

Abstract
Corticosteroids (CS) regulate the expression of numerous genes at the mRNA and protein levels. The time course of CS pharmacogenomics and proteomics were examined in livers obtained from adrenalectomized rats given a 50-mg/kg bolus dose of methylprednisolone. Microarrays and mass spectrometry-based proteomics were employed to quantify hepatic transcript and protein dynamics. One-hundred, sixty-three differentially expressed mRNA and their corresponding proteins (163 genes) were clustered into two dominant groups. The temporal profiles of most proteins were delayed compared with their mRNA, attributable to synthesis delays and slower degradation kinetics. On the basis of our fifth-generation model of CS, mathematical models were developed to simultaneously describe the emergent time patterns for an array of steroid-responsive mRNA and proteins. The majority of genes showed time-dependent increases in mRNA and protein expression before returning to baseline. A model assuming direct, steroid-mediated stimulation of mRNA synthesis was applied. Some mRNAs and their proteins displayed down-regulation following CS. A model assuming receptor-mediated inhibition of mRNA synthesis was used. More complex patterns were observed for other genes (e.g., biphasic behaviors and opposite directionality in mRNA and protein). Models assuming either stimulation or inhibition of mRNA synthesis coupled with dual secondarily induced regulatory mechanisms affecting mRNA or protein turnover were derived. These findings indicate that CS-regulated gene expression manifested at the mRNA and protein levels are controlled via mechanisms affecting key turnover processes. Our quantitative models of CS pharmacogenomics were expanded from mRNA to proteins and provide extended hypotheses for understanding the direct, secondary, and downstream mechanisms of CS actions.
Introduction
Corticosteroids (CS) are a class of pleiotropic immunosuppressive agents used in the treatment of various inflammatory and autoimmune diseases, such as asthma (Barnes, 1998) and rheumatoid arthritis (Kirwan and Gunasekera, 2017). Their potent immunosuppressive properties also form the basis for their use in preventing rejection of solid organ transplants (Taylor et al., 2005). Beneficial effects derived from immunosuppression are accompanied by numerous metabolic side effects, which upon long-term steroid usage are manifested as osteoporosis, insulin resistance, diabetes, and obesity (Schacke et al., 2002). The ubiquitously expressed glucocorticoid receptor (GR) is the principal target in tissues mediating both therapeutic and adverse CS outcomes. Upon binding GR, CS produce effects that are either rapid in onset (immune cell trafficking and adrenal suppression) or delayed (genomic regulation of mediators) (Jusko, 1995). Pharmacogenomic CS mechanisms involve a series of intracellular transduction steps, including drug-receptor binding in the cytosol, GR dimerization, nuclear translocation, DNA binding (i.e., gene regulation), and consequent alterations in mRNA and protein expression. Although immune regulation by CS is mediated by both genomic and nongenomic mechanisms (Cain and Cidlowski, 2017), metabolic actions in tissues such as liver are largely receptor/gene-mediated.
Systems pharmacodynamic modeling that integrates “horizontal” and “vertical” aspects of drug actions are critical for gaining quantitative insights into drugs such as CS with complex mechanisms (Jusko, 2013). Since hundreds of steroid-target genes are regulated in an organ, the “horizontal” can be captured by studying large-scale gene expression changes within the tissue. The “vertical” is reflected by the intermediary mechanisms linking CS pharmacokinetics (PK) to resulting pharmacogenomic changes (Ramakrishnan et al., 2002b), and ultimately, to clinically relevant pharmacodynamic (PD) outcomes (Ayyar et al., 2018). We previously examined the entire temporal profiles of about 8000 genes in rat liver after a single 50-mg/kg dose of the synthetic CS methylprednisolone (MPL) (Almon et al., 2003). This led to “vertical” model-based integration of MPL PK, receptor binding, and dynamics, and consequent primary and secondary drug-mediated transcriptional effects, which captured the emergent time patterns for six clusters of CS-responsive mRNAs (143 different genes) (Jin et al., 2003).
Although highly useful in understanding the genomic mechanisms of CS regulation, sole use of transcriptomic approaches are limited in that changes in mRNA expression may not directly correlate with protein expression and, hence, may not fully reflect drug effects (Maier et al., 2009; Payne, 2015). Although the central dogma (Crick, 1970) tightly coupled flow of molecular information from DNA to mRNA to protein, complexities in gene regulation and dynamics arising during transcription, post-transcriptional processing, and translation complicate the interpretation of the relationship between mRNA and protein abundances, especially in mammalian systems. That protein levels at steady-state are primarily determined by their mRNA has been established in experimental systems (Vogel and Marcotte, 2012; Liu et al., 2016b). However, such steady-state dynamics are perturbed upon acute or long-term exposure to biologic or pharmacological stressors (Vogel, 2013; Cheng et al., 2016; Liu et al., 2016b). Since proteins, or protein networks, are more direct mediators of pharmacological actions, integration of -omics information within systems models can yield a deeper understanding of molecular drug actions (Kamisoglu et al., 2017).
Upon examining the relationship of protein and mRNA dynamics in vertebrate embryonic development, Peshkin et al. (2015) demonstrated that mutual information is contained across both scales. A simple model of protein turnover considering mass action kinetics as a basis predicted protein dynamics from mRNA concentrations for a large number of dynamically varying genes. For modeling drug actions, both direct and secondarily regulated mechanisms that alter steady-state mRNA and protein turnover must also be considered (Jin et al., 2003). Secondarily induced gene regulatory mechanisms, such as hormones, cytokines, transcription factors, and microRNAs (miRNA), can impact gene regulation at the transcriptional, post-transcriptional, and translational stages (Jin et al., 2004; Valencia-Sanchez et al., 2006; Cho et al., 2015).
We conducted a time-course animal study similar to our previous microarray studies (Almon et al., 2003; Jin et al., 2003) and applied ion-current-based quantitative nano–liquid chromatography–mass spectrometry (LC/MS) methods to examine the temporal proteomic response of rat liver (Nouri-Nigjeh et al., 2014). The major application for this investigation was to develop mechanism-based PK/PD models that expanded our quantitative “horizontal” and “vertical” assessments of pharmacogenomic MPL actions from the liver transcriptome to the proteome. In this report, our current models of CS gene regulation were expanded from mRNA to proteins, which simultaneously explain emergent time patterns within the liver transcriptome and proteome as observed within our studies.
Materials and Methods
Experimental Procedures
Animal Experiments.
The data for this study were obtained from two sets of animal experiments performed in adrenalectomized (ADX) male Wistar rats (Harlan Sprague Dawley Inc., Indianapolis, IN). Our research protocol adheres to the Principles of Laboratory Animal Care (NIH publication 85–23, revised in 1985) and was approved by the University at Buffalo Institutional Animal Care and Use Committee. The first set of experiments consisted of 43 animals (group 1) given a single intravenous dose of 50 mg/kg of MPL succinate and euthanized at 16 different time points after dosing (0.25, 0.5, 0.75, 1, 2, 4, 5, 5.5, 6, 7, 8, 12, 18, 30, 48, or 72 hours). Four untreated rats were sacrificed at 0 hours as controls. The second set of experiments consisted of 55 animals (group 2) given a single intramuscular dose of 50 mg/kg of the same drug. The livers from group 2 were perfused with cold heparinized saline (5 ml of heparin/1 l saline) before sacrifice to remove blood (as necessary for accurate proteomic profiling) and the animals were euthanized at successive times after dosing (0.5, 1, 2, 4, 5.5, 8, 12, 18, 30, 48, and 66 hours). Five untreated rats were sacrificed at random times as controls. Liver harvested from animals from both experiments were flash frozen in liquid nitrogen and stored at −80°C until further analysis. More information about the animal procedures can be obtained from previous reports (Almon et al., 2002; Nouri-Nigjeh et al., 2014).
Transcriptomics.
