

Abstract
An activated renin-angiotensin system (RAS) within the central nervous system has been implicated in sympathoexcitation during various disease conditions including congestive heart failure (CHF). In particular, activation of the RAS in the paraventricular nucleus (PVN) of the hypothalamus has been recognized to augment sympathoexcitation in CHF. We observed a 2.6-fold increase in angiotensinogen (AGT) in the PVN of CHF. To elucidate the molecular mechanism for increased expression of AGT, we performed in silico analysis of the 3′-untranslated region (3′-UTR) of AGT and found a potential binding site for microRNA (miR)-133a. We hypothesized that decreased miR-133a might contribute to increased AGT in the PVN of CHF rats. Overexpression of miR-133a in NG108 cells resulted in 1.4- and 1.5-fold decreases in AGT and angiotensin type II (ANG II) type 1 receptor (AT1R) mRNA levels, respectively. A luciferase reporter assay performed on NG108 cells confirmed miR-133a binding to the 3′-UTR of AGT. Consistent with these in vitro data, we observed a 1.9-fold decrease in miR-133a expression with a concomitant increase in AGT and AT1R expression within the PVN of CHF rats. Furthermore, restoring the levels of miR-133a within the PVN of CHF rats with viral transduction resulted in a significant reduction of AGT (1.4-fold) and AT1R (1.5-fold) levels with a concomitant decrease in basal renal sympathetic nerve activity (RSNA). Restoration of miR-133a also abrogated the enhanced RSNA responses to microinjected ANG II within the PVN of CHF rats. These results reveal a novel and potentially unique role for miR-133a in the regulation of ANG II within the PVN of CHF rats, which may potentially contribute to the commonly observed sympathoexcitation in CHF.
NEW & NOTEWORTHY Angiotensinogen (AGT) expression is upregulated in the paraventricular nucleus of the hypothalamus through posttranscriptional mechanism interceded by microRNA-133a in heart failure. Understanding the mechanism of increased expression of AGT in pathological conditions leading to increased sympathoexcitation may provide the basis for the possible development of new therapeutic agents with enhanced specificity.
Keywords: congestive heart failure, microRNA, miR-133a, angiotensin II, paraventricular nucleus, sympathoexcitation
exaggerated sympathoexcitation is associated with many cardiovascular diseases including congestive heart failure (CHF) (12, 47), hypertension, obesity, and insulin resistance (26). The prognosis of patients with elevated sympathoexcitation in cardiovascular diseases is dismal (9, 12, 45). Despite major advances in therapy directed to alleviating sympathoexcitation, morbidity and mortality are still very high for cardiovascular diseases (53, 64). There is accumulating evidence that suggests that excessive activation of the renin-angiotensin system (RAS) triggers the expression of ANG II, further enhancing the progression of cardiovascular maladies like CHF (36, 73) and hypertension (25). The increased sympathoexcitation is thought to originate from the central nervous system (CNS) (51). One particular site that has been identified to be of particular interest is the paraventricular nucleus (PVN) of the hypothalamus (36, 49, 68). A number of groups have demonstrated that the sympathetic nervous system and the RAS are markedly activated in the late stages of CHF, contributing to the pathophysiology of CHF (14, 36, 67, 74).
The primary effector biomolecule of RAS is an octapeptide, ANG II, formed by a cascade of enzymatic reactions involving three components: angiotensinogen (AGT), renin, and angiotensin-converting enzyme (ACE). Although studies regarding CHF and its therapy have focused on ACE and its inhibitors, the roles of initiating and rate-limiting steps in the angiotensin biosynthesis pathway from AGT have not been distinctly identified. Production of ANG II from AGT is initiated by the enzyme renin via cleavage from the amino-terminus of AGT, making this gene product a fundamental requirement for commencement of this pathway. Originally, the RAS was regarded as a circulating system, but in recent years many organs expressing components of the RAS, most notably the heart, kidney, and brain, have been identified (11, 17). There is growing evidence for neuronal synthesis of AGT and intraneuronal formation of ANG II, particularly in hypothalamic nuclei that are known to be involved in cardiovascular regulation (17). Overexpression of the AGT gene predominantly in brain and liver in transgenic mice has been shown to increase blood pressure (23), whereas AGT knockout mice express overt hypotension (59, 61) due to the reduction in plasma AGT and ANG II levels. Furthermore, AGT expression is elevated in many brain areas involved in the cardiovascular regulation of spontaneously hypertensive rats compared with normotensive Wistar-Kyoto rats, suggesting that regulation of the brain RAS is altered during the development of hypertension (54). The elucidation of mechanism(s) by which AGT gene expression is regulated is potentially important for understanding the molecular basis for increased activation of the RAS in the brain.
MicroRNAs (miRNAs) are endogenous, conserved, and short (20–23-nucleotides) noncoding RNAs that regulate gene expression by binding to the 3′-untranslated region (3′-UTR) using a partial base-pairing mechanism resulting in translational inhibition and/or mRNA destabilization (1). Interactions between miRNA “seed” sequence (positions 2–8) and complementary “seed match” sequence regions within the 3′-UTR of mRNA target are believed to be important for determining miRNA targets (33). Recent computational predictions in mammals have indicated that miRNAs may regulate 60% of human protein-coding genes (16) involved in many pathological processes such as cancer (10), schizophrenia (52), and various cardiovascular diseases (41, 63). We performed an online analysis of rat AGT 3′-UTR of protein-coding transcript with the miRNA search system miRDB (http://mirdb.org/cgi-bin/search.cgi) and TargetScan (http://www.targetscan.org/) databases and found the seed match sequence for miR-133a. Furthermore, it has recently been shown that increased cardiac AGT expression in diabetes is mitigated by overexpression of miR-133a (8).
In the present study, we hypothesized that decreased expression of miR-133a may be responsible for an increased expression of AGT, which subsequently leads to increased ANG II-mediated sympathetic tone via the PVN in rats with CHF. The 3′-UTR of the protein-coding transcript for AGT is targeted by miR-133a, leading to translational inhibition of AGT, suggesting a possible molecular switch for the regulation of AGT in the PVN during CHF. In this study, we determined whether altered miR-133a within the PVN contributes to the increased angiotensinergic tone resulting in an overactivation of the sympathetic drive commonly observed during CHF.
METHODS
Neuronal culture.
Neuroblastoma × glioma (NG108-15) hybrid cells were purchased from the American Type Culture Collection (ATCC HB-12317) and grown in DMEM (Sigma D) without pyruvate and supplemented with 0.1 mM hypoxanthine, 400 nM aminopterin, 0.016 mM thymidine, 10% FBS, and 10% penicillin-streptomycin solution (GIBCO). Cultures were maintained in a humidified atmosphere of 95% air and 5% CO2 at 37°C. The medium was changed every 2 days, and the cultures were split at 1:6 ratios once a week.
Plasmids and constructs.
miR-133a (pEZX-MR03-miR-133a, catalog no. MmiR3445-MR03), scramble (catalog no. CmiR0001-MR03), and AGT 3′-UTR clone (catalog no. RmiT048999-MT01) were purchased from GeneCopoeia (Rockville, MD).
Animal model of CHF.
CHF was induced by ligation of the left coronary artery of male Sprague-Dawley rats weighing ~220–240 g (Sasco Breeding Laboratories, Omaha, NE), which has been described previously (58) and has been extensively used by our laboratory (28, 49–51, 68, 72). Rats were fed and housed according to approved guidelines of the University of Nebraska Medical Center Institutional Animal Care and Use Committee (IACUC), which conform with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. The experimental protocols involved in the animal studies were approved by the University of Nebraska Medical Center IACUC under the provisions of the Animal Welfare Assurance (A3294-01), and experiments were performed following the relevant guidelines and regulations. Rats were randomly assigned to either the sham-operated group or the CHF group and housed according to institutional guidelines. This myocardial infarct model mimics the most common cause of CHF in humans. Left ventricular dysfunction and failure were assessed using hemodynamic and anatomic criteria (27). Transthoracic echocardiography (VEVO 770, Visual Sonics, Toronto, ON, Canada) was performed 6 wk after ligation of the coronary artery under light isoflurane anesthesia in a subset of rats. Before euthanization, left ventricular end-diastolic pressure (LVEDP) and left ventricular pressure (dP/dt) were measured (PowerLab, AD Instruments, Colorado Springs, CO) using a conductance catheter (Mikro-Tip catheter, SPR 524, size: 3.5-Fr, Millar Instruments, Houston, TX) inserted into the left ventricle via the right carotid artery in all the rats. To anatomically assess the extent of CHF, the infarct size of the left ventricle (in %) was estimated by dividing the size of the infected area by the total size of the left ventricle using ImageJ software (NIH) in all the rats. Rats with elevated LVEDP (>15 mmHg), an infarct size of >35% of the total left ventricular wall, and significant reductions in dP/dtmax were considered to be in CHF. Experiments were performed on the rats 8–10 wk after coronary artery ligation.
