transcription of protein coding genes independent of its putative role at distal cis-regulatory elements. Furthermore, to exclude off-target or indirect effects, we performed transient depletion of ARID1A using a different shRNA sequence and rescued its protein levels with exogenous ARID1A (wt RMG-1 cells were co-transfected with shRNAs and either the pLVX backbone vector or the pLVX-ARID1A vector). We assayed expression of candidate genes downregulated in our inducible system and found that their modulation is ARID1A-dependent (Fig. 2H). Furthermore, we analyzed the effect of short-term (72h) ARID1A depletion on two additional cell lines, the ovarian cystadenocarcinoma OVCA429 and the embryonic derived HEK293T. While OVCA429 recapitulated the effect observed in RMG-1 cells (Supplemental Fig. S2A), we could not observe major changes in gene expression when performing the same experiment in HEK293T (Supplemental Fig. S2B), suggesting that this phenotype may be specific to ovarian epithelial cells. Taken together, our data suggest that ARID1A modulates widespread transcriptional activation with a mechanism independent of chromatin remodeling.
ARID1A loss affects RNAPII-dependent transcription
Global variations of steady-state RNA, especially at such broad scale, may be attributed to dysregulation of RNA processing and stability. To determine whether loss of ARID1A directly impacts nascent transcription, we performed Global Run-On sequencing (GRO-seq; Core et al., 2008; Gardini, 2017) and RNA-seq of the chromatin fraction (CHROM-RNA-seq) (Lai et al., 2015). Depletion of ARID1A led to significant decrease (Wilcoxon rank sum test p < 2.2 × 10−16) of nascent transcription (GRO-seq) both at protein coding genes and intergenic enhancers (Fig. 3A). Notably, ARID1A loss did not affect either RNAPI-dependent rRNA genes or RNAPIII-transcribed genes (Fig. 3A), while the vast majority of RNAPII-dependent genes are downregulated (Fig. 3B). To investigate the nature of this transcriptional impairment, we used GRO-seq to gauge pausing indexes (PI) for all transcribed genes (N=6,281). Pausing index (Core et al., 2008) is an estimate of the ratio of RNA Polymerase II (RNAPII) that is found in pausing conformation vs its elongating form. We observed that pausing of RNAPII (−50/+300 bp from the TSS) diminished across all active genes in the absence of ARID1A (Fig. 3D; Wilcoxon rank sum test p < 2.2 × 10−16). Next, we performed pausing index analysis using chromatin-bound RNA-seq (CHROM-RNA-seq) that is highly enriched for primary transcripts. Coherently, CHROM-RNA-seq revealed a decrease in pausing indexes across RNAPII genes (Fig. 3E; Wilcoxon rank sum test p < 2.2 × 10−16). Collectively, our analysis of nascent and primary transcripts suggests that loss of ARID1A has a direct bearing on real-time transcription and may affect the dynamics of RNAPII.
Figure 3 – ARID1A depletion causes attenuation of nascent transcription.
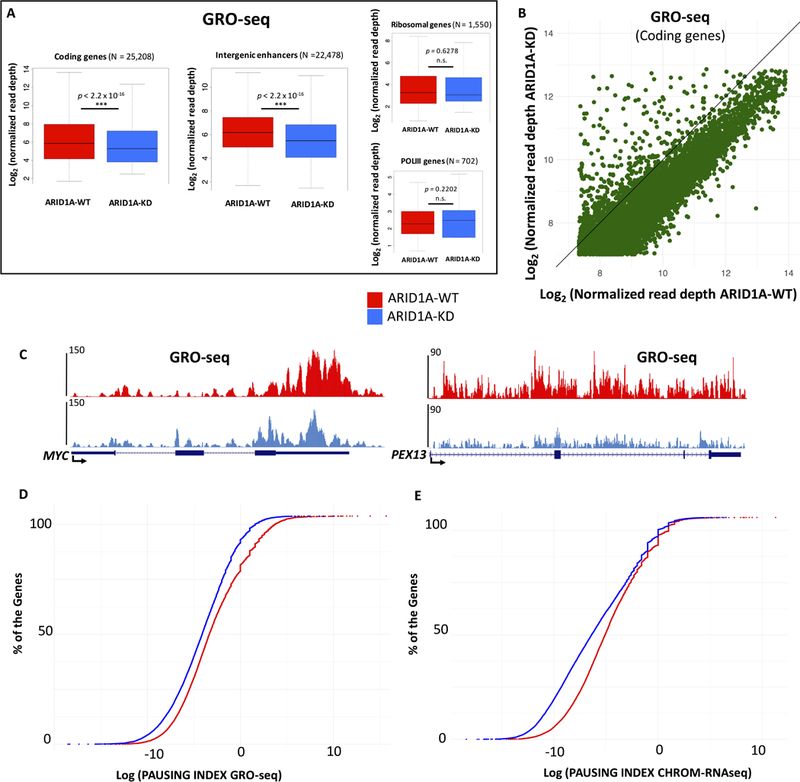
(A) Box plots compare nascent transcription (GRO-seq) before and after ARID1A depletion. Normalized read depth were calculated for the entire set of active protein coding genes and enhancers (intergenic H3K27ac peaks). Additionally, transcription at all ribosomal genes (rRNA transcript type in v17 of GENCODE) and RNAPIII dependent genes is shown. (B) GRO-seq correlation plot for all transcribed coding genes reveals global transcriptional downregulation upon ARID1A-KD. (C) GRO-seq profiles at MYC and PEX13 loci. (D and E) GRO-seq and chromatin-enriched RNA-seq (CHROM-RNAseq) data were used to calculate Pausing Indexes of all expressed genes. Most genes show a decrease in their pausing index (proximal promoter reads/gene body reads) upon depletion of ARID1A (ARID1A-KD), suggesting a defect in pausing of RNAPII.