Powdered liver (100 mg) from each animal (group 1) was added to 1 ml of TRIzol Reagent (Invitrogen/Thermo Fisher Scientific, Carlsbad, CA). Total RNA extractions were carried out according to manufacturer’s instructions and were further purified by passage through RNeasy columns (QIAGEN, Valencia, CA). RNAs were quantified spectrophotometrically, and purity and integrity were assessed by agarose gel electrophoresis. Isolated RNA from each liver was used to prepare target according to manufacturer’s protocols. The biotinylated cRNAs were hybridized to 47 individual Affymetrix GeneChips Rat Genome U34A (Affymetrix, Inc., Santa Clara, CA) containing 7000 probe sets. More information about this data set can be obtained from Gene Expression Omnibus (GEO) database (GSE490).
Proteomics.
A total of 80 mg of powdered liver from each animal (group 2) was extracted, digested, and analyzed using a nano-LC/MS instrument. The Nano Flow Ultra-High Pressure LC system (nano-UPLC) consisted of a Spark Holland Endurance autosampler and an ultra-high pressure Eksigent Nano-2D nano-LC system, with a LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Waltham, MA) used for detection. Separation was performed on a long column (100 cm long and 50-μm inner diameter) with small particles (Pepmap 2-μm C18, 100 Å) under high pressure (∼9000–11,000 psi with heating at 52°C). The LC/MS raw data were searched against the UniProt-reviewed rat protein database (released October 2012) with 7853 protein entries using SEQUEST-based Proteome Discoverer (version 1.2.0.208; Thermo Fisher Scientific). The false discovery rate was estimated by a target-decoy search strategy, using a concatenated database containing both forward and reversed sequences. Protein quantification used the area under the curve (AUC) of the ion-current peaks as a basis. A more detailed description of the analytical methodology was published (Nouri-Nigjeh et al., 2014; Tu et al., 2014).
Data Mining and Cluster Analysis.
Individual probe-set intensity (microarray data) and protein AUC (proteomics data) were normalized as a ratio to the mean of the controls, which had a distribution around 1. Proteins and transcripts with differential temporal profiles were determined by using the extraction and analysis of gene expression (EDGE) software (Storey et al., 2005; Leek et al., 2006). Within-class differential expression was employed to identify proteins that showed a differential expression profile over time. Only mRNA and proteins that varied significantly over time (P value <0.05 and q-value <0.01) were employed in the subsequent analysis (Kamisoglu et al., 2015; Ayyar et al., 2017). Temporal data for the differentially expressed genes identified at both transcriptomic and proteomic levels were concatenated and subjected to hierarchical clustering using the clustergram function in the Bioinformatics Toolbox of MATLAB (Mathworks, Natick, MA) as described previously (Kamisoglu et al., 2015). Proteins showing similar expression patterns after MPL dosing were identified using a quality-threshold clustering algorithm in GeneSpring 4.1 (Silicon Genetics, Redwood City, CA), employing Pearson’s correlation as the similarity measurement. Common coregulatory proteins and transcription factors were extracted within each cluster on the basis of the informatics analysis using the Ingenuity Pathway Analysis package (IPA; Ingenuity Systems/QIAGEN, Redwood City, CA). Experimentally validated microRNA-target gene interactions in murine and rodent models were extracted from the miRTarBase database (release 7.0) (Chou et al., 2018). Only microRNA-gene interactions confirmed under “strong evidence” (i.e., reporter assays, Western blots, and quantitative polymerase chain reaction) were retained for further analysis. The PubMed database was searched for literature-based evidence of glucocorticoid or CS regulation of the miRNA across species and experimental systems (date accessed: 04/05/2018). The miRNA-mRNA interaction network was visualized using Cytoscape (version 3.6.1) (Shannon et al., 2003).
Pharmacokinetic/Pharmacodynamic/Pharmacogenomic/Pharmacoproteomic Models
MPL Pharmacokinetics.
Plasma concentrations of MPL following intravenous and intramuscular dosing were modeled simultaneously. A two-compartment model with linear elimination was used to describe the biexponential disposition of plasma MPL. In addition, two absorption components from the injection site was used to describe the absorption kinetics of MPL following intramuscular dosing (Hazra et al., 2007c). Equation and initial conditions (IC) describing the model are:
| (1) |
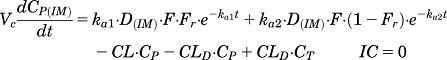 |
(2) |
| (3) |
where C and D represent the concentration and dose of MPL in the corresponding plasma (P) and tissue (T) compartments, Fr and (1 − Fr) are fractions of dose absorbed through the absorption pathways described by first-order rate constants ka1 and ka2, CL is clearance from the central compartment, CLD is the distribution clearance, F is the overall bioavailability of MPL after intramuscular injection, and Vc and VT are the central and peripheral volumes of distribution.
Receptor Dynamics.
The molecular receptor-mediated mechanisms governing CS pharmacodynamics as depicted by our fifth-generation model of receptor dynamics was employed for developing the pharmacokinetic/pharmacodynamic/pharmacogenomic/pharmacoproteomic model. The dynamics of drug-receptor complex and feedback inhibition of receptor mRNA production was used as previously described (Ramakrishnan et al., 2002a). The equations describing the receptor dynamics are:
| (4) |
| (5) |
| (6) |
| (7) |
where symbols represent the free cytosolic glucocorticoid receptor (R), cytosolic drug-receptor complex (DR), nuclear-translocated drug-receptor complex (DRn), and receptor mRNA (GRm) concentrations. The ks,GR and kd,GR are first-order rate constants for the production of free receptor from the translation of GR mRNA and the degradation of the free receptor, kon is the second-order rate constant for formation of drug-receptor complex (DR) by the binding of free ligand and receptor in the cytosol, and kt is the first-order rate constant for translocation of the drug-receptor complex from cytosol (DR) into the nucleus (DRn). Part of DRn may recycle back to the cytosol controlled by the rate constant Rf ⋅kre with the remainder degraded by rate constant (1 − Rf)⋅kre, and ks,GRm and kd,GRm are rate constants for the production and degradation of the receptor mRNA. The IC50,GRm is the concentration of DRn at which the synthesis rate of GR mRNA is reduced to 50% of its baseline.
Equations (4) and (7) yield the following baselines:
| (8) |
| (9) |
where 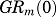 and
and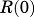 are the baseline values of receptor mRNA and free cytosolic GR density. These baseline values were fixed as the mean values obtained in liver from the control animals (Hazra et al., 2007b). Parameters from our previous report (Hazra et al., 2007a) were used to simulate receptor dynamics and produce the driving force for genomic CS actions in the present study.
are the baseline values of receptor mRNA and free cytosolic GR density. These baseline values were fixed as the mean values obtained in liver from the control animals (Hazra et al., 2007b). Parameters from our previous report (Hazra et al., 2007a) were used to simulate receptor dynamics and produce the driving force for genomic CS actions in the present study.
Pharmacogenomic and Proteomic Models.
The diverse cellular and molecular mechanisms that govern the pharmacodynamic and pharmacogenomic effects of CS are depicted in Fig. 1. Binding of CS with the receptor leads to activation and translocation of the receptor into the nucleus. Activated GR binds to recognition sites known as glucocorticoid response elements (GREs) in the promoter region of target genes and activates or inhibits target gene transcription. Part of the nuclear receptors are recycled back into the cytoplasm after exerting their effects. Furthermore, the CS cause homologous down-regulation of their own receptors via decreased transcription. Growing evidence indicates that CS modulate many transcription factors such as CCAAT-enhancer binding protein (C/EBP) and hepatocyte nuclear factor-1α (HNF-1α), as well as miRNAs (Suh and Rechler, 1997; Phuc Le et al., 2005; Smith et al., 2013; Clayton et al., 2018). The altered expression of these transcription factors and miRNAs can in turn affect the expression of other genes during transcriptional and translational processing of mRNA and peptides (Valinezhad Orang et al., 2014). Secondary effects of CS on other hormones, such as cyclic adenosine monophosphate and insulin, also influence gene regulation (Jin et al., 2004). Since the exact regulatory mechanisms or networks for each CS-responsive gene at the mRNA and protein levels has not been clarified thus far, models in the present report collectively refer to such mediators as secondary regulators. Thus, a target mRNA or protein may be regulated either directly by DRn, or through a secondary regulator, or by both. Various pharmacogenomic models are proposed to describe diverse hepatic mRNA and protein expression profiles following acute MPL dosing. The DRn is assumed as the driving force influencing the dynamics of mRNA expression, which then translates to changes in proteins. In the absence of drug, the expression of mRNA is described by a turnover model with a zero-order production rate (ks,mRNA) and a first-order degradation rate (kd,mRNA), whereas protein turnover is controlled by a first-order production rate (ks,protein) dependent on mRNA expression and with a power coefficient  and a first-order degradation rate (kd,protein):
and a first-order degradation rate (kd,protein):
| (10) |
| (11) |
Since endogenous glucocorticoid production in ADX rats is negligible, steady-state gene expression was assumed before drug administration. The following baseline conditions are derived:
| (12) |
| (13) |
where 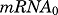 and
and 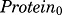 are the baseline targets for mRNA and protein expression levels. Since all data were normalized as ratios to the baseline, their values were fixed to 1, except in some cases in which estimation yielded significant improvement of model fitting. Figure 2 depicts, along with the core fifth-generation CS model, six mathematical models proposed to explain the observed gene expression profiles.