Micropunch of the PVN area.
Rats were euthanized by an overdose of pentobarbital (65 mg/kg ip), and the brain was removed and swiftly frozen on dry ice. Six serial coronal sections (100 μm) were cut using a cryostat. The Palkovits and Brownstein (46) technique was used to punch the PVN area bilaterally using a diethylpyrocarbonate-treated blunt 18-gauge needle attached to a syringe, as previously described (28, 37, 58).
Real-time PCR.
Total RNA for real-time PCR was isolated from PVN punches by the Tri Reagent (Molecular Research Center) method (57, 58) and from NG108 cells using a Direct-zol RNA MiniPrep Kit (Zymo Research) as per the manufacturers' instructions. After the extraction of RNA, samples underwent reverse transcription using a Bio-Rad iScript cDNA Synthesis Kit (catalog no. 170-8841) as per the manufacturer's instructions. Briefly, the reaction was performed with priming at 25°C for 5 min, reverse transcription at 42°C for 5 min, and inactivation at 85°C for 5 min. Real-time measurements were made using the iCycler iQ Multicolor Real-Time Detection System with output to a computer-based acquisition system (Bio-Rad). The protocol comprised denaturation (95°C for 10 min), amplification and quantification repeated 45 times (95°C for 1 min, 55°C for 30 s), denaturation at 72°C for 1 min, reannealing at 55°C for 1 min, and a melt curve (55–95°C with a heating rate of 0.5°C per 10 s). The reaction mixture consisted of SYBR Green Supermix (Bio-Rad), 10 nM forward primer, 10 nM reverse primer, nuclease-free H2O, and the cDNA template of interest. The sequences of primers used for real-time PCR are shown in Table 1. Relative expression of genes was calculated using the comparative cycle threshold (2−ΔΔCT) method, which relates the expression of the target gene to the expression of a reference gene (38).
Table 1.
Real-time PCR primer sequences
Analysis of miRNA.
For the miRNA assay, total RNA was isolated from the PVN punches as described above (57, 58). The quality of RNA was checked by NanoDrop 2000c (Thermo Scientific, Waltham, MA), and RNA with 260/280 > 1.8 and 260/230 > 1.8 were used for the miR-133a assay using TaqMan probes. miRNA RT-PCR followed by Taqman quantitative PCR (qPCR) were performed using miR-133a (assay ID 002246) and U6 small nuclear (sn)RNA (assay ID 001973, Life Technologies)-specific primers following the manufacturer's instructions as previously published (42). The miRNA PCR mixtures were amplified with the Bio-Rad CFX qPCR instrument, and data were analyzed by integrated Bio-Rad CFX Manager 3.0 software (Bio-Rad).
Western blot analysis of AGT and ANG II type 1 receptor.
Western blot analysis was performed from PVN punches and NG108 cells as previously described (56). Briefly, lysates were prepared by homogenizing the tissue/cells in 100-μl radioimmunoprecipitation assay buffer (no. 9806, Cell Signaling Technology) containing complete protease inhibitor cocktail (no. 5871, Cell Signaling Technology). Protein lysates (20–30 μg) were fractionated on a 7.5% polyacrylamide gel. The fractionated proteins were subsequently transferred to a polyvinylidene difluoride membrane (Millipore). Blockade of the membranes was performed using nonfat dry milk [5% (wt/vol)] in Tris-buffered saline with Tween 20 (10 mM Tris, 150 mM NaCl, and 0.05% Tween 20) at ambient temperature for 1 h. The membrane was then incubated with the appropriate primary antibody overnight [anti-AGT (rabbit, ab108334, Abcam), anti-ANG II type 1 receptor (anti-AT1R; rabbit, ab18801, Abcam), and anti-actin (mouse, sc-47778, Santa Cruz Biotechnology)] followed by the corresponding peroxidase-conjugated secondary antibody for 1 h. An enhanced chemiluminescence substrate (Pierce Chemical, Rockford, IL) was used to visualize the signals, which were detected by a Worklab digital imaging system. ImageJ (NIH) was used to quantify the signal. The expression of the protein was calculated as the ratio of the intensity of the target protein to the intensity of the housekeeping β-actin band.
Transient transfection of NG108 cells.
Transient transfection was performed in NG108 cells (2 × 105 cells) at 60–80% confluences using Lipofectamine 3000 (Invitrogen, Carlsbad, CA) as per the manufacturer's instructions. Briefly, miR-133a plasmid or scramble was mixed with 150 μl Opti-MEM (Invitrogen). In a second tube, 10 μl Lipofectamine was mixed with 150 μl Opti-MEM and incubated at room temperature for 5 min. These two solutions were gently mixed and then incubated for 30 min at 37°C. This lipid-DNA formulation was layered on cells. After 4 h of incubation, cells were replated into the complete growth medium with FBS, and RNA was then isolated after 48 h posttransfection as described above.
3′-UTR reporter luciferase assay for miR-133a.
NG108 cells were seeded at 2 × 105 cells/well in six-well plates containing 2 ml of complete growth media without antibiotics. All transfections were optimized and performed using Lipofectamine 3000 transfection reagent as per the manufacturer's instructions and as outlined above. The cotransfection was performed with 1 μg of AGT (NM_134432.2) reporter gene construct (hLuc/hRLuc) containing the 3′-UTR of the AGT gene promoter, which was custom cloned by GeneCopoeia in pEZX-MT01 vector, and 0, 1, and 2 μg of miR-133a plasmid or scrambled control. After 48 h of incubation, cells were washed with PBS, and a luciferase reporter assay was performed using the Dual-Glow Luciferase Assay System (catalog no. E2920, Promega) following the manufacturer's instructions (6). The experiment was performed five times, and the results are presented as means ± SE.
Immunocytochemistry.
In vitro immunocytochemistry staining was performed for AGT expression in NG108 cells transfected with miR-133a or scrambled plasmid using the standard protocol. Briefly, adherent cells grown on laminin-coated 6-mm Transwell-Clear inserts (Costar, Corning) were incubated with primary antibody against AGT (1–5 μg/ml) overnight at 4°C, after fixation (4% paraformaldehyde, 10 min), permeabilization (0.2% Triton X-100, 20 min), and blocking (10% goat serum, 1 h). On the next day, cells were incubated with fluorescence-conjugated secondary antibodies for 1 h at room temperature. Coverslips were then mounted onto frosted glass microscope slides using Fluoromount G (Southern Biotechnology) and observed under Olympus fluorescence microscope with the corresponding filters. Images were captured and subsequently analyzed by ImageJ software (NIH).
Lentivirus injections into the PVN.
Eight weeks after ligation surgery, lentiviral vectors carrying miR-133a (LV-miR-133a) or green fluorescent protein (LV-GFP; as a control vector) were injected bilaterally into the PVN by microinjection (1 × 108 plaque-forming units/ml, 100 nl) under the guidance of a stereotaxic instrument (Kent Scientific) in urethane (0.75 g/kg ip)- and α-chloralose (70 mg/kg ip)-anesthetized rats. The following four groups of rats were used in this study: sham-LV-GFP, sham-LV-miR-133a, CHF-LV-GFP, and CHF-LV-miR-133a. Experiments were performed 2–3 wk after viral injections.
Renal sympathetic nerve activity recordings.