ARID1A loss causes global reduction of paused RNAPII
To investigate the impact of ARID1A loss on RNAPII dynamics, we performed ChIP-seq, in two independent biological replicates, before and after depletion of ARID1A. We used antibodies raised against Rpb1 (POLR2A) that recognize all isoforms of RNAPII, irrespective of phosphorylation status. ChIP-seq revealed significant decrease of RNAPII occupancy at most TSS of protein coding genes (Wilcoxon rank sum test p < 2.2 × 10−16; Fig. 4A–D). There is broad consensus that most of the promoter-proximal RNAPII signal represents the paused form of polymerase (Adelman and Lis, 2012; Henriques et al., 2013; Levine, 2011; Williams et al., 2015). After transcription initiation, an early RNAPII elongation complex begins mRNA synthesis, only to arrest within the first 30–50bp. The early elongating RNAPII is highly phosphorylated at Ser5 of the CTD (C-terminal domain), and is under the control of the negative elongation complex, NELF and DSIF. Productive elongation requires the recruitment of pTEFb, which phosphorylates NELF, DSIF, and Ser2 of the CTD to ultimately release RNAPII from the promoter-proximal region (Adelman and Lis, 2012; Levine, 2011). Notably, we observed a sharp reduction of RNAPII at the TSS of coding genes (Fig. 4A–D; Supplemental Fig. S3A) upon depletion of ARID1A. We performed genome-wide analysis of phospho-RNAPII and observed a significant reduction of initiating and elongating polymerase, however, phopsho-Ser5 boasted the largest decrease (Supplemental Fig. S3B). Importantly, total levels of RNAPII in chromatin extracts remained unchanged (Fig. 4E; Quantitative blots in Supplemental Fig. S3C). To further investigate RNAPII dysregulation upon loss of ARID1A, we analyzed the travelling ratio of polymerase to assess changes in the fraction of paused vs. elongating RNAPII (Rahl et al., 2010). Depletion of ARID1A elicited a marked increase in travelling ratio at active genes (N=6,281; Wilcoxon rank sum test p < 2.2 × 10−16; Fig. 4F), consistent with pausing index analysis of nascent transcription (Fig. 3D-E), and suggestive of a role for ARID1A in maintaining RNAPII pausing in ovarian epithelial cells. To confirm that ARID1A is directly implicated in regulation of pausing, we performed a rescue experiment in parental RMG-1 cells. Using a different shRNA for ARID1A, we were able to successfully restore RNAPII occupancy upon co-expression of exogenous ARID1A (Fig. 4G).
Figure 4 – Accumulation of paused RNAPII in OCCC cells depends on ARID1A.
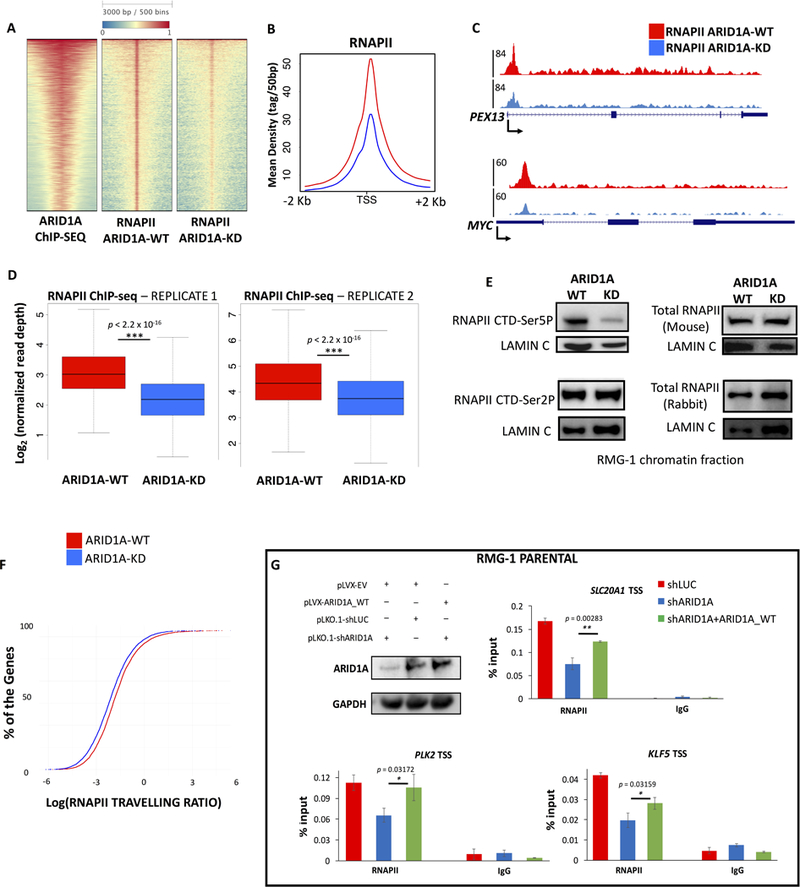
(A) ARID1A depletion (ARID1A-KD) affects RNAPII levels at the majority of active TSS. The heatmap shows 5,914 TSS targeted by SWI/SNF, ranked by mean intensity of ARID1A. (B) Average profiles of RNAPII ChIP-seq (ARID1A-WT, KD) around the TSS of 5,914 genes presented in panel (A). (C) UCSC Genome Browser screenshot of RNAPII ChIP-seq profiles (ARID1A-WT, KD) at PEX13 and MYC loci. There is a significant reduction of RNAPII occupancy at the promoter proximal regions. (D) Box plots showing read depth of RNAPII ChIP-seq at the TSS of ARID1A target genes. RNAPII occupancy is significantly reduced (p<2.2×10−16) after depletion of ARID1A, in two independent replicates. (E) Immunoblot analysis of the chromatin fraction of OCCC cells, before and after ARID1A depletion. While total RNAPII levels (assayed with two different antibodies, see methods) and phosphorylation of Ser2 remain unchanged, phosphorylation of Ser5 is reduced. Lamin C is shown as loading control. Quantitative immunoblots are shown in Supplemental Fig. S3B. (F) Travelling ratio analysis reveals changes in RNAPII occupancy at proximal promoter vs. gene body. Incremental plots demonstrate that ARID1A depletion elicits a reduction of pausing genome-wide. (G) Parental RMG-1 cells were infected with shRNAs against ARID1A and rescued with exogenously expressed full-length ARID1A. Quantitative PCR at three candidate genes shows that downregulation of RNAPII at TSS is significantly reversed (n=3) by ARID1A overexpression.