are the baseline targets for mRNA and protein expression levels. Since all data were normalized as ratios to the baseline, their values were fixed to 1, except in some cases in which estimation yielded significant improvement of model fitting. Figure 2 depicts, along with the core fifth-generation CS model, six mathematical models proposed to explain the observed gene expression profiles.
Fig. 1.

General schematic of molecular and cellular mechanisms of corticosteroid action regulating mRNA and protein expression. CBG, corticosteroid-binding globulin; hsp 70/90, heat shock protein 70/90; FKBP, FK506 binding protein; nGRE, negative-glucocorticoid response element; RNAP, RNA polymerase.
Fig. 2.

Pharmacogenomic models for CS effects on mRNA and protein expression via diverse mechanisms. (A–F) Models A to F are defined further in the text [eqs. (14–27)]. The dotted lines and rectangles indicate stimulation (open bar) and inhibition (solid bar) of the various processes via indirect mechanisms.
Model A (Fig. 2A) assumes enhancement of gene transcription by DRn (i.e., stimulation of mRNA production), modeled as
| (14) |
| (15) |
where  is a linear stimulation constant by which
is a linear stimulation constant by which 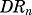 increases the synthesis of the target mRNA.
increases the synthesis of the target mRNA.
Model B (Fig. 2B) assumes repression of gene transcription by DRn (i.e., inhibition of mRNA production), represented as
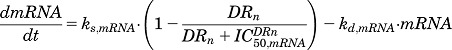 |
(16) |
| (17) |
where 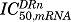 is the concentration of
is the concentration of 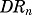 at which mRNA synthesis rate drops to 50% of its baseline value.
at which mRNA synthesis rate drops to 50% of its baseline value.
Genes described by model C (Fig. 2C) were characterized by a stimulatory effect on gene transcription by 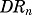 along with delayed, induced mRNA degradation by a secondary regulator. In the proposed and subsequent “dual-effect” models, the secondary regulator (SR) represents
along with delayed, induced mRNA degradation by a secondary regulator. In the proposed and subsequent “dual-effect” models, the secondary regulator (SR) represents  -induced changes of a transcription factor, miRNA, or other mediator from its original baseline. Equations reflecting these joint effects are
-induced changes of a transcription factor, miRNA, or other mediator from its original baseline. Equations reflecting these joint effects are
| (18) |
 |
(19) |
| (20) |
where  is a linear stimulation constant by which
is a linear stimulation constant by which  increases the synthesis of the target mRNA. The intermediate step SR is described in a simplified manner using a linear transduction model (Sun and Jusko, 1998). The SR variable represents the absolute change of regulator level from baseline produced by
increases the synthesis of the target mRNA. The intermediate step SR is described in a simplified manner using a linear transduction model (Sun and Jusko, 1998). The SR variable represents the absolute change of regulator level from baseline produced by 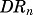 via a first-order rate constant (kSR). The initial condition of eq. (18) was fixed to 0.
via a first-order rate constant (kSR). The initial condition of eq. (18) was fixed to 0.
Model D (Fig. 2D) assumes a combinatorial effect of both DRn and SR on the synthesis of mRNA, where DRn inhibits gene transcription, whereas SR stimulates the same. This model is given by the equations
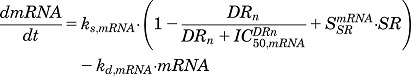 |
(21) |
| (22) |
where the SR is described by eq. (18).
Model E (Fig. 2E) describes the inhibition of gene transcription by DRn occurring in combination with repressed mRNA degradation by a secondary regulator, given by the equations
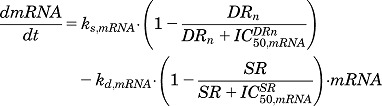 |
(23) |
| (24) |
where the SR is described by eq. (18) and 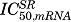 is the concentration of SR when the inhibitory effect of SR on the mRNA reaches half of its maximum.
is the concentration of SR when the inhibitory effect of SR on the mRNA reaches half of its maximum.
Model F (Fig. 2F) characterizes genes which show opposite patterns in their mRNA and protein expression. This model assumes an inhibition of mRNA synthesis by DRn, in combination with inhibition of protein degradation mediated by DR, represented as
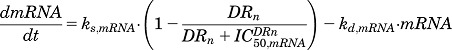 |
(25) |
| (26) |
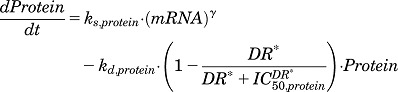 |
(27) |
where the 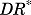 represents the activated intracellular receptor interacting with protein and
represents the activated intracellular receptor interacting with protein and  is the concentration of
is the concentration of 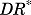 at which the inhibitory effect of
at which the inhibitory effect of 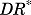 on a target protein reaches half maximum.
on a target protein reaches half maximum.
Data Analysis
Data taken from individual rats (n = 2–4 per time point) were pooled at each time. Mean mRNA and protein time profiles for each gene were modeled simultaneously. Mean transcriptomic and proteomic data were employed for model fitting for practical ease and feasibility in data handling. The ADAPT 5 software was used for all data fitting and simulation of model equations (D’Argenio et al., 2009). The maximum likelihood method was applied for fitting the data. The variance model specified was:
where Vi is the variance of the ith data point, σ1 and σ2 are the variance parameters, and Yi is the model-predicted concentration or response. Variance parameters σ1 and σ2 were estimated along with model parameters during fittings. The goodness-of-fit was assessed by system convergence, visual inspection of the fitted curves, objective function values such as Akaike Information Criterion (AIC), improved likelihood, examination of residuals, and precision (%CV) of the estimated parameters.
Results
Clustering and Gene Ontology Analysis
The temporal profiles of MPL-regulated transcriptomics and proteomics are shown in Fig. 3. Time-course data for 163 common genes that were available from both transcriptomic and proteomic data sets were concatenated and hierarchically clustered as described previously (Kamisoglu et al., 2015). Functional description and discussion of the proteomic data has been reported (Ayyar et al., 2017). On the basis of the analysis, cluster 1 was populated with 80 genes for which corresponding mRNA and protein expression profiles were essentially parallel in direction, whereas for 83 genes in cluster 2 the directionality between mRNA and protein was reversed (Fig. 3). The collective dynamics of the mRNA in cluster 1 revealed peak expression around 4–8 hours after MPL, whereas the proteins in the same cluster peaked around 8 hours after dosing. Conversely, mRNA in cluster 2 were down-regulated by about 45% between 4 and 8 hours, and several corresponding proteins peaked around 6–8 hours.
Fig. 3.

Changes in mRNA and corresponding protein expression of CS-responsive genes in liver as a function of time after 50-mg/kg intravenous and intramuscular injections of MPL in ADX rats. Each green (mRNA) and yellow (protein) line represents connection of mean values for one gene or protein from two to four animals at each time point. Each solid pink and blue line depict the mean profile of all mRNAs and proteins within each cluster.