For general surgery for hemodynamic and renal sympathetic nerve activity (RSNA) measurements, rats were anesthetized with urethane (0.75 g/kg ip) and α-chloralose (70 mg/kg ip). The left femoral vein was cannulated with polyethylene tubing (PE-50) for injection of supplemental anesthesia. RSNA was monitored in four groups of rats, namely, sham-LV-GFP, sham-LV-miR-133a, CHF-LV-GFP, and CHF-LV-miR-133a, as previously described (35, 36, 58, 71). RSNA recorded at the end of the experiment (after cutting the central end) was defined as background noise. Basal nerve discharge was defined by subtraction of the background noise from the actual nerve discharge before administration of drugs into the PVN. The value of RSNA was calculated by subtracting the background noise from the actual recorded value, and changes found in the integrated nerve discharge during the experiment were expressed as a percentage of basal value. The peak response of RSNA to the administration of drugs into the PVN during the experiment (averaged over 20–30 s) was subsequently expressed as the percent change from baseline. The maximum RSNA was determined by temporary occlusion (30–60 s) of the trachea at the end of the experiment (19). Previously, nasopharyngeal stimulus has been demonstrated to evoke a similar large response that is highly reproducible (3). Using this stimulus, where mean RSNA can increase severalfold, it has been estimated that resting nerve activity may only comprise the activation of 10–20% of the nerves in the bundle (40), consistent with the temporary occlusion technique used in this protocol. There was no significant difference in maximum RSNA between the groups. The left femoral artery was also connected via a pressure transducer (Gould P23 1D) to a computer-based data-recording and -analyzing program (PowerLab) to record mean arterial blood pressure (MAP) and heart rate (HR) by previously established methods (36, 58).
Microinjections into the PVN.
The anesthetized rat [urethane (0.75 g/kg) and α-chloralose (70 mg/kg ip)] was placed in a stereotaxic apparatus (David Kopf Instruments, Tujanga, CA). The bregma was exposed after a longitudinal incision was made on the head, and a small burr hole was made in the skull to allow access to the PVN for the placement of microinjection cannulas. The coordinates for the PVN, determined with the Paxinos and Watson atlas (51a), were 1.5 mm posterior to the bregma, 0.4 mm lateral to the midline, and 7.8 mm ventral to the dura. A thin needle (0.2-mm outer diameter) connected to a 0.5-ml microsyringe (Hamilton, Reno, NV) was lowered into the PVN. ANG II was injected into the PVN in two doses (100 and 200 pmol) in random order. Succeeding injections were made at least 20 min after the prior dose to allow MAP, HR, and RSNA to return to basal levels.
Statistical analyses.
Data are expressed as means ± SE. Differences between groups and among groups were assessed by t-test and one- or two-way ANOVA followed by a Holm-Sidak multiple-comparison test for post hoc analysis of significance (Prism 7, GraphPad Software). P values of <0.05 were indicative of statistical significance.
RESULTS
3′-UTR AGT mRNA is targeted by miR-133a.
The 3′-UTR of AGT (Fig. 1A) was uploaded into multiple target prediction databases (e.g., TargetScan and miRDB). AGT 3′-UTR binding to miR-133a is conserved among several species (Fig. 1B). The predicted consequential pairing of the 3′-UTR sequence of rat and mouse AGT and miR-133a is shown in Fig. 1C. Since miRNAs bind to the nucleotide sequence located in the 3′-UTR of mRNA, it appears to modulate its expression by either posttranscriptional or posttranslational mechanisms. We further validated the putative direct binding of miR-133a to the 3′-UTR of AGT mRNA through a dual-luciferase reporter assay. NG108 cells were cotransfected with reporter gene construct (hLuc/hRLuc) containing the 3′-UTR of AGT and increasing doses of miR-133a mimic (pEZX-MR03-miR-133a) or scrambled plasmid. The results of the luciferase assay revealed that the activity of firefly luciferase in the miR-133a-transfected group was downregulated to 50% at 1 μg and 63% at 2 μg compared with 0 μg control (both P < 0.005; Fig. 1D), respectively, whereas no significant changes were observed in the scramble-transfected group. Taken together, these results suggest that AGT is directly regulated by miR-133a.
Fig. 1.

A: 3′-untranslated region (3′-UTR) of angiotensinogen (AGT). B: AGT 3′ UTR:microRNA (miR)-133a-binding prediction among species. C: schematic diagram of in silico analysis of putative miR-133a binding sites within the 3′-UTR of AGT. CD, coding sequence; MRE, microRNA response element; rno, Rattus norvegicus; mmu, Mus musculus. D: luciferase assay to demonstrate the binding of miR-133a on the 3′-UTR of AGT in NG108 cells transfected with the 3′-UTR clone of AGT (1 μg) and miR-133a overexpression plasmid/scramble (Scr) with different doses (0–2 μg). Changes in luciferase expression were measured using the Dual-Glo Luciferase Assay System and are shown as changes in relative luciferase activity. Values are means ± SE of quadruplicate analyses. *P < 0.05 vs. the scrambled control.
AGT expression is reduced by miR-133a.
Since miR-133a selectively binds to the 3′-UTR of AGT, we next wanted to examine whether miR-133a overexpression affects AGT levels in NG108 cells, which have endogenous expression of AGT, renin, and AT1R. To perform this experiment, we expressed miR-133a in NG108 cells using GFP-tagged miR-133a mimic plasmid (Fig. 2A) and analyzed AGT and AT1R mRNA levels (Fig. 2B). As expected, the AGT mRNA level was reduced by 1.4-fold compared with the scrambled control. Interestingly, the AT1R mRNA level was also reduced by 1.5-fold. Furthermore, a 42% reduction in AGT levels (Fig. 2C) was observed in NG108 cells overexpressing miR-133a. Immunocytochemistry imaging also revealed a decreased AGT level in miR-133a-overexpressing cells, as shown by solid arrowheads, compared with neighboring nontransfected cells (Fig. 2, D and E).
Fig. 2.

Transient transfection of miR-133a overexpression plasmid decreases AGT expression in NG108 cells. A: representative pictures of green fluorescent protein (GFP)-tagged miR-133a overexpression plasmid transfection after 24 h in NG108 cells. GFP, green; bright field, gray. B: real-time PCR of AGT and ANG II type 1 receptor (AT1R) relative to actin in NG108 cells transfected with miR-133a overexpression plasmid. C: Western blot analysis of AGT and actin in NG108 cells transfected with miR-133a overexpression plasmid. Values are means ± SE of four independent experiments. D: immunocytochemical staining of AGT in NG108 cells transfected with GFP-miR-133a overexpression plasmid or scrambled control. AGT, red; nuclei stained by 4′,6-diamidino-2-phenylindole (DAPI), blue; GFP, green. Solid arrowheads represent miR-133a-overexpressing cells, and open arrowheads represent neighboring nontransfected cells. Scale bars = 50 μm. E: cumulative data of AGT protein expression intensity in arbitrary units in NG108 cells transfected with miR-133a overexpression plasmid or scrambled control. *P < 0.05 vs. the scrambled control.
Characterization of CHF model.
Table 2 shows the salient morphological and hemodynamics characteristics of the sham and CHF groups used in the present studies. The hearts of the CHF group showed an average infarcted area of ~38% of the endocardial surface, whereas there was no visible damage to the myocardium in the sham group. LVEDP was significantly elevated in the CHF group compared with the sham group; dP/dtmax was significantly decreased in the CHF group compared with the sham groups, indicating reduced contractility. Taken together, the >30% infarct size, increased LVEDP, and decreased dP/dtmax indicate that rats in the CHF group were experiencing cardiac dysfunction.
Table 2.
Baseline characteristics of sham-operated and CHF rats
| Sham | CHF | |
|---|---|---|
| Body weight, g | 386.7 ± 13.5 | 386.7 ± 7.7 |
| Lung weight, %body wt | 0.4 ± 0.02 | 0.6 ± 0.07* |
| Infarct size, %left ventricle | 0 | 37.9 ± 2.0* |
| MAP, mmHg | 113.5 ± 3.3 | 100.4 ± 2.4 |
| HR, beats/min | 363.8 ± 12.8 | 317.2 ± 7.4 |
| LVEDP, mmHg | 3.9 ± 0.6 | 21.4 ± 2.7* |
| dP/dtmax, mmHg/s | 8,796.7 ± 670.4 | 4,966.3 ± 361* |
| dP/dtmin, mmHg/s | −7,686.8 ± 539.4 | −4,323 ± 526.1* |
Values are means ± SE; n = 6 rats/group. CHF, congestive heart failure; MAP, mean arterial pressure; HR, heart rate; LVEDP, left ventricular end-diastolic pressure; dP/dt, change in pressure over change in time.
P < 0.05 vs. sham.
AGT expression in the PVN.
AGT expression at the transcriptional level was determined by real-time PCR in the PVN of CHF and sham rats (Fig. 3). The relative AGT mRNA level was appreciably higher: 12-fold in PVN tissues from the CHF group compared with the sham group (Fig. 3A). Consistent with the increased mRNA level, AGT protein expression in the PVN is also enhanced 2.5-fold in CHF rats compared with sham rats (Fig. 3B).