ARID1A depletion does not affect transcriptional initiation
Our data suggest that ARID1A loss prevents accumulation of paused RNAPII at the proximal promoter. We hypothesize that ARID1A mediates RNAPII regulation directly at the chromatin level. Accordingly, bulk levels of RNAPII and its elongating form (Ser-2P) do not change in chromatin extracts (Fig. 4E; Quantitative blots in Supplemental Fig. S3B), whereas the initiation/early elongation form of RNAPII (Ser-5P) diminishes. Furthermore, protein levels of pausing regulators NELF and SPT5 are unchanged. However, widespread dampening of transcription at RNAPII genes may be also attributed to initiation defects and/or impaired loading of the pre-initiation complex, all of which would result in marked loss of RNAPII at the TSS (Fig. 4A). First, we analyzed a group of specialized RNA genes (RNAPII-dependent UsnRNAs) that are not known to rely on polymerase pausing to regulate their expression. U snRNAs are highly transcribed short genes constitutively expressed and boast the highest levels of RNAPII by ChIP-seq. Our data indicate that depletion of ARID1A does not affect loading and initiation of RNAPII at these genes (Supplemental Fig. SD). Next, we investigated the ability of ARID1A-depleted cells to properly initiate transcription, by leveraging the CDK9 inhibitor flavopiridol (inhibitor of pTEFb kinase activity). Flavopiridol prevents transcriptional elongation within hours of treatment and leads to accumulation of initiated RNAPII that is unable to clear the proximal promoter region (Rahl et al., 2010). We treated both ARID1A-WT and ARID1A-KD cells with flavopiridol for 2 hours, before performing ChIP-seq of RNAPII. Treatment of ARID1A-depleted cells with flavopiridol completely restored accumulation of RNAPII at the proximal promoter, to an extent comparable, if not higher, than that of ARID1A-competent cells treated with flavopiridol (Fig. 5A–C). Importantly, treatment with flavopiridol also abolished the global decrease of travelling ratio determined by ARID1A depletion (Fig. 5D). Collectively, these data indicate that ARID1A loss hinders accumulation of RNAPII at proximal promoters without affecting the process of initiation or the integrity of the pre-initiation complex.
Figure 5 – ARID1A depletion impairs RNAPII pausing without affecting transcriptional initiation in OCCC cells.
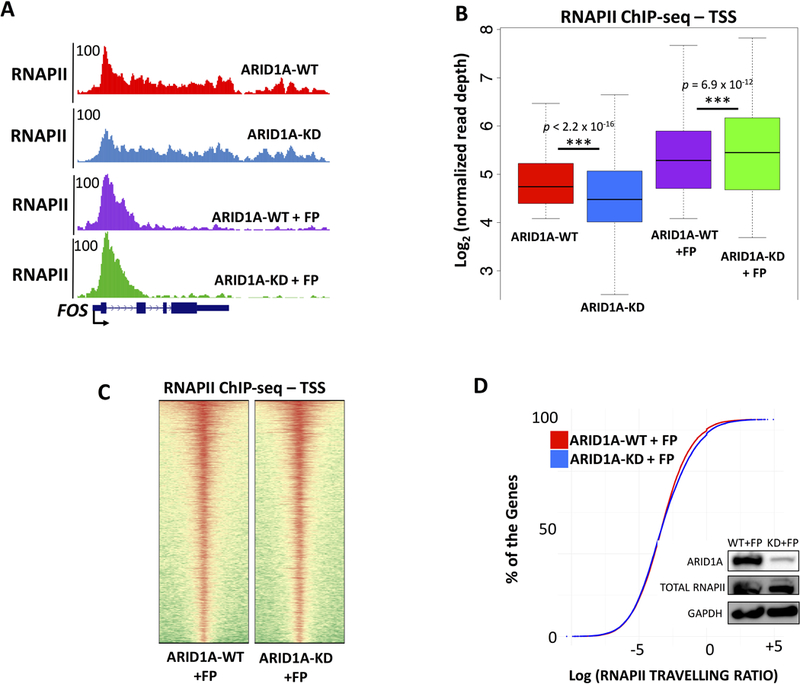
(A) UCSC Genome Browser screenshot of RNAPII at the FOS locus. ARID1A-KD reduces paused RNAPII at the 5’ of FOS, treatment with flavopiridol (2h) inhibits elongation and fully restores accumulation of RNAPII at the TSSs, similar to ARID1A-WT. (B) Box plot of RNAPII ChIP-seq binding at the TSS of 6,281 transcribed genes. We compare ARID1A-WT and ARID1A-KD conditions, with or without flavopiridol (FP). Upon elongation inhibition elicited by FP, accumulation of RNAPII in ARID1A-KD+FP is comparable or even higher than that of ARID1A-WT+FP, suggesting that ARID1A loss does not affect integrity of the pre-initiation complex but hinders accumulation of RNAPII at proximal promoters. (C) Comparison of RNAPII ChIP-seq at 6,281 TSS of flavopiridol treated wild type RMG-1 (ARID1A-WT + FP) and flavopiridol treated ARID1A depleted RMG-1 (ARID1A-KD + FP). The heatmap suggests similar accumulation of RNAPII at the proximal promoter in ARID1A-WT+FP and ARID1A-KD+FP cells. (D) Travelling ratio measures changes in RNAPII occupancy at proximal promoter vs. gene body (Fig. 4). Flavopiridol treated wild type RMG-1 (ARID1A-WT + FP) are compared to flavopiridol treated ARID1A depleted RMG-1 (ARID1A-KD + FP). We observe minor differences between the two conditions, further suggesting that flavopiridol fully restores physiological levels of pausing.
Next, we assessed changes in the chromatin status of OCCC cells. First, we profiled H3K36me3, a methylation mark that is co-transcriptionally deposited and correlates with transcriptional elongation (Fong et al., 2017; Zhang et al., 2015), to determine if ARID1A may have an additional impact in regulating release of paused RNAPII and downstream elongation. In fact, levels of H3K36me3 are known to mirror productive elongation of RNAPII throughout the gene body. Our analysis suggests that loss of ARID1A does not impair elongation, since average levels of H3K36me3 moderately increase across the gene body of active loci in ARID1A-KD (Suppl. Fig. 4A, B). Next, to determine if ARID1A depletion changes the chromatin status of its target genes and enhancers, we performed ChIP-seq of H3K27ac. Previous reports suggest that ARID1A depletion leads to loss of acetylation at enhancers (Alver et al., 2017; Kelso et al., 2017; Mathur et al., 2017). Consistent with these findings, we observed moderate but significant decrease of H3K27ac across enhancers (p < 2.2 × 10−16; Supplemental Fig. S4C, D). However, the sharpest decrease of acetylation was found at weak enhancers, which may lose acetylation as a mere consequence of decreased accessibility (Fig. 2, Supplemental Fig. S4C). Furthermore, enhancers associated to the top 200 downregulated genes upon ARID1A depletion did not display changes in acetylation (Supplemental Fig. S2C). We next examined the acetylation status of promoters. Notably, ARID1A target promoters displayed a significant increase of H3K27ac at their +1 nucleosome (Supplemental Fig. S4E, F), which was validated by ChIP-qPCR at select loci upon depletion of ARID1A with a different shRNA sequence (Supplemental Fig. S4G).
Collectively, our analysis of the chromatin status further supports the hypothesis that ARID1A directly favors accumulation of paused RNAPII, rather than supporting transcription indirectly through deposition of activating chromatin marks.