Quality-threshold clustering of both up- and down-regulated proteins was performed, by which proteins with highly similar temporal profiles were clustered (Fig. 4A). Coregulatory mechanisms for proteins within the same cluster were evaluated on the basis of a common transcription factor identification strategy, as employed previously in analyzing CS-induced mRNA expression (Jin et al., 2003; Nguyen et al., 2010). The common transcription factors of the clusters, extracted using the IPA package, are shown in Fig. 4A. Links between CS and identified transcription factors are supported by previous reports. For example, hepatocyte nuclear factor-4α (HNF-4α) plays a crucial role in the transcriptional regulation of hepatic gluconeogenesis (Suh and Rechler, 1997). HNF-4α interacts with the GR and may inhibit CS-enhanced transcription involved in liver glucose metabolism (Pierreux et al., 1999; Yamamoto et al., 2004). Nuclear factor (erythroid-derived 2)-like 2, NFE2L2, is a key transcription factor regulating detoxifying enzymes and antioxidant genes involved in hepatic drug metabolism (Kratschmar et al., 2012). Among the common transcription factors involved only in down-regulation, SWI/SN-related regulator of chromatin (SMARCB1) is responsible for the nucleosome disruption that may lead to repressing basal transcription of a number of genes (Ostlund Farrants et al., 1997). The CS are known to stimulate the nucleosome-disrupting activity for the SWI/SN complex (Ostlund Farrants et al., 1997).
Fig. 4.

Representative temporal clusters of drug-altered proteins (A). Four quality-threshold up-regulated protein clusters, one down-regulated protein cluster, and the main common contributing transcription factors to those protein clusters are shown. Interaction network for microRNA and drug-responsive target genes (B). Yellow boxes represent specific microRNA, and green boxes depict target genes. The blue asterisks on yellow boxes denote glucocorticoid-regulated microRNAs.
Glucocorticoid-induced regulation of miRNA expression as well as the miRNA-mediated regulation of glucocorticoid-inducible genes have been documented. The MPL-regulated genes from Fig. 3 were analyzed for experimentally validated interactions with specific miRNAs using miRTarBase (Chou et al., 2018). As depicted in Fig. 4B, 20 genes were found to interact with at least one miRNA in murine or rodent models. From a total of 25 interacting miRNAs, the expression of 16 were reported to be altered by glucocorticoids either in vitro or in vivo (Fig. 4B; Supplemental Table 1). For example, miR-155 was shown to increase by 2.4-fold in preadipocytes upon dexamethasone treatment (Peshdary and Atlas, 2018). The miR-210 is up-regulated by hypoxia in an HNF-1α-dependent manner in cardiomyocytes (Martinez et al., 2017). Of interest, HNF-1α was identified as an important coregulatory transcription factor for MPL actions (Fig. 4A), suggesting possibly intertwined mechanisms of gene regulation.
Pharmacokinetics and Receptor Dynamics
The pharmacokinetics of MPL for both intramuscular and intravenous studies were simulated using previously estimated parameters (Ayyar et al., 2018). The simulated curves shown reflect biexponential disposition of MPL (Supplemental Fig. 1). The parameter values are listed (Supplemental Table 2). The receptor mRNA, free cytosolic receptors, and the nuclear drug-receptor complex concentrations, which serves as the driving force for MPL actions, were simulated (Supplemental Fig. 1) using parameter values obtained from a previous report (Hazra et al., 2007a). Parameter values are provided (Supplemental Table 2).
Pharmacogenomics and Proteomics
Model A.
In total, 32 mRNA and their corresponding proteins were well captured by model A, assuming induced transcription by DRn, suggesting that all these genes are regulated by CS via similar mechanisms. Figure 5 shows representative fittings of 16 genes that were well described by model A. Genes described by this model included prototypic hepatic biomarkers of CS, including tyrosine aminotransferase (Tat), cytosolic aspartate aminotransferase (Got1), and tryptophan 2,3-dioxygenase (Tdo2). The peak of enhancement for the mRNA of these genes (5 hours) exhibited a marked time shift compared with the profile of the driving force DRn (Supplemental Fig. 1; peak at 2.2 hours), whereas their associated protein peaked at later times (8 hours and beyond). Table 1 lists the estimated parameters for all 64 mRNA and proteins described by model A. The mRNA and protein degradation rate constants (kd,mRNA and kd,protein) represents the drug-independent property of the physiologic system. The kd,mRNA exhibited a limited range between 0.08 and 0.8 hours−1, indicating that these mRNA have similar stability, with half-lives ranging from 1 to 8 hours. The estimated kd,protein varied to a greater extent, ranging from 0.008 to 1.5 hours−1, indicating a greater diversity in protein stability, with half-lives ranging from about 0.5 to 86 hours. The linear stimulation factor represents the drug-specific property of the message. The limited range of  estimates from 0.001 to 0.008 (fmol/mg protein)−1 implies that the transcriptional machineries of these genes have similar sensitivity to CS action. The estimated γ coefficient describing the efficiency of mRNA-to-protein translation showed a mean estimated value of 1.7, indicating a modest difference in the magnitude of protein enhancement from mRNA.
estimates from 0.001 to 0.008 (fmol/mg protein)−1 implies that the transcriptional machineries of these genes have similar sensitivity to CS action. The estimated γ coefficient describing the efficiency of mRNA-to-protein translation showed a mean estimated value of 1.7, indicating a modest difference in the magnitude of protein enhancement from mRNA.
Fig. 5.

Representative fittings of genes described by model A. Solid circles are the mean gene array data, and the open circles depict the mean protein data. Solid lines are fittings for each mRNA and dashed line for each protein after MPL. Estimated parameter values for each mRNA and protein are listed in Table 1.
TABLE 1.
Pharmacodynamic parameters for genes fitted by model A
| No. | Gene Name | Symbol |
kd,mRNA |
SDRn(mRNA) |
kd,protein |
γ |
||||
|---|---|---|---|---|---|---|---|---|---|---|
| Estimate | %CV | Estimate | %CV | Estimate | %CV | Estimate | %CV | |||
| h–1 | fmol/mg | h–1 | ||||||||
| 1 | 26S Proteasome regulatory subunit 8 | Pscm5 | 0.24 | 66 | 0.001 | 45 | 0.54 | 122 | 1.4 | 43 |
| 2 | 40S Ribosomal protein S5 | Rs5 | 0.33 | 66 | 0.001 | 40 | 1.1 | 159 | 1.9 | 33 |
| 3 | 40S Ribosomal protein S7 | Rs7 | 0.16 | 55 | 0.001 | 38 | 1.2 | 156 | 2.4 | 28 |
| 4 | 60S Ribosomal protein l23a | Rl23a | 0.09 | 54 | 0.003 | 49 | 0.35 | 218 | 0.5 | 78 |