Fig. 3.

A: mRNA expression of AGT relative to actin in the paraventricular (PVN) measured by real-time PCR in sham and congestive heart failure (CHF) groups. B: representative Western blot and cumulative data of AGT expression in the PVN. C: miR-133a expression relative to U6 small nuclear (sn)RNA in the PVN. D: mRNA expression of AGT and AT1R relative to actin in the PVN measured by real-time PCR in sham and CHF groups. Values are means ± SE of analyses on 6−8 rats in each group. *P < 0.05 vs. sham.
Expression of miR-133a in the PVN.
Estimation of miR-133a showed a 1.92-fold decrease in the expression of miR-133a in the PVN of CHF rats compared with sham rats (Fig. 3C). This suggests that perhaps because of limited availability of miR-133a, there is decreased binding of miR-133a to the 3′-UTR of the AGT transcript, which may, in turn, lead to increased AGT expression in the PVN of the CHF group, eventually resulting in elevated ANG II levels. Additionally, to validate and corroborate our previous observations, qPCR was performed in the same PVN lysates used for AT1R (Fig. 3D). Consistent with our previous findings of increased AT1R expression (70), we observed increased AT1R expression in the PVN of CHF rats (Fig. 3D). These data indicate that there is an increased angiotensinergic mechanism with a concomitant decrease in miR-133a in the PVN of CHF rats.
Overexpression of miR-133a in the PVN.
To determine whether the overexpression of miR-133a downregulates AGT expression in vivo, we measured AGT (Fig. 4A) and AT1R expression (Fig. 4B) after lentiviral transduction of miR-133a within the PVN. The results showed that in the CHF-LV-miR-133a group, relative AGT expression was significantly downregulated compared with the CHF-LV-GFP (0.34 ± 0.02 vs. 0.55 ± 0.05) group but it was not different from the sham-LV-GFP (0.34 ± 0.04) or sham-LV-miR-133a (0.37 ± 0.03) groups. These data show that restoration of miR-133a within the PVN of rats with CHF was able to abrogate the enhanced expression of AGT in the PVN. Similar to AGT, the significantly increased AT1R expression in the CHF-LV-GFP (0.88 ± 0.05) group compared with the sham-LV-GFP (0.61 ± 0.06) group was attenuated in the CHF-LV-miR-133a group (0.64 ± 0.07), possibly because of the downstream effect of reduced AGT leading to decreased ANG II formation. ANG II has been documented to increase AT1R expression in the PVN (67). Basal levels of RSNA, MAP, HR, and hemodynamic characteristics are shown in Table 3. LVEDP was improved very slightly but was not statistically different; however, dP/dtmax was significantly improved in rats of the CHF-LV-miR-133a group compared with the CHF-LV-GFP group. An illustration of raw recordings of baseline RSNA, MAP, and HR and those after ANG II (200 pmol) microinjection in the PVN of the four groups is shown in Fig. 5A. Basal RSNA (calculated as the percentage of maximum or absolute change) was significantly higher in CHF rats compared with sham rats (Fig. 5B). Lentiviral transduction of miR-133a significantly reduced basal RSNA in the CHF-LV-miR-133a group compared with the CHF-LV-GFP group. Interestingly, the sham-LV-miR-133a group also showed a trend toward a significant decrease in basal RSNA compared with the sham-LV-GFP group. Furthermore, the administration of ANG II into the PVN elicited an increase in RSNA, MAP, and HR in sham-LV-GFP, CHF-LV-GFP, sham-LV-miR-133a, and CHF-LV-miR-133a rats (Fig. 6, A–C) at two different doses. These responses were significantly enhanced in the CHF-LV-GFP group. Lentiviral transduction of miR-133a significantly reduced RSNA (10 ± 1 vs. 16 ± 1%, P < 0.05), MAP (6 ± 1 vs. 10 ± 2 mmHg, P < 0.05), and HR (7 ± 1 vs. 14 ± 1 beats/min, P < 0.05) responses to 100 pmol ANG II in the CHF-LV-miR-133a group compared with the CHF-LV-GFP group. There were no significant differences in any of the parameters monitored between the sham-LV-GFP and sham-LV-miR-133a groups.
Fig. 4.

Upregulation of miR-133a decreases AGT and AT1R expression in the PVN of CHF rats. A: representative AGT gel (top) and cumulative data of AGT protein expression normalized to β-actin (bottom). B: representative AT1R gel (top) and quantification of densitometry analysis of AT1R normalized to β-actin (bottom) in PVN punches of sham-LV-GFP, sham-LV-miR-133a, CHF-LV-GFP, and CHF-LV-miR-133a. Values are means ± SE of analyses on 4−5 rats in each group. *P < 0.05 vs. sham-LV-GFP; #P < 0.05 vs. CHF-LV-GFP.
Table 3.
Baseline characteristics of sham-LV-GFP, sham-LV-miR-133a, CHF-LV-GFP, and CHF-LV-miR-133a rats
| Sham-LV-GFP | CHF-LV-GFP | Sham-LV-miR-133a | CHF-LV-miR-133a | |
|---|---|---|---|---|
| Body weight, g | 386.7 ± 6.2 | 380.6 ± 5.1 | 384.1 ± 6.0 | 376.4 ± 5.3 |
| Infarct size, %left ventricle | 0 | 34.9 ± 1.9* | 0 | 34.3 ± 2.0 |
| MAP, mmHg | 86.6 ± 8.3 | 88.3 ± 6.9 | 83.5 ± 4.1 | 84.7 ± 3.4 |
| HR, beats/min | 262.4 ± 15.3 | 319.2 ± 30.4 | 285.6 ± 5.6 | 288.0 ± 25.4 |
| LVEDP, mmHg | 4.5 ± 0.4 | 13.6 ± 2.3* | 3.1 ± 0.8 | 11.8 ± 2.2 |
| RSNA, ∝V·s | 3.4 ± 0.5 | 4.1 ± 0.6* | 2.6 ± 0.8‡ | 2.7 ± 0.1† |
| dP/dtmax, mmHg/s | 6,537.5 ± 608.5 | 4,284.2 ± 333.4* | 6,993.4 ± 974.8 | 5,868.8 ± 211.9† |
| dP/dtmin, mmHg/s | −5,871.8 ± 563.8 | −4,028.6 ± 169.1* | −6,419.4 ± 978.5 | −4,543.6 ± 261.8 |
Values are means ± SE; n = 5 rats/group. RSNA, renal sympathetic nerve activity.
P < 0.05 vs. the sham-LV-GFP group;
P < 0.05 vs. the CHF-LV-GFP group;
P = 0.09 vs. the sham-LV-GFP group.
Fig. 5.

A: segment of an original recording from individual rats demonstrating the starting baseline parameters and peak changes in renal sympathetic nerve activity (RSNA), mean arterial pressure (MAP), and heart rate (HR) after microinjection of ANG II (200 pmol) into the PVN of the following four groups of rats: sham-LV-GFP, sham-LV-miR-133a, CHF-LV-GFP, and CHF-LV-miR-133a. B: basal RSNA calculated as a percentage of maximum. Int., integrated; bpm, beats/min. *P < 0.05 vs. sham-LV-GFP; #P < 0.05 vs. CHF-LV-GFP.
Fig. 6.