ARID1B restores paused RNAPII and transcription in ovarian cancer cells
To investigate how ovarian epithelial cells cope with the global defects in transcription that follow ARID1A loss, we examined its paralog, ARID1B. ARID1B represents a specific vulnerability in ARID1A–mutated tumors, and their synthetic lethality has been previously described (Helming et al., 2014). We surmised that OCCC cells could leverage ARID1B to recover from the widespread transcriptional repression that follows loss of ARID1A. Hence, we induced depletion of ARID1A through an extended time course experiment (day 0–7 of DOX). Ovarian epithelial cells express low levels of ARID1B and gradually increase its expression in response to the knock-down of ARID1A (Fig 6A, Suppl. Fig. 4H). Furthermore, ChIP-qPCR showed a gradual and steady increase of ARID1B occupancy at the TSS of genes typically bound by ARID1A, peaking at day 7 (Fig. 6B). The peak of ARID1B occupancy (day 7) correlated with rescue of promoter proximal paused-RNAPII (Fig 6B), and restoration of mRNA levels (Fig. 6B). Collectively, these data suggest that upregulation of ARID1B compensates for the loss of ARID1A and re-establishes physiological levels of paused RNAPII. To validate the function of ARID1B as regulator of transcription and RNAPII dynamics in OCCC, we performed a complementation experiment in wild type RMG-1 cells. We depleted ARID1A while overexpressing exogenous ARID1B (Fig. 6C) and determined that ARID1B is sufficient to rescue paused RNAPII and restore gene expression at a set of candidate genes (Fig. 6C). These data suggest that ARID1A loss induces ARID1B upregulation in ovarian epithelial cells, which re-enforces paused polymerase genome-wide. However, the oncogenic mechanism triggered by ARID1A loss remains unclear. Genetic and biochemical evidences suggest that ARID1A and ARID1B are only partly redundant. We hypothesize that a set of genes may be strictly dependent on ARID1A and escape rescue by ARID1B. To determine differences between ARID1A- and ARID1B-regulated transcriptomes, we performed RNA-seq at day 0 and 7 of DOX induction. Notably, the global repression of transcription determined by ARID1A depletion (day 3; Fig. 2E) is relieved by ARID1B upregulation (day 7; Fig. 6D). Nonetheless, we detected 1,817 genes differentially expressed between day 0 and 7 (DESeq2; 906 upregulated, and 911 downregulated; FDR < 5%; Table S2). These transcripts represent potential ARID1A-dependent genes that may drive tumorigenesis in OCCC. Ingenuity Pathway Analysis revealed that dysregulated genes are significantly enriched in the following functional categories: Cancer (p = 2.48 × 10−7), DNA Repair (p = 2.94 × 10−12), Cell Cycle (p = 1.06 × 10−14), and Cell Death and Survival (p = 4.45 × 10−12). For instance, the PARP binding protein PARI prevents genomic instability by inhibiting inappropriate homologous recombination (Moldovan et al., 2012), and its expression is not rescued by ARID1B. Similarly, the centrosomal proteins CENPE and CENPK are persistently downregulated at day 7 of DOX induction, and may underlie defects in chromosome alignment and segregation that lead to genomic instability.
Figure 6 – Upregulation of ARID1B restores RNAPII pausing and transcription at most genes.
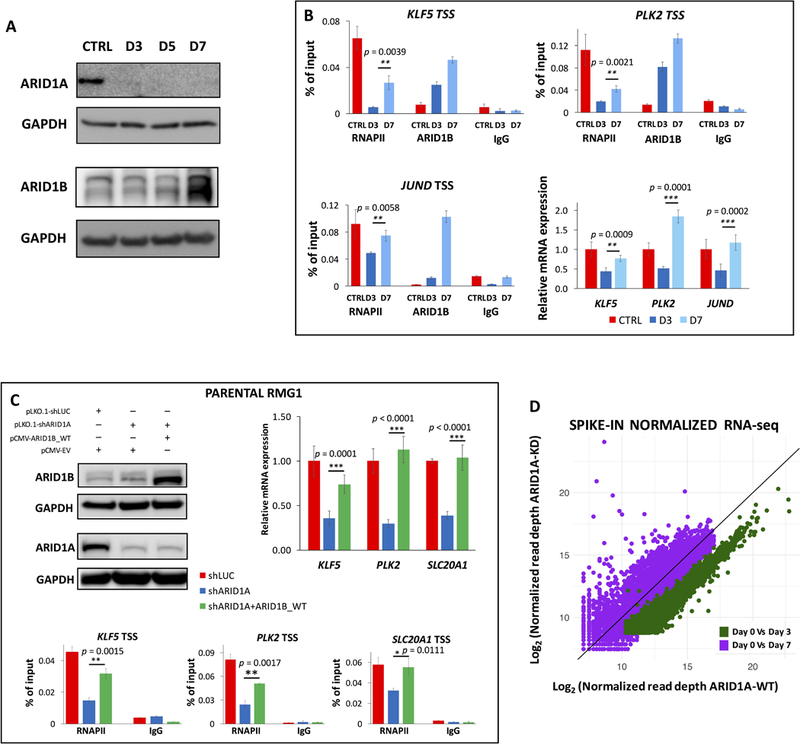
(A) Immunoblot analysis of ARID1A and ARID1B during a time course of doxycycline (days 3, 5, 7). ARID1B is increasingly upregulated in response to ARID1A depletion, peaking at day 7. Quantitative immunoblots are shown in Supplemental Fig. S4H. (B) ChIP-qPCR analysis of RNAPII and ARID1B binding for a panel of ARID1A target genes at 0 hours (CTRL), 3 days, and 7 days of treatment with doxycycline. ARID1B gradually replaces ARID1A at the TSS, whilst RNAPII proximal-promoter occupancy is restored. Similarly, qRT-PCR for the same genes (fold enrichment compared to CTRL, normalized to 18S rRNA) suggests that transcription resumes its physiological level at day 7, concurrent with highest occupancy of ARID1B. (C) Parental RMG-1 cells were transduced with shRNAs against ARID1A and a full-length ARID1B vector. ChIP-qPCR for RNAPII, and qRT-PCR at three candidate loci, show that ARID1A-dependent transcriptional regulation is complemented (p<0.0111) by ARID1B (qRT-PCR fold change compared to shLUC, normalized by 18s rRNA). (D) Correlation plot (spike-in normalized RNA-seq) for all expressed coding genes shows that global transcriptional downregulation provoked by ARID1A depletion (day 3 of DOX induction, green) is largely rescued by ARID1B upregulation (day 7 of DOX induction, purple). Genes that are persistently dysregulated at day 7 have been functionally evaluated (IPA) and may represent ARID1A-specific targets.