| 5 | 60S Ribosomal protein L3 | Rl3 | 0.17 | 59 | 0.001 | 43 | 0.65 | 120 | 1.9 | 31 |
| 6 | ADP ribosylation factor 4 | Arf4 | 0.82 | 30 | 0.002 | 18 | 0.32 | 51 | 1.0 | Fixed |
| 7 | Argininosuccinate lyase | Asl | 0.74 | 30 | 0.003 | 19 | 0.05 | 141 | 0.7 | 105 |
| 8 | Argininosuccinate synthase 1 | Ass1 | 0.06 | 40 | 0.008 | 42 | 0.027 | 61 | 3.3 | 34 |
| 9 | Aspartate aminotransferase | Got1 | 0.34 | 37 | 0.006 | 14 | 0.024 | 57 | 2.1 | 21 |
| 10 | Aspartate–tRNA ligase, cytoplasmic | Sdac | 0.49 | 52 | 0.001 | 26 | 0.10 | 45 | 3.8 | 28 |
| 11 | Aspartyl-tRNA Synthetase | Dars | 0.37 | 60 | 0.001 | 33 | 0.81 | 123 | 1.6 | 36 |
| 12 | CCAAT-binding transcription factor i | Ybx1 | 0.07 | 34 | 0.005 | 32 | 0.1 | 54 | 1.5 | 27 |
| 13 | Cytochrome P450 27A1 | Cyp27a1 | 0.12 | 48 | 0.002 | 38 | 0.58 | 98 | 1.0 | 28 |
| 14 | Cytochrome P450 reductase | Por | 0.11 | 32 | 0.008 | 30 | 0.036 | 55 | 1.8 | 31 |
| 15 | Galectin-9 | Leg9 | 0.07 | 30 | 0.007 | 30 | 0.019 | 57 | 3.9 | 28 |
| 16 | Heat Shock 70 kDa protein 5 | Hspa5 | 0.90 | 42 | 0.001 | 15 | 0.3 | Fixed | 0.4 | 53 |
| 17 | Heat shock cognate 71 kDa protein | Hspa8 | 0.19 | 50 | 0.002 | 35 | 0.04 | 97 | 1.5 | 60 |
| 18 | Heat shock protein d (hsp60) 1 | Hspd1 | 0.10 | 38 | 0.003 | 32 | 0.06 | 98 | 0.7 | 67 |
| 19 | Karyopherin subunit beta 1 | Kpnb1 | 0.20 | 31 | 0.003 | 24 | 0.3 | Fixed | 0.6 | 36 |
| 20 | NADPH:P450 oxidoreductase | Ncpr | 0.10 | 38 | 0.007 | 26 | 0.045 | 56 | 1.7 | 27 |
| 21 | Nucleolin | Ncl | 0.40 | 38 | 0.004 | 17 | 0.06 | 44 | 1.0 | Fixed |
| 22 | Nucleophosmin | Npm | 0.28 | 29 | 0.006 | 10 | 0.041 | 51 | 1.4 | 21 |
| 23 | Oligosaccharyltransferase subunit 48 | Ddost | 0.46 | 37 | 0.001 | 21 | 0.3 | Fixed | 1.0 | Fixed |
| 24 | Phosphoglucomutase-1 | Pgm1 | 1.56 | 37 | 0.002 | 16 | 0.3 | Fixed | 0.2 | 59 |
| 25 | Proteasome 26S subunit, ATPase 2 | Psmc2 | 0.27 | 43 | 0.002 | 27 | 0.10 | 183 | 0.7 | 107 |
| 26 | Ribosomal protein l4 | Rpl4 | 0.20 | 50 | 0.001 | 32 | 0.07 | 61 | 2.1 | 35 |
| 27 | Signal activator of transcription 3 | Stat3 | 0.33 | 24 | 0.005 | 11 | 0.12 | 22 | 1.9 | 9.3 |
| 28 | Tryptophan di-oxygenase | Tdo2 | 0.16 | 25 | 0.004 | 21 | 1.5 | 65 | 2.0 | 15 |
| 29 | Tubulin alpha 1 | Tuba1c | 0.09 | 49 | 0.004 | 45 | 0.3 | Fixed | 0.2 | 175 |
| 30 | Tubulin beta 4b class iv | Tubb4b | 0.07 | 43 | 0.004 | 41 | 0.023 | 123 | 1.8 | 84 |
| 31 | Tudor domain-containing protein 11 | Snd1 | 0.14 | 54 | 0.001 | 40 | 0.031 | 96 | 2.3 | 64 |
| 32 | Tyrosine aminotransferase | Tat | 0.26 | 55 | 0.002 | 34 | 0.17 | 50 | 5.0 | 26 |
Model B.
Seven expressed genes were described reasonably well by model B assuming an inhibition on the mRNA. Figure 6 shows representative fittings of four genes that were captured by model B. Included within this group are such genes as Mug-1/2 and Cyp2c18 that have been reported previously to be down-regulated by steroids (Northemann et al., 1989; de Morais et al., 1993). Table 2 lists the estimated parameters for the mRNA and proteins described by model B. In general, message expression for the genes in this group were down-regulated by 30%–50%, reaching a nadir around 6–8 hours after dosing, which can be attributed to similar kd,mRNA values. Protein expression was down-regulated in accordance with the time course of mRNA but showed a delayed nadir around 20 hours. This may be explained by the longer half-lives for proteins compared with their mRNA (around 21 hours). The  of the transcripts described by model B ranged from 130 to 780 fmol/mg protein.
of the transcripts described by model B ranged from 130 to 780 fmol/mg protein.
Fig. 6.

Representative fittings of genes described by model B. Solid circles are the mean gene array data, and the open circles depict the mean protein data. Solid lines are fittings for each mRNA and dashed line for each protein after MPL. Estimated parameter values for each mRNA and protein are listed in Table 2.
TABLE 2.
Pharmacodynamic parameters for genes fitted by model B
| No. | Gene Name | Symbol |
kd,mRNA |
IC50, DRn(mRNA) |
kd,protein |
γ |
||||
|---|---|---|---|---|---|---|---|---|---|---|
| Estimate | %CV | Estimate | %CV | Estimate | %CV | Estimate | %CV | |||
| h−1 | fmol/mg | h−1 | ||||||||
| 1 | Alanine–glyoxylate aminotransferase 2 | Agxt2 | 0.13 | 15 | 130.7 | 28 | 0.074 | 36 | 1.0 | Fixed |
| 2 | Aldehyde dehydrogenase 1A7 | Aldh1a7 | 0.50 | 85 | 622.0 | 46 | 0.3 | Fixed | 1.0 | Fixed |
| 3 | Amine oxidase A | Maoa | 0.90 | 18 | 587.2 | 7 | 0.013 | 63 | 1.0 | Fixed |
| 4 | Carboxylesterase 1E | Ces1e | 0.41 | 33 | 272.8 | 25 | 0.033 | 31 | 5.0 | Fixed |
| 5 | Cytochrome P450 2C18 | Cyp2c18 | 0.50 | Fixed | 416.8 | 17 | 0.018 | 43 | 5.0 | Fixed |
| 6 | Glutathione peroxidase 1 | Gpx1 | 1.50 | 50 | 779.4 | 21 | 0.020 | 88 | 1.1 | 7 |
| 7 | Murinoglobulin-1 | Mug1/2 | 0.24 | 27 | 164.6 | 34 | 0.038 | 35 | 5.0 | Fixed |
Model C.
In total, eight expressed genes were well captured by model C. The mRNA and proteins described by this model showed rapid increases after MPL, followed by an immediate decline below baseline, an acute tolerance/rebound phenomenon. Both mRNA and protein returned to baseline beyond 30 hours. Model C assumed an initial enhancement produced by the transcriptional control of MPL via DRn, whereas the hypothetical secondary regulator SR produced by linear transduction was responsible for the decrease in mRNA, which explained the acute tolerance phenomenon. Figure 7 depicts representative fittings of six genes that were captured by model C. The estimated parameters for the mRNA and proteins described by model C are provided in Table 3. The mean estimated kd,mRNA of 0.37 hours−1 within this group was like that estimated for mRNA described by model A (0.31 hours−1), and the kd,protein values for most proteins within this cluster ranged between 0.01 and 0.6 hours−1. The mean degradation rate constant for the secondary regulator (kSR), excluding that for Rnp2, was 0.11 hours−1, which is slightly slower compared with the first-order constants for target mRNA. The Rnp2 gene displayed a distinct profile with mRNA and protein peaking slightly earlier than the DRn (not shown), which explains the much faster rate constants kSR and kd,protein for Rnp2. Both 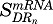 and
and 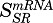 averaged about 0.008 (fmol/mg protein)−1, which is comparable to the sensitivity constants for genes described by model A.
averaged about 0.008 (fmol/mg protein)−1, which is comparable to the sensitivity constants for genes described by model A.
Fig. 7.

Representative fittings of genes described by model C. Solid circles are the mean gene array data, and the open circles depict the mean protein data. Solid lines are fittings for each mRNA and dashed line for each protein after MPL. Estimated parameter values for each mRNA and protein are listed in Table 3.
TABLE 3.