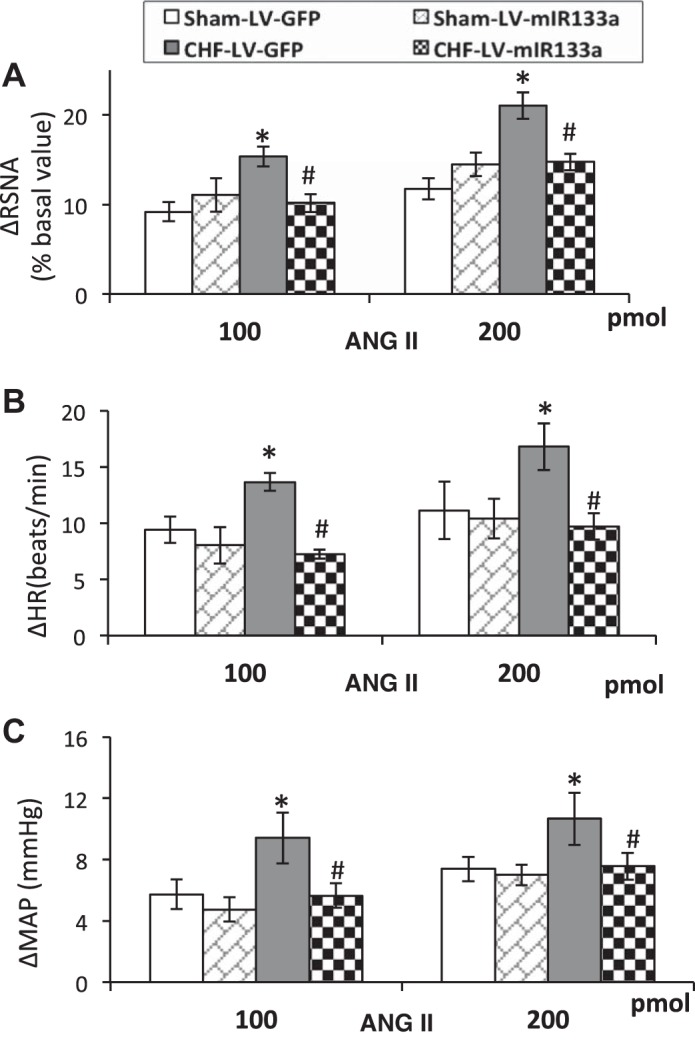
Mean changes in functional responses to microinjection of increasing doses of ANG II into the PVN in sham-LV-GFP, sham-LV-miR-133a, CHF-LV-GFP, and CHF-LV-miR-133a. A: RSNA. B: MAP (in mmHg). C: HR (in beats/min). For RSNA, values represent the percent change from baseline. The groups after ANG II microinjection are analyzed as separate groups. Values are means ± SE of 5−6 rats in each group. *P < 0.05 vs. sham-LV-GFP; #P < 0.05 vs. CHF-LV-GFP.
DISCUSSION
The novel finding of this study is that miR-133a downregulates AGT expression in the PVN during CHF, posttranscriptionally. Our in silico analysis of the 3′-UTR of AGT and the luciferase reporter assay validated miR-133a binding to the 3′-UTR of AGT. Lentiviral transduction of miR-133a significantly reduced the basal level of RSNA as well as RSNA, MAP, and HR responses to ANG II microinjections within the PVN of rats with CHF. This study shows that altered miR-133a at the level of the PVN may contribute to the increased angiotensinergic tone and subsequent increased sympathetic drive commonly observed during CHF.
In the present study, we used the coronary artery ligation model of CHF, which is a reliable and consistent simulation of the CHF condition (49–51, 68, 72) and has advantages over other models of CHF, such as ventricular pacing. Ligation of the coronary artery simulates the blockage of an artery, as commonly seen in patients with CHF. To delineate the molecular pathways and further substantiate our in vivo whole animal results in a controlled situation, we used the NG108 cell line for in vitro studies. NG108 cells have neuronal properties, are immunoreactive to renin, and have AGT, ACE, and AT1R and ANG II type 2 receptor subtypes (13, 32), making this cell line an appropriate surrogate in vitro model for our studies (57, 58, 72).
Cardiovascular disorders represent one of the leading causes of human morbidity and mortality, underscoring the need for innovative new therapies and diagnostics. Increased levels of ANG II exert effects on the CNS in pathological states such as heart failure (70), hypertension (65), and diabetes (20) to cause an increase in sympathoexcitation (36). These physiological and pathological actions of ANG II are thought to be mediated mainly through AT1R. Activation of AT1R has been suggested to instigate hypertension and myocardial infarction, two predominant conditions leading to CHF. Previously, we have demonstrated that altered expressions of genes for the RAS are associated with changes in renal sympathetic nerve discharge (48, 70). As a corollary, ANG II receptor blockers (36) as well as ACE inhibitors (24) have been shown to be effective in reducing sympathoexcitation during CHF, thus suggesting that any molecular mechanism that activates or deactivates the RAS may be essential in the regulation of sympathoexcitation in normal and disease conditions. Previously, Wang et al. (66) demonstrated a role for central AGT in the development of sympathetic hyperactivity, left ventricular remodeling, and dysfunction after myocardial infarction using transgenic rats expressing an antisense RNA against AGT. The production of ANG II in the brain is reduced in these transgenic rats, supporting the concept that AGT-mediated production of ANG II centrally plays a critical role in the regulation of sympathoexcitatory effects (21, 66).
Present in almost all eukaryotic cells, miRNAs are noncoding, regulatory RNAs (1). More than one-third of the protein-coding genes in the human genome are regulated by miRNAs (39). They are known as endogenous negative modulators of gene expression acting posttranscriptionally by promoting the degradation of target mRNA or inhibiting translation (30). They were formerly identified as moderate biological modifiers, but now miRNAs have emerged as potent regulators of different cellular mechanisms with an important potential role in disease management (62). Microarray analysis and other online tools have enabled researchers to correlate the dysregulation of miRNAs and the progression of various diseases in both animals and humans (62). A number of studies have shown the expression of miRNA in neurons and distinct expression patterns within the developing CNS, implying the potential importance of miRNA in brain function and development (29, 55).
Despite growing evidence that miRNAs play a significant role in brain development and various brain disorders, the role of miRNA in the regulation of the RAS centrally is very limited and unclear (22). By combining microarray expression profiling and miR-133a-specific qPCR, we found the expression of miR-133a in the PVN. Furthermore, bioinformatics analysis of the 3′-UTR of AGT has revealed the consensus binding site for miR-133a, and a luciferase reporter assay and in vitro studies in NG108 cells further confirmed that miR-133a regulates AGT (Figs. 1C and 2). Consistent with this concept, we found decreased levels of miR-133a with a concomitant increase in AGT expression in the PVN of CHF rats, suggesting a potentially crucial role for miR-133a in the regulation of AGT expression within this site in the brain. Since the neuronal synthesis of AGT and intraneuronal formation of ANG II, particularly in hypothalamic nuclei such as the PVN (17, 54), are potentially critical for sympathetic outflow, thus miR-133a may be a vital regulator of AGT expression that is altered in various cardiovascular disease conditions. This study provides insights into the possible molecular mechanism by which AGT gene expression is regulated in a key site, the PVN, in the CHF condition. The question as to whether other cell types, such as glial and inflammatory microglia, which also synthesize AGT within the CNS, are affected remains to be explored.
Intriguingly, upregulation of miR-133a in NG108 cells caused a significant reduction in AGT as well as AT1R mRNA levels. Since NG108 cells harbor a complete RAS including renin, perhaps the activation of intracellular renin may have generated ANG II locally via AGT cleavage intracellularly (13) and reduced AT1R levels after miR-133a upregulation. It is likely that the downstream effect of decreased ANG II levels is a consequence of less availability of AGT, but there is a possibility of a yet-unidentified direct effect of miR-133a on AT1R expression that remains to be determined.
It is of interest to note that miR-133a was originally thought to be expressed in the heart and skeletal muscle only but now it is established that this miRNA was also expressed in the CNS and thus categorized into CNS-heart-shared-specific miRNAs (31, 60). In the heart, miR-133a has also been shown to regulate genes in the β1-adrenergic receptor signaling cascade (5) and cardiac remodeling and is considered to be cardioprotective because it alleviates hypertrophy (4) and fibrosis (8). Recently, we reported that miR-133a is present in the neural tissue of diabetic hearts and regulates norepinephrine biosynthesis at the neural-cardiac junction that stimulates β-adrenergic receptors to elicit cardiac contractility (44). However, the molecular mechanisms by which miR-133a transcription is regulated in the CNS or heart are not known. Perhaps an activated cytokine system (15, 18) or increased levels of ANG II (28, 73) (via a feedforward mechanism) in CHF may contribute toward a decrease in transcription of miR-133a in the PVN, a possibility that remains to be elucidated.