Lastly, TP53 (p = 6.32 × 10−7) and the estrogen receptor ESR1 (p = 1.96 × 10−8) resulted among the top enriched upstream regulators of ARID1A-dependent genes. Notably, p53 is rarely mutated in ARID1A-deficient ovarian tumors, suggesting that the impairment of its function may occur transcriptionally at its genomic targets. Overall, these data suggest that a subset of genes, strictly dependent on ARID1A for their fine-tuned expression, may underlie cell transformation in ovarian epithelial cells.
Discussion
A model for ARID1A function in normal and malignant cells
Here we present a comprehensive genome-wide profiling of ARID1A activity in ovarian epithelial cells and propose a mechanistic model that explains both the physiological role of ARID1A in transcription and the oncogenic consequences of its genetic ablation, which occurs in nearly 60% of clear cell carcinomas (OCCC). In summary, ARID1A targets active promoters and ensures that physiological levels of paused RNA polymerase II accumulate after initiation. When ARID1A is the predominant ARID component of the BAF complex, its loss results in widespread transcriptional repression due to ineffective pausing. Upregulation of ARID1B relieves the transcriptional crunch, however, a set of genes that are strictly dependent on ARID1A do not recover their prior expression level and may pave the way to cellular transformation.
Distribution of ARID1A and its role in remodeling
First, we provide an accurate profiling of ARID1A genome-wide in ovarian cells. We find that ARID1A targets most active promoters and enhancers, and is required to sustain transcription of both protein coding genes and noncoding enhancer RNAs. Notably, we observe a very distinct profile of ARID1A at promoters (nucleosome-associated) and enhancers (centered on the nucleosome depleted region). This finding may reflect different functions of the SWI/SNF complex or underlie a different chromatin organization at these regulatory regions, and is in accordance with previous profiling of the catalytic BRG1 subunits in melanoma cells (Laurette et al., 2015).
Previous work suggested a preference for ARID1A and SWI/SNF towards binding enhancer elements (Alver et al., 2017; Mathur et al., 2017, Kelso et al. 2017), however, other studies point out a more diffuse and broad distribution of the ARID1 proteins and other SWI/SNF subunits, similar to our observations (Laurette et al., 2015; Morris et al., 2014; Raab et al., 2015). While the SWI/SNF complex is a universal chromatin remodeler in eukaryotes, the role for ARID1A in maintaining accessibility at promoters and enhancers in OCCC is marginal, suggesting that its role in nucleosome positioning may be negligible. Previous studies describe a role for SWI/SNF components (especially BRG1) as modulators of accessibility at enhancers (Bao et al., 2015; Laurette et al., 2015), but we find that the contribution of ARID1A is limited to the weakest enhancer regions, displaying the lowest enrichment for ARID1A by ChIP-seq. Similarly, promoter accessibility is regulated by the BRG1 and SNF subunits of SWI/SNF (Tolstorukov et al., 2013), but is not affected by ARID1A according to our ATAC-seq analysis. Coherently, a previous DNAse-seq study suggested that very limited changes in hypersensitive sites occurred in ARID1A KO ovarian cells (Kim et al., 2016), however, MNAse-seq studies in mouse ES cells yielded different results (Lei et al., 2015). It is possible that ARID1A contributes to nucleosome remodeling in embryonic stem cells but not in somatic cells. In our system, the most active promoters and enhancers display very limited changes in accessibility after 3 days of ARID1A depletion, in spite of massive transcriptional repression (assessed by RNA-seq, GRO-seq, and CHROM-RNA-seq). It must also be noted that accessibility may not always reflect changes in nucleosome positioning (Mueller et al., 2017). Our studies may be extended in the future using MNase digestion followed by H3 ChIP, before excluding any role for ARID1A as adjuvant of nucleosome remodeling in somatic cells.
A new role for ARID1A in regulation of RNAPII
Our data suggest that ARID1A endows the SWI/SNF (BAF) complex with regulatory activity towards RNAPII pausing, determining the physiological rate of transcription for a large fraction of active genes in ovarian epithelial cells. The mechanism of RNAPII pausing has emerged, over the last decade, as a fundamental regulatory step of transcription in higher eukaryotes (Adelman and Lis, 2012). It must be noted that pausing of RNAPII is not simply a brake for transcription, as paused RNAPII in ES cells is required to maintain cell homeostasis and properly respond to environmental stimuli and cues (Henriques et al., 2013; Shao and Zeitlinger, 2017; Williams et al., 2015). In the absence of properly functioning pausing machinery, such as upon depletion of NELF, thousands of genes were shown to respond with transcriptional attenuation (Williams et al., 2015). In fact, polymerase that fails to pause appears not to be released into productive elongation but is lost from the body of the gene (Gilchrist et al., 2010). These findings are consistent with our observation that failure to accumulate paused RNAPII at the proximal promoter after ARID1A depletion results in general attenuation of transcription. Importantly, we show that protein levels of RNAPII, NELF, and SPT5 are not affected by depletion of ARID1A, and the elongation block induced with flavopiridol fully restores proximal paused RNAPII, thereby excluding defects in the initiation step.
Pausing of RNAPII is governed by protein complexes such as NELF, PAF, Mediator, and Integrator (Chen et al., 2015; Galbraith et al., 2013; Gardini et al., 2014; Goldman et al., 2016; Patel et al., 2013), as well as chromatin structure (Adelman and Lis, 2012). Interestingly, depletion of ARID1A elicited a marked increase of H3K27 acetylation at the +1 nucleosome of most protein coding genes. Recently, an association between acetylation levels of the +1 nucleosome and the pausing activity of RNAPII was proposed (Boija et al., 2017). In particular, the authors suggest that fine-tuned levels of H3K27ac on the first nucleosome downstream of the TSS are required to maintain a proper balance between paused RNAPII and productive elongation. We speculate that ARID1A may be required for the correct acetylation balance, with increased acetylation resulting from ARID1A loss preventing accumulation of paused RNAPII at the proximal promoter.