Pharmacodynamic parameters for genes fitted by model C
| No. | Gene Name | Symbol |
kSR |
SSR(mRNA)
|
kd,mRNA |
SDRn(mRNA)
|
kd,protein |
γ |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Est | %CV | Est | %CV | Est | %CV | Est | %CV | Est | %CV | Est | %CV | |||
| h−1 | (fmol/mg)−1 | h−1 | (fmol/mg)−1 | h−1 | ||||||||||
| 1 | Chaperonin containing TCP1 subunit 3 | Cct3 | 0.01 | 174 | 0.022 | 97 | 0.1 | 70 | 0.005 | 51 | 0.66 | 165 | 0.3 | 43 |
| 2 | Eukaryotic translation initiation factor 4A2 | Eif4a2 | 0.06 | 73 | 0.006 | 45 | 0.2 | 63 | 0.004 | 51 | 0.042 | 51 | 2.1 | 32 |
| 3 | Heterogeneous nuclear ribonucleoprotein A/B | Hnrnpab | 0.21 | 50 | 0.019 | 191 | 0.03 | 182 | 0.025 | 191 | 3.8 | 380 | 0.4 | 33 |
| 4 | Polypyrimidine tract-binding protein 1 | Ptbp1 | 0.18 | 98 | 0.002 | 37 | 0.3 | - | 0.002 | 50 | 0.65 | 113 | 1.7 | 37 |
| 5 | PAPS synthase 2 | Papss2 | 0.18 | 47 | 0.005 | Fixed | 0.5 | 28 | 0.007 | 27 | 0.24 | 68 | 0.7 | 39 |
| 6 | Ribonuclease P protein | Rnp2 | 3.04 | 27 | 0.086 | 107 | 0.04 | 107 | 0.106 | 106 | 2.6 | 69 | 1.0 | Fixed |
| 7 | RNA-binding protein 8A | Tars | 0.09 | 83 | 0.003 | 44 | 1.4 | 52 | 0.003 | 31 | 0.28 | 111 | 0.5 | 50 |
| 8 | UDP-glucose 6-dehydrogenase | Ugdh | 0.05 | 92 | 0.004 | 64 | 0.4 | 80 | 0.002 | 49 | 0.012 | 74 | 6.8 | 64 |
Est, estimate.
Models D and E.
Some genes in cluster 1 showed a fast and prolonged decline in mRNA followed by a further delayed sustained induction. Their corresponding proteins showed either modest early decreases or remained unchanged before increasing above baseline. This pattern suggests that, as with genes described by model C, two mechanisms might be involved in CS action. To describe the observed patterns, various models were tested, including two competing models (models D and E) developed by Jin et al. (2003) to describe such transcript patterns. Model D assumed repressed transcription by DRn followed by an enhanced transcription that was mediated by a steroid-enhanced SR. Model E assumed an inhibition of mRNA synthesis by DRn and an inhibition of mRNA degradation by the steroid-enhanced regulator SR. Figure 8 depicts representative fittings of four genes that were described by models D and E. On the basis of the goodness-of-fit and the precision of the estimated parameters, model E performed better than model D in capturing the mRNA and proteins. The estimated parameters for the mRNA and proteins described by models D and E are provided in Tables 4 and 5. Some parameter estimates were associated with relatively high %CV, especially for model D, implying that the models might be somewhat overparameterized. Like findings by Jin et al., mRNA described by model E yielded high kd,mRNA estimates ranging from 1.1 to 3 hours−1, suggesting that the messages may have relatively low stability with half-lives ranging from 15 to 45 minutes. Furthermore, the steep initial slopes of mRNA down-regulation in this cluster can be attributed to the high kd,mRNA values. The generally low  for genes fitted by model E implied that the transcriptional machineries of these genes are sensitive to CS repression. The low kd,protein values, averaging 0.031 hours−1, indicates a mean half-life of about 23 hours, which suggests that these proteins are quite stable. The slower degradation kinetics of these proteins also explain their shallower, prolonged temporal profiles.
for genes fitted by model E implied that the transcriptional machineries of these genes are sensitive to CS repression. The low kd,protein values, averaging 0.031 hours−1, indicates a mean half-life of about 23 hours, which suggests that these proteins are quite stable. The slower degradation kinetics of these proteins also explain their shallower, prolonged temporal profiles.
Fig. 8.

Representative fittings of genes described by models D and E. Solid circles are the mean gene array data, and the open circles depict the mean protein data. Solid lines are fittings for each mRNA and dashed line for each protein after MPL. Lines colored in blue are fits by model D and black lines are fits by model E. Estimated parameter values for each mRNA and protein are listed in Table 4 (model D) and Table 5 (model E).
TABLE 4.
Pharmacodynamic parameters for genes fitted by model D
| No. | Gene Name | Symbol |
kSR |
SSR(mRNA)
|
kd,mRNA |
IC50, DRn(mRNA)
|
kd,protein |
γ |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Est | %CV | Est | %CV | Est | %CV | Est | %CV | Est | %CV | Est | %CV | |||
| h−1 | (fmol/mg)−1 | h−1 | fmol/mg | h−1 | ||||||||||
| 1 | Adenosine kinase | Adk | 1.4 | 42 | 0.005 | 62 | 0.75 | 78 | 98.6 | 280 | 0.03 | 95 | 1.0 | Fixed |
| 2 | Arginase 1 | Arg1 | 0.04 | 40 | 0.017 | 54 | 1.2 | 52 | 100 | Fixed | 0.01 | 95 | 1.0 | Fixed |
| 3 | Cystathionine-beta-synthase | Cbs | 0.22 | 49 | 0.005 | 113 | 0.5 | 164 | 139 | 540 | 0.08 | 68 | 1.4 | 43 |
| 4 | Heat shock protein HSP 90β | Hsp90ab | 0.38 | 46 | 0.003 | 102 | 0.76 | 146 | 262 | 292 | 0.04 | 176 | 1.0 | Fixed |
| 5 | Sulfotransferase 1A1 | Sult1a1 | 0.07 | 36 | 0.01 | 28 | 1.2 | 63 | 64.6 | 280 | 0.05 | 94 | 1.0 | Fixed |
See eqs. (10), (14), and (18) for definition of parameters. Est, estimate.
TABLE 5.
Pharmacodynamic parameters for genes fitted by model E
| No. | Gene Name | Symbol |
kSR |
IC50, SR(mRNA) |
kd,mRNA |
IC50, DRn(mRNA) |
kd,protein |
γ |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Est | %CV | Est | %CV | Est | %CV | Est | %CV | Est | %CV | Est | %CV | |||
| h−1 | fmol/mg | h−1 | fmol/mg | h−1 | ||||||||||
| 1 | Adenosine kinase | Adk | 0.32 | 94 | 154.6 | 75 | 1.4 | 42 | 369.2 | 120 | 0.02 | 116 | 1.0 | Fixed |
| 2 | Arginase 1 | Arg1 | 0.14 | 76 | 76.3 | 47 | 2.8 | 35 | 162.0 | 79 | 0.01 | 93 | 1.0 | Fixed |
| 3 | Cystathionine-beta-synthase | Cbs | 0.19 | 71 | 191.5 | 31 | 1.1 | 69 | 799.6 | 79 | 0.1 | 73 | 1.5 | 46 |
| 4 | Heat shock protein HSP 90-beta | Hsp90ab1 | 0.07 | 72 | 274.0 | 52 | 2.1 | 207 | 4063 | 135 | 0.04 | 115 | 1.0 | Fixed |
| 5 | Sulfotransferase 1A1 | Sult1a1 | 0.17 | 43 | 54.0 | 39 | 2.4 | 19 | 154.7 | 55 | 0.03 | 47 | 1.0 | Fixed |
Est, estimate.
Model F.