The tonic activity of the RAS is dependent on the circulating concentrations of AGT. The binding of renin to intracellular (pro)renin receptors (PRRs) in presympathetic neurons of the PVN (34) plays a pivotal role in brain ANG II formation via AGT, which is subsequently secreted into the extracellular space. Alternatively, extracellular prorenin binds to PRRs on the neuronal membrane and metabolizes extracellular AGT secreted by astrocytes or neurons to generate ANG II (69). AGT abundance is regulated at the transcriptional level through changes in cardiovascular and electrolyte status (7) and via hormonal and cell type-specific regulators (2) in a graded fashion. Human (h)AGT alleles have been associated with hypertensive phenotypes through a mechanism that may involve enhanced transcription. C/A polymorphism at nucleoside 11525 located in the 3′-UTR is modestly associated with increased blood pressure, and binding of miR-31 and miR-584 downregulates the hAGT levels, suggesting a new therapeutic approach to ameliorate hypertension (43). In the present study, the lentivirus-mediated restoration of miR-133a in the PVN during CHF resulted in AGT downregulation (Fig. 4A). Although the 3′-UTR and miR-133a seed sequences are identical in the mouse and rat, this binding site does not match perfectly in humans (Fig. 1B). When miRNA seed sequence correctly binds to the 3′-UTR of an mRNA, it degrades the mRNA. However, in most cases, the miRNA seed sequence does not perfectly bind to the 3′-UTR, and only a few bases of 3′-UTR bind to the seed sequence of miRNA. In this situation, miRNA binding does not allow the translational machinery to pass through the mRNA for protein synthesis, leading to decreased protein synthesis.
The brain RAS is pivotal in the generation of central sympathoexcitation in CHF (36, 67, 74). In the current study, the basal RSNA and RSNA response to PVN microinjection of ANG II were potentiated in rats with CHF. Lentiviral transduction of miR-133a reduced both basal RSNA as well as RSNA responses to ANG II in CHF rats. These data strongly suggest that in the PVN, the endogenous angiotensin system contributes as the driving force for the sympathoexcitation during the CHF condition. The finding that lentiviral transduction of miR-133a reduced sympathetic nerve activity in animals with CHF to a greater extent suggests that miR-133a attenuates the endogenous angiotensinergic tone within the PVN. This may be via downregulation of the expression for AGT by miR-133a. Conversely, silencing of AGT via small interfering RNA or antisense within the PVN would further elucidate and confirm the specificity of the relationship between miR-133a and AGT.
Previously, we have shown an enhanced expression of AT1R (70) within the PVN due to increased levels of ANG II in rats with CHF. The evidence presented here suggests that centrally activated AGT expression, probably mediated by decreased levels of miR-133a, is likely involved in increased central levels of ANG II (21, 73), contributing to increased sympathoexcitation commonly observed in cardiovascular diseases such as CHF (Fig. 7). Augmentation of central AGT production with ANG II stimulation might drive further RAS activation in a positive feedforward loop, a possibility that remains to be examined. Understanding the molecular mechanism of miR-133a-mediated inhibition of ANG II levels via targeting AGT in the PVN may provide the basis for the development of new therapeutic agents with enhanced specificity for the treatment of pathophysiological conditions associated with increased sympathoexcitation such as heart failure, diabetes, and hypertension.
Fig. 7.
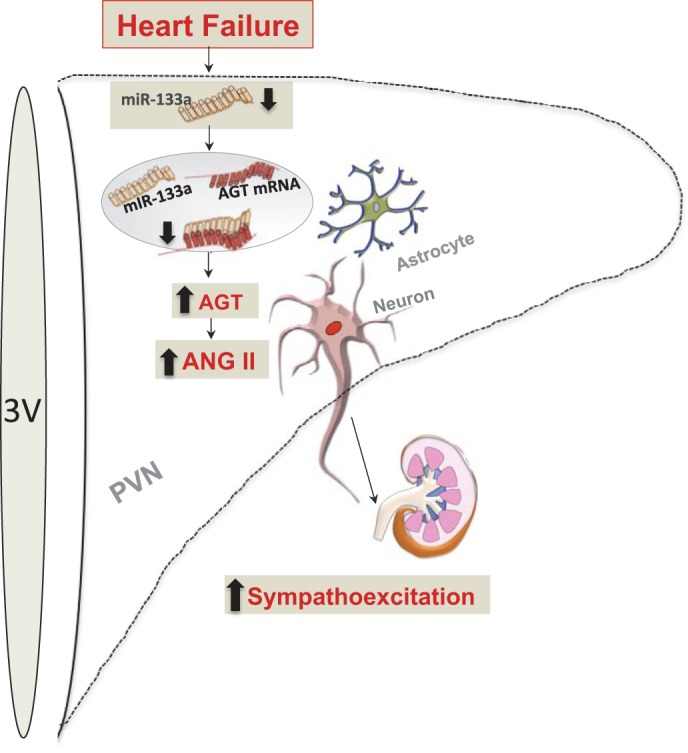
Schematic representing the proposed model for the upregulation of AGT in either an astrocyte or a neuron within the PVN during CHF. 3V, third ventricle. Levels of miR-133a are decreased in the PVN in CHF. The decreased miR-133a leads to decreased binding of miR-133a to the 3′-UTR of AGT, resulting in reduced miR-133a-mediated inhibition of AGT, resulting in increased AGT levels, which might be responsible for the increased ANG II levels in the PVN, contributing to increased neuronal activation, leading to sympathoexcitation.
GRANTS
This work was supported by funding from American Heart Association Grant 14SDG19980007 and National Heart, Lung, and Blood Institute Grants HL-124104 and HL-62222.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
N.M.S., P.K.M., and K.P.P. conceived and designed research; N.M.S., S.S.N., and H.Z. performed experiments; N.M.S., S.S.N., and K.P.P. analyzed data; N.M.S., S.S.N., H.Z., P.K.M., and K.P.P. interpreted results of experiments; N.M.S. and S.S.N. prepared figures; N.M.S., H.Z., and K.P.P. drafted manuscript; N.M.S., H.Z., P.K.M., and K.P.P. edited and revised manuscript; N.M.S., S.S.N., H.Z., P.K.M., and K.P.P. approved final version of manuscript.
ACKNOWLEDGMENTS
The technical assistance of Dr. Xuefei Liu is greatly appreciated.
REFERENCES
- 1.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell : 281–297, 2004. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 2.Brasier AR, Han Y, Sherman CT. Transcriptional regulation of angiotensinogen gene expression. Vitam Horm : 217–247, 1999. doi: 10.1016/S0083-6729(08)60645-7. [DOI] [PubMed] [Google Scholar]
- 3.Burke SL, Head GA. Method for in vivo calibration of renal sympathetic nerve activity in rabbits. J Neurosci Methods : 63–74, 2003. doi: 10.1016/S0165-0270(03)00121-3. [DOI] [PubMed] [Google Scholar]
- 4.Carè A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Høydal M, Autore C, Russo MA, Dorn GW II, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G. MicroRNA-133 controls cardiac hypertrophy. Nat Med : 613–618, 2007. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 5.Castaldi A, Zaglia T, Di Mauro V, Carullo P, Viggiani G, Borile G, Di Stefano B, Schiattarella GG, Gualazzi MG, Elia L, Stirparo GG, Colorito ML, Pironti G, Kunderfranco P, Esposito G, Bang ML, Mongillo M, Condorelli G, Catalucci D. MicroRNA-133 modulates the β1-adrenergic receptor transduction cascade. Circ Res : 273–283, 2014. doi: 10.1161/CIRCRESAHA.115.303252. [DOI] [PubMed] [Google Scholar]
- 6.Castoldi G, Di Gioia CR, Bombardi C, Catalucci D, Corradi B, Gualazzi MG, Leopizzi M, Mancini M, Zerbini G, Condorelli G, Stella A. MiR-133a regulates collagen 1A1: potential role of miR-133a in myocardial fibrosis in angiotensin II-dependent hypertension. J Cell Physiol : 850–856, 2012. doi: 10.1002/jcp.22939. [DOI] [PubMed] [Google Scholar]
- 7.Chang E, Perlman AJ. Multiple hormones regulate angiotensinogen messenger ribonucleic acid levels in a rat hepatoma cell line. Endocrinology : 513–519, 1987. doi: 10.1210/endo-121-2-513. [DOI] [PubMed] [Google Scholar]
- 8.Chen S, Puthanveetil P, Feng B, Matkovich SJ, Dorn GW II, Chakrabarti S. Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes. J Cell Mol Med : 415–421, 2014. doi: 10.1111/jcmm.12218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohn JN, Levine TB, Olivari MT, Garberg V, Lura D, Francis GS, Simon AB, Rector T. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med : 819–823, 1984. doi: 10.1056/NEJM198409273111303. [DOI] [PubMed] [Google Scholar]
- 10.Dalmay T. MicroRNAs and cancer. J Intern Med : 366–375, 2008. doi: 10.1111/j.1365-2796.2008.01926.x. [DOI] [PubMed] [Google Scholar]
- 11.Danser AH. Local renin-angiotensin systems. Mol Cell Biochem : 211–216, 1996. doi: 10.1007/BF00227900. [DOI] [PubMed] [Google Scholar]