Role of ARID1A at enhancers
ARID1A has been recently proposed to control enhancer acetylation via recruitment of the EP300 acetyltransferase (Alver et al., 2017; Mathur et al., 2017). Our data, however, indicate that the highest occupancy of ARID1A anticorrelates with changes in H3K27ac levels, which are more likely to occur at weak enhancers. Weak enhancers likely comprise inactive or poised enhancers that are not actively regulating growth and homeostasis of RMG-1 cells. We also find that the most active, ARID1A-bound, regulatory elements (strong enhancers) are significantly enriched in AP-1 binding sites. In the context of ovarian epithelial cells, the AP-1 transcription factor may sustain open conformation and acetylation of enhancers with the support of the catalytic activity of SWI/SNF (Vierbuchen et al., 2017), leaving ARID1A dispensable for such role. Nonetheless, we hypothesize that enhancer activity may be compromised in ARID1A-deficient cells, since GRO-seq data revealed widespread transcription attenuation at enhancers. GRO-seq assays detect reads from the enhancer-associated bi-directional noncoding RNAs (eRNAs) (Gardini and Shiekhattar, 2015). Several studies suggest that enhancer activity is functionally linked to transcription of eRNAs and their accumulation at chromatin (Bose et al., 2017; Gardini and Shiekhattar, 2015; Lai et al., 2015). We speculate that transcription attenuation at strong enhancers will impact their activity. Intriguingly, enhancers may be subjected to the same RNAPII pausing mechanism enforced at protein coding genes (Henriques et al., 2018), suggesting a similar role for ARID1A. Future research on the enhancer-promoter network of ovarian epithelial cells will clarify the precise role of ARID1A at these regulatory elements.
Tumorigenesis mediated by ARID1A-loss
Finally, in this work we show that upregulation of ARID1B offsets loss of ARID1A at protein coding genes and restores physiological transcription, establishing a mechanistic model for the synthetic lethality of ARID1A and ARID1B (Helming et al., 2014). Consistently, ARID1B was reported upregulated in mouse embryonic fibroblasts that lack one or both ARID1A alleles (Helming et al., 2014). However, genetic and biochemical evidence suggest that ARID1A and ARID1B are only partly redundant. First, the two paralogs seem to associate with different sets of transcription factors (Raab et al., 2015). Second, genetic mouse models suggest that the two proteins are functionally distinct (Celen et al., 2017; Goldman et al., 2016; Krosl et al., 2010; Vasileiou et al., 2015; Zhai et al., 2016). As a matter of fact, we found a subset of 1,817 genes that are not entirely rescued by ARID1B expression and remain dysregulated. While ARID1B restores RNAPII pausing at the majority of protein coding genes, some loci may be preferentially modulated by ARID1A and de facto trigger tumorigenesis once normal epithelial cells biallelically lose ARID1A. Importantly, a subset of permanently dysregulated genes are TP53 targets. Given that TP53 is rarely mutated in ARID1A-deficient OCCCs, our data may account for its functional impairment in this type of tumor. Lastly, while our studies are conducted in ovarian cancer cells, ARID1A is the most mutated chromatin modifier in cancers of various origin. Future studies will help determine the role of impaired RNAPII pausing and partial complementation by ARID1B in additional tumor types with recurrent (>30%) ARID1A loss-of-function mutations such as hepatocellular carcinoma, uterine carcinosarcoma, and stomach adenocarcinoma.
Experimental Procedures
Cell lines
RMG-1 cells were obtained from JCRB Cell Bank and maintained as described in Supplemental Experimental Procedures. RMG1 cells were infected with lentivirus carrying the inducible tet-pLKO-shARID1A (sequence: GCCTGATCTATCTGGTTCAAT; Supplemental Experimental Procedures). For validation experiments, ARID1A was depleted using a different shRNA (CCTCTCTTATACACAGCAGAT). For rescue experiments, parental RMG-1 cells were simultaneously infected with a lentivirus carrying the shARID1A vector, and with a lentivirus carrying the pLVX-ARID1A vector. The pLVX empty vector was used as a control. Infected cells were selected with 2 µg/ml Puromycin for 72 hours.
For ARID1B rescue experiments, a commercial plasmid was used (Supplemental Experimental Procedures). OVCA429 cells were lentivirally infected with pLKO.1-shARID1A (TRCN0000059090) and selected with 2 µg/ml Puromycin. shRNA against luciferase was used as control.
Western Blot
Western Blots were performed as described in Supplemental Experimental Procedures.
Antibodies
Used antibodies are listed in Supplemental Experimental Procedures.
ChIP-seq and ChIP-qPCR sample processing
Samples from different conditions were processed together to prevent batch effects. For each replicate, 10 million RMG-1 cells were used to perform ChIP-seq experiments as described in Supplemental Experimental Procedures. For flavopiridol experiments, ARID1A-WT and ARID1A-KD cells were treated with flavopiridol (2µM) for two hours before being harvested. For ChIP-qPCR, on day 1 the sonicated lysate was aliquoted into single immunoprecipitations of 2.5 × 106 cells each. On day3, ChIP eluates and input were assayed by real-time quantitative PCR.
ChIP-seq analyses
Sequences were aligned to the reference hg19, using Burrows Wheeler Alignment tool (BWA), with the MEM algorithm (Li, 2013). Detailed settings are described in Supplemental Experimental Procedures.
We called peaks for each individual using MACS2, at 5% FDR, with default parameters (Zhang et al., 2008). Heatmaps were generated with ChAsE v. 1.0.11 (Younesy et al., 2016), average profiles with seqMINER v. 1.3.4 (Zhan and Liu, 2015)
ATAC-seq sample processing
Two independent biological replicates of 50,000 cells per condition were processed as described in the original ATAC-seq protocol paper (Buenrostro et al., 2013).
ATAC-seq analyses
ATAC-seq data were processed with the same pipeline described for ChIP-seq, with one modification: all mapped reads were offset by +4 bp for the forward-strand and −5 bp for the reverse-strand. After peak calling, peaks replicated in both of the two ARID1A-WT samples were used for downstream analyses. EdgeR (Robinson et al., 2010) was used to test for regions with differential accessibility between ARID1A-KD and ARID1A-WT using a likelihood ratio test in a generalized linear model with the following parameters: glmLRT(fit,coef=2), based on quantile-normalized read counts.
RNA-seq sample processing
Total RNA-seq was performed on two independent biological replicates per condition. Samples from different conditions were processed together to prevent batch effects. For each sample, 10 million cells were used to extract total RNA and produce RNA-seq libraries (Supplemental Extended Procedures).
RNA-seq analyses
Reads were aligned to hg19 using STAR v2.5 (Dobin et al., 2013). Detailed parameters are in Supplemental Extended Procedures. We filtered bam files based on alignment quality (q = 10) using Samtools v0.1.19 (Li et al., 2009). We used the latest annotations obtained from GENCODE to build reference indexes for the STAR alignment. FeatureCounts (Liao et al., 2014) was used to count reads mapping to each gene. RSEM (Li and Dewey, 2011) was used to obtain FPKM (Fragments Per Kilobase of exon per Million fragments mapped). We analyzed differential gene expression with DESeq2 (Love et al., 2014), with the following model: design = ~condition, where condition indicates either ARID1A-WT or ARID1A-KD.