A significant number of genes in cluster 2 (Fig. 3) showed atypical profiles: Transcripts and proteins changed in opposite directions. In particular, several genes within this cluster displayed a time-dependent down-regulation of mRNA (characteristic of genes described by model B) but an up-regulation in corresponding protein expression. Some of the proteins returned to baseline, whereas others displayed biphasic regulation. To describe this pattern, model F assumed an inhibition of mRNA production by DRn in conjunction with a secondary, DR-mediated process inhibiting the rate of target protein degradation. The basis of this putative nongenomic, post-translational model of CS action is a recently identified molecular mechanism mediated by dexamethasone (Kong et al., 2017). The mRNA and protein expressions of 10 mRNA and their corresponding proteins were well described by this model. Figure 9 depicts the fittings of six representative genes. The mRNA within this group were down-regulated to nadir by 3 to 4 hours after MPL, whereas protein expression peaked around 5 to 6 hours post-dosing. The estimated parameters for the genes described by model F are listed in Table 6. The system parameters kd,mRNA and kd,protein averaged around 0.9 and 0.3 hours−1, indicating quicker turnover of message compared with protein. The mean  of the mRNA captured by this model was 459 fmol/mg protein, quite like that for mRNA described by model B (425 fmol/mg protein). To reduce the overall number of estimated parameters, the rate constant for DR to interact with protein was assumed to be equal to the nuclear translocation rate constant of DR (kt). The cytoplasmic concentrations of DR* peaked sharply around 25 minutes (∼4.5 fmol/mg protein) after MPL, and returned to baseline by 12 hours (not shown). The estimated
of the mRNA captured by this model was 459 fmol/mg protein, quite like that for mRNA described by model B (425 fmol/mg protein). To reduce the overall number of estimated parameters, the rate constant for DR to interact with protein was assumed to be equal to the nuclear translocation rate constant of DR (kt). The cytoplasmic concentrations of DR* peaked sharply around 25 minutes (∼4.5 fmol/mg protein) after MPL, and returned to baseline by 12 hours (not shown). The estimated 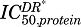 for the target proteins described by this model ranged from 0.06 to 2 fmol/mg protein.
for the target proteins described by this model ranged from 0.06 to 2 fmol/mg protein.
Fig. 9.

Representative fittings of genes described by models F. Solid circles are the mean gene array data, and the open circles depict the mean protein data. Solid lines are fittings for each mRNA and dashed line for each protein after MPL. Estimated parameter values for each mRNA and protein are listed in Table 6 (model F).
TABLE 6.
Pharmacodynamic parameters for genes fitted by model F
| No. | Gene Name | Symbol |
IC50DR*(protein) |
kd,mRNA |
IC50, DRn(mRNA) |
kd,protein |
||||
|---|---|---|---|---|---|---|---|---|---|---|
| Est | %CV | Est | %CV | Est | %CV | Est | %CV | |||
| fmol/mg | h−1 | fmol/mg | h−1 | |||||||
| 1 | 17β-Hydroxysteroid dehydrogenase type II | Hsd17β2 | 0.056 | 82 | 0.31 | 9 | 8.9 | 35 | 0.35 | 42 |
| 2 | Acetyl-coenzyme A acyltransferase 2 | Acaa2 | 2 | 48 | 0.26 | 52 | 508.2 | 43 | 0.34 | 53 |
| 3 | Acyl-CoA dehydrogenase long chain | Acadl | 1.1 | 37 | 0.96 | 41 | 729.1 | 25 | 0.33 | 41 |
| 4 | Alcohol dehydrogenase 4 | Adh4 | 1.3 | 62 | 0.59 | 49 | 543.9 | 31 | 0.24 | 67 |
| 5 | Aldehyde dehydrogenase 3 member A2 | Aldh3a2 | 0.54 | 74 | 0.98 | 34 | 368 | 22 | 0.1 | 84 |
| 6 | Cytochrome P450 2A12 | Cyp2a12 | 0.7 | 33 | 0.61 | 22 | 280.8 | 15 | 0.28 | 36 |
| 7 | Cytochrome P450 2C40 | Cyp2c40 | 0.72 | 29 | 1.3 | 58 | 971.3 | 24 | 0.52 | 30 |
| 8 | Glutathione S-transferase A3 | Gsta3 | 0.93 | 41 | 0.17 | 28 | 267.9 | 37 | 0.21 | 32 |
| 9 | Isocitrate dehydrogenase 1 | Idh1 | 0.63 | 91 | 0.87 | 17 | 66.7 | 13 | 0.2 | 92 |
| 10 | Nucleotide pyrophosphatase 1 | Enpp1 | 0.33 | 30 | 2.5 | 47 | 845.7 | 25 | 0.45 | 22 |
Est, estimate.
Discussion
This report examined the temporal relationships between the liver transcriptome and proteome following MPL dosing and used pharmacokinetic/pharmacodynamic systems modeling to assess CS pharmacogenomics at the mRNA and protein levels in livers harvested from intact animals. This area has been of general interest in molecular and systems pharmacology, as protein expression is often cited as being more complementary to drug efficacy and toxicity than is mRNA expression. Our previous studies using microarrays provided the basis to model the possible receptor-mediated mechanisms controlling the time course of several mRNAs (Jin et al., 2003). Together, our bioinformatics (Kamisoglu et al., 2015) and current model-based analysis indicate that transcript expression recapitulated protein dynamics for approximately 45%–50% of the genes for which both transcript and protein information were available within the –omics data sets. The present models serve to provide mechanistic hypotheses on how mRNA and protein turnover are controlled by primary and secondary drug effects occurring during transcriptional, post-transcriptional, translational, and post-translational processing. These models confirm known mechanisms at both mRNA and protein levels for some of the genes studied, but in some cases represent possibilities with general molecular mechanisms as a basis, and thus require further exploration with gene-specific experiments.
Numerous factors affect the temporal profiles of drug-responsive proteins, such as early receptor signaling, transcriptional effects, and post-transcriptional factors, including miRNA. Additionally, the kinetics of mRNA and protein turnover also govern their temporal responses. Common hepatic transcription factors such as HNF-4α, NFE2L2, and SMARCB1 were posed as factors contributing to the common temporal characteristics of the clusters (Fig. 4A). Of emerging interest is the role of miRNAs as mediators of glucocorticoid signaling and response (Clayton et al., 2018). On the basis of this analysis, 20 genes were found to interact with at least one miRNA in murine or rodent models. Additionally, the expression of 16 interacting miRNAs were reported to be glucocorticoid-regulated (Fig. 4B; Supplemental Table 1). Our analysis is limited in that it considered studies that reported glucocorticoid-dependent regulation of miRNAs across any type of cell line and tissue (i.e., not liver-specific). However, these findings provide some basis to warrant further investigation of miRNAs as mediators in hepatic glucocorticoid actions.
A large group of genes that showed time-dependent increases in transcript and protein expression were well captured by model A, which assumed a nuclear complex-mediated stimulation of mRNA synthesis rate. Genes described by this model included well studied biomarkers of CS such as Tat, Got1, and Tdo2, as well as several genes related to such cell regulatory processes as transcription and translation (e.g., nucleolin and nucleophosmin). Enhancement of the mRNAs and proteins of genes involved in amino acid breakdown, such as Tat, Got1, and Tdo2, by MPL can be confirmed by the presence of at least one GRE sequence within each of their promoter regions (Jantzen et al., 1987; Comings et al., 1995; Garlatti et al., 1996). It was recently demonstrated that hepatic cytochrome P450 reductase (Por) mRNA and protein were modestly up-regulated following a single 1.5 mg/kg i.p. dose of dexamethasone in rats (Hunter et al., 2017). This is comparable to our findings. The similar magnitude of induction despite a much lower dose was possibly attributable to the higher potency of dexamethasone compared with MPL.
Some genes exhibited a time-dependent down-regulation at both mRNA and protein levels after MPL dosing. For example, Mug-1/2 mRNA and protein expression was reduced by about 50% in our studies, consistent with a previous report demonstrating transient down-regulation in Mug-1/2 mRNA at 4 hours after a 4-mg/kg dexamethasone injection in rats (Northemann et al., 1989). The mechanism for the direct, receptor-mediated down-regulation of Cyp2c18 mRNA by MPL is supported by the presence of GREs within its sequence (de Morais et al., 1993). In contrast with our microarray data set, a relatively lower proportion of down-regulated proteins were mined from the proteomics data set. This could be because low-abundance proteins were not detectable, especially upon down-regulation by MPL. Furthermore, some proteins may not have met cut-off criteria during mining (e.g., quantification at all time points).