- 12.Ferguson DW, Berg WJ, Sanders JS, Kempf JS. Clinical and hemodynamic correlates of sympathetic nerve activity in normal humans and patients with heart failure: evidence from direct microneurographic recordings. J Am Coll Cardiol : 1125–1134, 1990. doi: 10.1016/0735-1097(90)90544-Y. [DOI] [PubMed] [Google Scholar]
- 13.Fishman MC, Zimmerman EA, Slater EE. Renin and angiotensin: the complete system within the neuroblastoma x glioma cell. Science : 921–923, 1981. doi: 10.1126/science.6272392. [DOI] [PubMed] [Google Scholar]
- 14.Francis GS. The relationship of the sympathetic nervous system and the renin-angiotensin system in congestive heart failure. Am Heart J : 642–648, 1989. doi: 10.1016/0002-8703(89)90291-3. [DOI] [PubMed] [Google Scholar]
- 15.Francis J, Chu Y, Johnson AK, Weiss RM, Felder RB. Acute myocardial infarction induces hypothalamic cytokine synthesis. Am J Physiol Heart Circ Physiol : H2264–H2271, 2004. doi: 10.1152/ajpheart.01072.2003. [DOI] [PubMed] [Google Scholar]
- 16.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res : 92–105, 2009. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grobe JL, Xu D, Sigmund CD. An intracellular renin-angiotensin system in neurons: fact, hypothesis, or fantasy. Physiology (Bethesda) : 187–193, 2008. doi: 10.1152/physiol.00002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guggilam A, Haque M, Kerut EK, McIlwain E, Lucchesi P, Seghal I, Francis J. TNF-alpha blockade decreases oxidative stress in the paraventricular nucleus and attenuates sympathoexcitation in heart failure rats. Am J Physiol Heart Circ Physiol : H599–H609, 2007. doi: 10.1152/ajpheart.00286.2007. [DOI] [PubMed] [Google Scholar]
- 19.Guild SJ, Barrett CJ, McBryde FD, Van Vliet BN, Head GA, Burke SL, Malpas SC. Quantifying sympathetic nerve activity: problems, pitfalls and the need for standardization. Exp Physiol : 41–50, 2010. doi: 10.1113/expphysiol.2008.046300. [DOI] [PubMed] [Google Scholar]
- 20.Harrison-Bernard LM, Imig JD, Carmines PK. Renal AT1 receptor protein expression during the early stage of diabetes mellitus. Int J Exp Diabetes Res : 97–108, 2002. doi: 10.1080/15604280214483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang BS, Ganten D, Leenen FH. Responses to central Na(+) and ouabain are attenuated in transgenic rats deficient in brain angiotensinogen. Hypertension : 683–686, 2001. doi: 10.1161/01.HYP.37.2.683. [DOI] [PubMed] [Google Scholar]
- 22.Kemp JR, Unal H, Desnoyer R, Yue H, Bhatnagar A, Karnik SS. Angiotensin II-regulated microRNA 483-3p directly targets multiple components of the renin-angiotensin system. J Mol Cell Cardiol : 25–39, 2014. doi: 10.1016/j.yjmcc.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kimura S, Mullins JJ, Bunnemann B, Metzger R, Hilgenfeldt U, Zimmermann F, Jacob H, Fuxe K, Ganten D, Kaling M. High blood pressure in transgenic mice carrying the rat angiotensinogen gene. EMBO J : 821–827, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kinugawa T, Ogino K, Kitamura H, Saitoh M, Omodani H, Osaki S, Hisatome I, Miyakoda H. Catecholamines, renin-angiotensin-aldosterone system, and atrial natriuretic peptide at rest and during submaximal exercise in patients with congestive heart failure. Am J Med Sci : 110–117, 1996. doi: 10.1016/S0002-9629(15)41774-4. [DOI] [PubMed] [Google Scholar]
- 25.Kishi T, Hirooka Y. Oxidative stress in the brain causes hypertension via sympathoexcitation. Front Physiol : 335, 2012. doi: 10.3389/fphys.2012.00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kishi T, Hirooka Y.. Cardiac complications in hypertension and diabetes: role of sympathetic nervous activity. Curr Hypertens Rev : 274–277, 2013. doi: 10.2174/157340210904140815122944. [DOI] [Google Scholar]
- 27.Kleiber AC, Zheng H, Schultz HD, Peuler JD, Patel KP. Exercise training normalizes enhanced glutamate-mediated sympathetic activation from the PVN in heart failure. Am J Physiol Regul Integr Comp Physiol : R1863–R1872, 2008. doi: 10.1152/ajpregu.00757.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kleiber AC, Zheng H, Sharma NM, Patel KP. Chronic AT1 receptor blockade normalizes NMDA-mediated changes in renal sympathetic nerve activity and NR1 expression within the PVN in rats with heart failure. Am J Physiol Heart Circ Physiol : H1546–H1555, 2010. doi: 10.1152/ajpheart.01006.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krichevsky AM, King KS, Donahue CP, Khrapko K, Kosik KS. A microRNA array reveals extensive regulation of microRNAs during brain development. RNA : 1274–1281, 2003. doi: 10.1261/rna.5980303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet : 597–610, 2010. doi: 10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- 31.Kumar M, Nerurkar VR. Integrated analysis of microRNAs and their disease related targets in the brain of mice infected with West Nile virus. Virology : 143–151, 2014. doi: 10.1016/j.virol.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laflamme L, Brechler V, Reudelhuber TL, Gallo-Payet N, Deschepper CF. The renin-angiotensin system in hybrid NG108-15 cells. Renin gene is from mouse neuroblastoma, angiotensinogen and angiotensin-converting enzyme genes are of rat glioma origin. Regul Pept : 9–15, 1998. [DOI] [PubMed] [Google Scholar]
- 33.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell : 15–20, 2005. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 34.Li W, Peng H, Seth DM, Feng Y. The prorenin and (pro)renin receptor: new players in the brain renin-angiotensin system? Int J Hypertens : 290635, 2012. doi: 10.1155/2012/290635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li YF, Cornish KG, Patel KP. Alteration of NMDA NR1 receptors within the paraventricular nucleus of hypothalamus in rats with heart failure. Circ Res : 990–997, 2003. doi: 10.1161/01.RES.0000102865.60437.55. [DOI] [PubMed] [Google Scholar]
- 36.Li YF, Wang W, Mayhan WG, Patel KP. Angiotensin-mediated increase in renal sympathetic nerve discharge within the PVN: role of nitric oxide. Am J Physiol Regul Integr Comp Physiol : R1035–R1043, 2006. doi: 10.1152/ajpregu.00338.2004. [DOI] [PubMed] [Google Scholar]
- 37.Li YF, Wang Y, Channon KM, Schultz HD, Zucker IH, Patel KP. Manipulation of neuronal nitric oxide synthase within the paraventricular nucleus using adenovirus and antisense technology. Methods Mol Med : 59–79, 2005. [DOI] [PubMed] [Google Scholar]
- 38.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCT) method. Methods : 402–408, 2001. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 39.Mallanna SK, Rizzino A. Emerging roles of microRNAs in the control of embryonic stem cells and the generation of induced pluripotent stem cells. Dev Biol : 16–25, 2010. doi: 10.1016/j.ydbio.2010.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malpas SC, Shweta A, Anderson WP, Head GA. Functional response to graded increases in renal nerve activity during hypoxia in conscious rabbits. Am J Physiol Regul Integr Comp Physiol : R1489–R1499, 1996. [DOI] [PubMed] [Google Scholar]
- 41.Mishra PK, Tyagi N, Kumar M, Tyagi SC. MicroRNAs as a therapeutic target for cardiovascular diseases. J Cell Mol Med : 778–789, 2009. doi: 10.1111/j.1582-4934.2009.00744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mishra PK, Tyagi N, Kundu S, Tyagi SC. MicroRNAs are involved in homocysteine-induced cardiac remodeling. Cell Biochem Biophys : 153–162, 2009. doi: 10.1007/s12013-009-9063-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mopidevi B, Ponnala M, Kumar A. Human angiotensinogen +11525 C/A polymorphism modulates its gene expression through microRNA binding. Physiol Genomics : 901–906, 2013. doi: 10.1152/physiolgenomics.00056.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nandi SS, Zheng H, Sharma NM, Shahshahan HR, Patel KP, Mishra PK. Lack of miR-133a decreases contractility in diabetic hearts: a role for novel cross-talk between tyrosine aminotransferase and tyrosine hydroxylase. Diabetes : 3075–3090, 2016. doi: 10.2337/db16-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Packer M. Neurohormonal interactions and adaptations in congestive heart failure. Circulation : 721–730, 1988. doi: 10.1161/01.CIR.77.4.721. [DOI] [PubMed] [Google Scholar]