GRO-seq
GRO-seq was performed on two independent biological replicates per condition as previously described (Gardini et al., 2014). GRO-seq data were analyzed with the same pipeline above described for total RNA-seq.
Other statistical and genomic analyses
All statistical analyses and plots were performed using R v3.3.1 and ggplot2 (R Core Team, 2016; Wickham, 2009). BEDtools v2.27.1 was used for genomic analyses (Quinlan and Hall, 2010). For travelling ratio analysis, TSS regions were extended −50/+300bp. Other thresholds were tested (up to +500 bp downstream), with no differences in the results. Pathway analysis was performed with Ingenuity Pathway Analysis Suite (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis)). Motif analyses were performed with Meme-ChIP (Bailey et al., 2009).
Supplementary Material
Acknowledgments
This work was supported by grant R01 CA202919 (R.Z.) from the National Institute of Health and funding from the W.W. Smith Charitable Trust to A.G. S.A.W. is supported by NIH training grant T32-GM071339. The authors thank Kavitha Sarma, Cristopher D. Brown, Minal Caliskan, and Benjamin Bitler for insightful discussions. Drosophila S2 cells used for spike-in were kindly provided by Maya Capelson (UPenn). All genome-wide data were generated and processed at the Wistar Institute with the outstanding technical support from all members of the Genomics Facility, especially Dr. Shashi Bala (NIH-NCI funded CCSG: P30-CA010815).
Footnotes
Declaration of interests
The authors declare no competing interests.
Data and Software Availability
Sequencing data are available in the Gene Expression Omnibus (GSE104545)
References
- Adelman K, and Lis JT (2012). Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet 13, 720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alver BH, Kim KH, Lu P, Wang X, Manchester HE, Wang W, Haswell JR, Park PJ, and Roberts CW (2017). The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat Commun 8, 14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayhan A, Mao TL, Seckin T, Wu CH, Guan B, Ogawa H, Futagami M, Mizukami H, Yokoyama Y, Kurman RJ, et al. (2012). Loss of ARID1A expression is an early molecular event in tumor progression from ovarian endometriotic cyst to clear cell and endometrioid carcinoma. Int J Gynecol Cancer 22, 1310–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, and Noble WS (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37, W202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X, Rubin AJ, Qu K, Zhang J, Giresi PG, Chang HY, and Khavari PA (2015). A novel ATAC-seq approach reveals lineage-specific reinforcement of the open chromatin landscape via cooperation between BAF and p63. Genome Biol 16, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, Schultz DC, Liu Q, Shih Ie M, Conejo-Garcia JR, et al. (2015). Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med 21, 231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitler BG, Wu S, Park PH, Hai Y, Aird KM, Wang Y, Zhai Y, Kossenkov AV, Vara-Ailor A, Rauscher FJ III, et al. (2017). ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat Cell Biol 19, 962–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boija A, Mahat DB, Zare A, Holmqvist PH, Philip P, Meyers DJ, Cole PA, Lis JT, Stenberg P, and Mannervik M (2017). CBP Regulates Recruitment and Release of Promoter-Proximal RNA Polymerase II. Mol Cell 68, 491–503 e495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose DA, Donahue G, Reinberg D, Shiekhattar R, Bonasio R, and Berger SL (2017). RNA Binding to CBP Stimulates Histone Acetylation and Transcription. Cell 168, 135–149 e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celen C, Chuang JC, Luo X, Nijem N, Walker AK, Chen F, Zhang S, Chung AS, Nguyen LH, Nassour I, et al. (2017). Arid1b haploinsufficient mice reveal neuropsychiatric phenotypes and reversible causes of growth impairment. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FX, Woodfin AR, Gardini A, Rickels RA, Marshall SA, Smith ER, Shiekhattar R, and Shilatifard A (2015). PAF1, a Molecular Regulator of Promoter-Proximal Pausing by RNA Polymerase II. Cell 162, 1003–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, and Lis JT (2008). Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322, 1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong N, Saldi T, Sheridan RM, Cortazar MA, and Bentley DL (2017). RNA Pol II Dynamics Modulate Co-transcriptional Chromatin Modification, CTD Phosphorylation, and Transcriptional Direction. Mol Cell 66, 546–557 e543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbraith MD, Allen MA, Bensard CL, Wang X, Schwinn MK, Qin B, Long HW, Daniels DL, Hahn WC, Dowell RD, et al. (2013). HIF1A employs CDK8-mediator to stimulate RNAPII elongation in response to hypoxia. Cell 153, 1327–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardini A (2017). Global Run-On Sequencing (GRO-Seq). Methods Mol Biol 1468, 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardini A, Baillat D, Cesaroni M, Hu D, Marinis JM, Wagner EJ, Lazar MA, Shilatifard A, and Shiekhattar R (2014). Integrator regulates transcriptional initiation and pause release following activation. Mol Cell 56, 128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardini A, and Shiekhattar R (2015). The many faces of long noncoding RNAs. Febs J 282, 1647–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist DA, Dos Santos G, Fargo DC, Xie B, Gao Y, Li L, and Adelman K (2010). Pausing of RNA polymerase II disrupts DNA-specified nucleosome organization to enable precise gene regulation. Cell 143, 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman AR, Bitler BG, Schug Z, Conejo-Garcia JR, Zhang R, and Speicher DW (2016). The Primary Effect on the Proteome of ARID1A-mutated Ovarian Clear Cell Carcinoma is Downregulation of the Mevalonate Pathway at the Post-transcriptional Level. Mol Cell Proteomics 15, 3348–3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helming KC, Wang X, Wilson BG, Vazquez F, Haswell JR, Manchester HE, Kim Y, Kryukov GV, Ghandi M, Aguirre AJ, et al. (2014). ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat Med 20, 251–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques T, Gilchrist DA, Nechaev S, Bern M, Muse GW, Burkholder A, Fargo DC, and Adelman K (2013). Stable pausing by RNA polymerase II provides an opportunity to target and integrate regulatory signals. Mol Cell 52, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques T, Scruggs BS, Inouye MO, Muse GW, Williams LH, Burkholder AB, Lavender CA, Fargo DC, and Adelman K (2018). Widespread transcriptional pausing and elongation control at enhancers. Genes Dev 32, 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Wang TL, Shih Ie M, Mao TL, Nakayama K, Roden R, Glas R, Slamon D, Diaz LA Jr., Vogelstein B, et al. (2010). Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 330, 228–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, and Crabtree GR (2013). Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 45, 592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelso TWR, Porter DK, Amaral ML, Shokhirev MN, Benner C, and Hargreaves DC (2017). Chromatin accessibility underlies synthetic lethality of SWI/SNF subunits in ARID1A-mutant cancers. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Lu F, and Zhang Y (2016). Loss of HDAC-Mediated Repression and Gain of NF-kappaB Activation Underlie Cytokine Induction in ARID1A- and PIK3CA-Mutation-Driven Ovarian Cancer. Cell Rep 17, 275–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortschak RD, Tucker PW, and Saint R (2000). ARID proteins come in from the desert. Trends Biochem Sci 25, 294–299. [DOI] [PubMed] [Google Scholar]
- Krosl J, Mamo A, Chagraoui J, Wilhelm BT, Girard S, Louis I, Lessard J, Perreault C, and Sauvageau G (2010). A mutant allele of the Swi/Snf member BAF250a determines the pool size of fetal liver hemopoietic stem cell populations. Blood 116, 1678–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai F, Gardini A, Zhang A, and Shiekhattar R (2015). Integrator mediates the biogenesis of enhancer RNAs. Nature 525, 399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurette P, Strub T, Koludrovic D, Keime C, Le Gras S, Seberg H, Van Otterloo E, Imrichova H, Siddaway R, Aerts S, et al. (2015). Transcription factor MITF and remodeller BRG1 define chromatin organisation at regulatory elements in melanoma cells. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei I, West J, Yan Z, Gao X, Fang P, Dennis JH, Gnatovskiy L, Wang W, Kingston RE, and Wang Z (2015). BAF250a Protein Regulates Nucleosome Occupancy and Histone Modifications in Priming Embryonic Stem Cell Differentiation. J Biol Chem 290, 19343–19352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M (2011). Paused RNA polymerase II as a developmental checkpoint. Cell 145, 502–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur R, Alver BH, San Roman AK, Wilson BG, Wang X, Agoston AT, Park PJ, Shivdasani RA, and Roberts CW (2017). ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat Genet 49, 296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldovan GL, Dejsuphong D, Petalcorin MI, Hofmann K, Takeda S, Boulton SJ, and D’Andrea AD (2012). Inhibition of homologous recombination by the PCNA-interacting protein PARI. Mol Cell 45, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SA, Baek S, Sung MH, John S, Wiench M, Johnson TA, Schiltz RL, and Hager GL (2014). Overlapping chromatin-remodeling systems collaborate genome wide at dynamic chromatin transitions. Nat Struct Mol Biol 21, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller B, Mieczkowski J, Kundu S, Wang P, Sadreyev R, Tolstorukov MY, and Kingston RE (2017). Widespread changes in nucleosome accessibility without changes in nucleosome occupancy during a rapid transcriptional induction. Genes Dev 31, 451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa S, Tsukazaki K, Sakayori M, Jeng CH, and Iizuka R (1988). Establishment of a human ovarian clear cell carcinoma cell line (RMG-I) and its single cell cloning--with special reference to the stem cell of the tumor. Hum Cell 1, 426–435. [PubMed] [Google Scholar]
- Patel MC, Debrosse M, Smith M, Dey A, Huynh W, Sarai N, Heightman TD, Tamura T, and Ozato K (2013). BRD4 coordinates recruitment of pause release factor P-TEFb and the pausing complex NELF/DSIF to regulate transcription elongation of interferon-stimulated genes. Mol Cell Biol 33, 2497–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. (2016). R: a language and environment for statistical computing R Foundation for Statistical Computing, Vienna, Austria: https://www.R-project.org/. [Google Scholar]
- Raab JR, Resnick S, and Magnuson T (2015). Genome-Wide Transcriptional Regulation Mediated by Biochemically Distinct SWI/SNF Complexes. PLoS Genet 11, e1005748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, and Young RA (2010). c-Myc regulates transcriptional pause release. Cell 141, 432–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryme J, Asp P, Bohm S, Cavellan E, and Farrants AK (2009). Variations in the composition of mammalian SWI/SNF chromatin remodelling complexes. J Cell Biochem 108, 565–576. [DOI] [PubMed] [Google Scholar]
- Shao W, and Zeitlinger J (2017). Paused RNA polymerase II inhibits new transcriptional initiation. Nat Genet 49, 1045–1051. [DOI] [PubMed] [Google Scholar]
- Tolstorukov MY, Sansam CG, Lu P, Koellhoffer EC, Helming KC, Alver BH, Tillman EJ, Evans JA, Wilson BG, Park PJ, et al. (2013). Swi/Snf chromatin remodeling/tumor suppressor complex establishes nucleosome occupancy at target promoters. Proc Natl Acad Sci U S A 110, 10165–10170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasileiou G, Ekici AB, Uebe S, Zweier C, Hoyer J, Engels H, Behrens J, Reis A, and Hadjihannas MV (2015). Chromatin-Remodeling-Factor ARID1B Represses Wnt/beta-Catenin Signaling. Am J Hum Genet 97, 445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ling E, Cowley CJ, Couch CH, Wang X, Harmin DA, Roberts CWM, and Greenberg ME (2017). AP-1 Transcription Factors and the BAF Complex Mediate Signal-Dependent Enhancer Selection. Mol Cell 68, 1067–1082 e1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2009). ggplot2: Elegant Graphics for Data Analysis Springer-Verlag; New York. [Google Scholar]
- Williams LH, Fromm G, Gokey NG, Henriques T, Muse GW, Burkholder A, Fargo DC, Hu G, and Adelman K (2015). Pausing of RNA polymerase II regulates mammalian developmental potential through control of signaling networks. Mol Cell 58, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilsker D, Patsialou A, Zumbrun SD, Kim S, Chen Y, Dallas PB, and Moran E (2004). The DNA-binding properties of the ARID-containing subunits of yeast and mammalian SWI/SNF complexes. Nucleic Acids Res 32, 1345–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younesy H, Nielsen CB, Lorincz MC, Jones SJ, Karimi MM, and Moller T (2016). ChAsE: chromatin analysis and exploration tool. Bioinformatics 32, 3324–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Y, Kuick R, Tipton C, Wu R, Sessine M, Wang Z, Baker SJ, Fearon ER, and Cho KR (2016). Arid1a inactivation in an Apc- and Pten-defective mouse ovarian cancer model enhances epithelial differentiation and prolongs survival. J Pathol 238, 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, and Liu DJ (2015). SEQMINER: An R-Package to Facilitate the Functional Interpretation of Sequence-Based Associations. Genet Epidemiol 39, 619–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Cooper S, and Brockdorff N (2015). The interplay of histone modifications - writers that read. EMBO Rep 16, 1467–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.