- 46.Palkovits M, Brownstein M. Microdissection of brain areas by the punch technique. In: Brain Microdissection Techniques, edited by Cuello AE. Chichester, UK: John Wiley & Sons, 1983, p. 1. –36. [Google Scholar]
- 47.Patel KP. Role of paraventricular nucleus in mediating sympathetic outflow in heart failure. Heart Fail Rev : 73–86, 2000. doi: 10.1023/A:1009850224802. [DOI] [PubMed] [Google Scholar]
- 48.Patel KP, Mayhan WG, Bidasee KR, Zheng H. Enhanced angiotensin II-mediated central sympathoexcitation in streptozotocin-induced diabetes: role of superoxide anion. Am J Physiol Regul Integr Comp Physiol : R311–R320, 2011. doi: 10.1152/ajpregu.00246.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patel KP, Zhang K. Neurohumoral activation in heart failure: role of paraventricular nucleus. Clin Exp Pharmacol Physiol : 722–726, 1996. doi: 10.1111/j.1440-1681.1996.tb01765.x. [DOI] [PubMed] [Google Scholar]
- 50.Patel KP, Zhang K, Kenney MJ, Weiss M, Mayhan WG. Neuronal expression of Fos protein in the hypothalamus of rats with heart failure. Brain Res : 27–34, 2000. doi: 10.1016/S0006-8993(00)02186-7. [DOI] [PubMed] [Google Scholar]
- 51.Patel KP, Zhang PL, Krukoff TL. Alterations in brain hexokinase activity associated with heart failure in rats. Am J Physiol Regul Integr Comp Physiol : R923–R928, 1993. [DOI] [PubMed] [Google Scholar]
- 51a.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Orlando, FL: Academic, 1986. [Google Scholar]
- 52.Perkins DO, Jeffries CD, Jarskog LF, Thomson JM, Woods K, Newman MA, Parker JS, Jin J, Hammond SM. microRNA expression in the prefrontal cortex of individuals with schizophrenia and schizoaffective disorder. Genome Biol : R27, 2007. doi: 10.1186/gb-2007-8-2-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Porter JP, Brody MJ. Neural projections from paraventricular nucleus that subserve vasomotor functions. Am J Physiol Regul Integr Comp Physiol : R271–R281, 1985. [DOI] [PubMed] [Google Scholar]
- 54.Printz MP, Healy DP. Changes in brain angiotensinogen during development of hypertension. Clin Exp Hypertens A : 1037–1046, 1983. [DOI] [PubMed] [Google Scholar]
- 55.Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol : R13, 2004. doi: 10.1186/gb-2004-5-3-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sharma NM, Llewellyn TL, Zheng H, Patel KP. Angiotensin II-mediated posttranslational modification of nNOS in the PVN of rats with CHF: role for PIN. Am J Physiol Heart Circ Physiol : H843–H855, 2013. doi: 10.1152/ajpheart.00170.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sharma NM, Zheng H, Li YF, Patel KP. Nitric oxide inhibits the expression of AT1 receptors in neurons. Am J Physiol Cell Physiol : C1162–C1173, 2012. doi: 10.1152/ajpcell.00258.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sharma NM, Zheng H, Mehta PP, Li YF, Patel KP. Decreased nNOS in the PVN leads to increased sympathoexcitation in chronic heart failure: role for CAPON and Ang II. Cardiovasc Res : 348–357, 2011. doi: 10.1093/cvr/cvr217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun Z, Cade R, Zhang Z, Alouidor J, Van H. Angiotensinogen gene knockout delays and attenuates cold-induced hypertension. Hypertension : 322–327, 2003. doi: 10.1161/01.HYP.0000050964.96018.FA. [DOI] [PubMed] [Google Scholar]
- 60.Tang X, Gal J, Zhuang X, Wang W, Zhu H, Tang G. A simple array platform for microRNA analysis and its application in mouse tissues. RNA : 1803–1822, 2007. doi: 10.1261/rna.498607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tanimoto K, Sugiyama F, Goto Y, Ishida J, Takimoto E, Yagami K, Fukamizu A, Murakami K. Angiotensinogen-deficient mice with hypotension. J Biol Chem : 31334–31337, 1994. [PubMed] [Google Scholar]
- 62.van Rooij E. The art of microRNA research. Circ Res : 219–234, 2011. doi: 10.1161/CIRCRESAHA.110.227496. [DOI] [PubMed] [Google Scholar]
- 63.van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA : 18255–18260, 2006. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vasan RS. Biomarkers of cardiovascular disease: molecular basis and practical considerations. Circulation : 2335–2362, 2006. doi: 10.1161/CIRCULATIONAHA.104.482570. [DOI] [PubMed] [Google Scholar]
- 65.Wang G, Anrather J, Huang J, Speth RC, Pickel VM, Iadecola C. NADPH oxidase contributes to angiotensin II signaling in the nucleus tractus solitarius. J Neurosci : 5516–5524, 2004. doi: 10.1523/JNEUROSCI.1176-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang H, Huang BS, Ganten D, Leenen FH. Prevention of sympathetic and cardiac dysfunction after myocardial infarction in transgenic rats deficient in brain angiotensinogen. Circ Res : 843–849, 2004. doi: 10.1161/01.RES.0000120864.21172.5A. [DOI] [PubMed] [Google Scholar]
- 67.Wei SG, Yu Y, Zhang ZH, Felder RB. Angiotensin II upregulates hypothalamic AT1 receptor expression in rats via the mitogen-activated protein kinase pathway. Am J Physiol Heart Circ Physiol : H1425–H1433, 2009. doi: 10.1152/ajpheart.00942.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu B, Zheng H, Patel KP. Enhanced activation of RVLM-projecting PVN neurons in rats with chronic heart failure. Am J Physiol Heart Circ Physiol : H1700–H1711, 2012. doi: 10.1152/ajpheart.00722.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu Q, Jensen DD, Peng H, Feng Y. The critical role of the central nervous system (pro)renin receptor in regulating systemic blood pressure. Pharmacol Ther : 126–134, 2016. doi: 10.1016/j.pharmthera.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zheng H, Li Y-F, Wang W, Patel KP. Enhanced angiotensin-mediated excitation of renal sympathetic nerve activity within the paraventricular nucleus of anesthetized rats with heart failure. Am J Physiol Regul Integr Comp Physiol : R1364–R1374, 2009. doi: 10.1152/ajpregu.00149.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zheng H, Li YF, Cornish KG, Zucker IH, Patel KP. Exercise training improves endogenous nitric oxide mechanisms within the paraventricular nucleus in rats with heart failure. Am J Physiol Heart Circ Physiol : H2332–H2341, 2005. doi: 10.1152/ajpheart.00473.2004. [DOI] [PubMed] [Google Scholar]
- 72.Zheng H, Liu X, Li Y, Sharma NM, Patel KP. Gene transfer of neuronal nitric oxide synthase to the paraventricular nucleus reduces the enhanced glutamatergic tone in rats with chronic heart failure. Hypertension : 966–973, 2011. doi: 10.1161/HYPERTENSIONAHA.111.176222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zucker IH, Wang W, Pliquett RU, Liu JL, Patel KP. The regulation of sympathetic outflow in heart failure. The roles of angiotensin II, nitric oxide, and exercise training. Ann N Y Acad Sci : 431–443, 2001. doi: 10.1111/j.1749-6632.2001.tb03696.x. [DOI] [PubMed] [Google Scholar]
- 74.Zucker IH, Xiao L, Haack KK. The central renin-angiotensin system and sympathetic nerve activity in chronic heart failure. Clin Sci (Lond) : 695–706, 2014. doi: 10.1042/CS20130294. [DOI] [PMC free article] [PubMed] [Google Scholar]