Models involving primary and secondarily induced mechanisms of actions were needed to describe select genes that showed more complex biphasic temporal patterns. For instance, the polypyrimidine tract binding protein 1 (Ptbp1) gene showed a profile in which its mRNA and protein were enhanced but then fell below baseline before returning to steady-state. As indicated by our miRNA analysis, miR-124 interacts with Ptbp1. The miRNA-dependent regulation of genes can occur through endonucleic cleavage of the target mRNA upon it base-pairing with the miRNA (Valencia-Sanchez et al., 2006). In addition, the expression of miR-124 can be induced by endogenous and exogenous glucocorticoids (Clayton et al., 2018). In summary, CS induces the expression of a regulator (miR-124) that mediates the destabilization of a primary gene (Ptbp1), secondary to the enhancement of Ptbp1 mRNA by CS. Although these molecular mechanisms have been elucidated primarily in neuronal systems (Makeyev et al., 2007), this mechanistic hypothesis is testable through measurement of miR-124 dynamics in liver, where expression of this miRNA has been confirmed (Liu et al., 2016a). Two other genes with evidence for secondary regulation by transcription factors are arginase 1 (Arg-1) and sulfotransferase 1A1 (Sult1a1), which are regulated by C/EBP (Gotoh et al., 1997; Jin et al., 2003) and the constitutive androgen receptor (CAR) (Duanmu et al., 2001; Fang et al., 2003).
Most genes in cluster 2 displayed patterns with down-regulated mRNA and up-regulated proteins. Our understanding of the biology and mechanisms behind this observation is limited. However, recent studies in macrophages by Kong et al. (2017) have provided key insights into a novel post-translational mechanism of glucocorticoid signaling. They demonstrated that dexamethasone-activated GR acts in a rapid, transcription-independent manner to interact with an inflammation-related cytoplasmic protein IRAK1 and thus interferes with protein-protein interactions between IRAK1 and β-TrCP (an E3 ligase), and subsequently suppresses K48 linkage-specific ubiquitination of IRAK1. In essence, the cytosolic drug-receptor complex rapidly acts to inhibit the degradation rate of IRAK1, and possibly other target proteins in a like manner, via inhibition of the ubiquitin-mediated proteasomal degradation of proteins. This formed the mechanistic basis for our mathematical model (model F) that captured several cluster 2 genes reasonably well. However, more detailed in vitro experiments in hepatocyte systems, possibly similar to those conducted by Kong, are necessary to validate the applicability of this model to describe specific genes.
Although the literature confirms that CS alters expression of many mRNAs and proteins that we observed, it is sometimes difficult to compare our results with previous work, especially for the genes with biphasic patterns found to be regulated differently at different times. Questions of drug, dose, time, in vitro/in vivo differences, and quantification methods arise (Jin et al., 2003). An extensive comparative analysis of our -omics data sets to those reported by others is also challenging, as most have investigated transcriptomics or proteomics at single time points after dosing. Nonetheless, changes in Tat protein were validated with measurements of enzyme activity in the same animals (Ayyar et al., 2018), and the pathways perturbed within our transcriptomic and proteomic data sets are, in terms of function, in agreement with recognized adverse and therapeutic effects of CS (Ayyar et al., 2017).
In this analysis, we modeled simultaneously the mRNA and protein dynamics corresponding to individual genes. The estimated rate and effect parameters for hypothetical regulators may represent a composite of multiple steps in the absence of the true biologic mediators. Further integrated models incorporating RNA-protein, protein-protein, and protein-DNA interactions and their inter-regulation will provide additional insights into signaling networks at molecular, cellular, and systemic levels. This type of approach was adopted in a more focused modeling analysis that integrated selected signaling pathways with physiologic PD endpoints of MPL efficacy and toxicity (Ayyar et al., 2018). The present models serve to analyze the time course of CS-regulated transcriptomics and proteomics as a whole to provide hypotheses on how mRNA and protein turnover is controlled by direct and secondary factors.
In addition to the technical limitations with use of microarrays (Jin et al., 2003), this study was limited by technical factors such as sensitivity limits in our proteomics methodology, use of nonperfused versus perfused livers for our transcriptomics versus proteomics animal studies, and limitations in the sensitivity of clustering analysis. Male ADX rats were used in our experiments to obviate endogenous effects of corticosterone, which could have altered the natural physiologic response to CS. Transcriptomics and proteomics were assessed at a single dose of MPL, which obliged use of linear stimulation constants instead of more appropriate nonlinear Hill-type functions. It has been recognized that chronic MPL dosing introduces added complexities in pharmacogenomic responses (Hazra et al., 2008). Certain parameters are associated with relatively high %CV, especially for describing genes with more complicated behaviors, suggestive of model overparameterization. This issue was limited when possible by fixing parameters to physiologically plausible values. For six genes (five described by model A and one by model B) yielding poor precision on kd, protein, values were fixed to 0.3 hours−1, which is the mean of all kd, protein values obtained for all other genes in cluster A that were estimated with reasonable precision by model fitting. The reasons for selecting the current high dose employed (50 mg/kg) were: 1) In conducting -omics assessments, it was our aim to evoke the largest number of changes of expressed transcripts and proteins possible within the tissue, and 2) we aimed to perturb drug-regulated mRNA and proteins toward a system-capacity from their baseline, which would allow for better resolution of their temporal properties, and consequently aid our modeling efforts.
In summary, we employed microarray technology with mass spectroscopy-based proteomics methods to jointly analyze temporal changes in steroid-regulated genes and proteins to evaluate underlying pharmacogenomic processes and to evolve our generalized mathematical models of receptor/gene/protein dynamics. This enhances our understanding on the global actions of CS in liver and provides some insights into how gene expression is controlled by turnover at various steps.
Acknowledgments
The authors thank Jiawei Zhou for technical assistance in data analysis.
Abbreviations
- ADX
adrenalectomized
- CS
corticosteroid(s)
- GR
glucocorticoid receptor
- GRE
glucocorticoid response element
- HNF-4α
hepatocyte nuclear factor-4α
- LC/MS
liquid chromatography–mass spectrometry
- miRNA
microRNA
- MPL
methylprednisolone
- PK
pharmacokinetic
- PD
pharmacodynamic
- SR
secondary regulator
Authorship Contributions
Participated in research design: Ayyar, Sukumaran, DuBois, Almon, Jusko.
Conducted experiments: Ayyar, DuBois.
Contributed new reagents or analytic tools: Ayyar, DuBois, Almon, Jusko.
Performed data analysis: Ayyar, Sukumaran, Jusko.
Wrote or contributed to the writing of the manuscript: Ayyar, DuBois, Almon, Jusko.
Footnotes
This work was supported by the National Institute of General Medical Sciences, National Institutes of Health [Grant GM24211].
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Almon RR, DuBois DC, Brandenburg EH, Shi W, Zhang S, Straubinger RM, Jusko WJ. (2002) Pharmacodynamics and pharmacogenomics of diverse receptor-mediated effects of methylprednisolone in rats using microarray analysis. J Pharmacokinet Pharmacodyn 29:103–129. [DOI] [PubMed] [Google Scholar]
- Almon RR, DuBois DC, Pearson KE, Stephan DA, Jusko WJ. (2003) Gene arrays and temporal patterns of drug response: corticosteroid effects on rat liver. Funct Integr Genomics 3:171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyar VS, Almon RR, DuBois DC, Sukumaran S, Qu J, Jusko WJ. (2017) Functional proteomic analysis of corticosteroid pharmacodynamics in rat liver: relationship to hepatic stress, signaling, energy regulation, and drug metabolism. J Proteomics 160:84–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyar VS, Sukumaran S, DuBois DC, Almon RR, Qu J, Jusko WJ. (2018) Receptor/gene/protein-mediated signaling connects methylprednisolone exposure to metabolic and immune-related pharmacodynamic actions in liver. J Pharmacokinet Pharmacodyn 45:557–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ. (1998) Efficacy of inhaled corticosteroids in asthma. J Allergy Clin Immunol 102:531–538. [DOI] [PubMed] [Google Scholar]
- Cain DW, Cidlowski JA. (2017) Immune regulation by glucocorticoids. Nat Rev Immunol 17:233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Teo G, Krueger S, Rock TM, Koh HWL, Choi H, Vogel C. (2016) Differential dynamics of the mammalian mRNA and protein expression response to misfolding stress. Mol Syst Biol 12:855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Park OH, Park J, Ryu I, Kim J, Ko J, Kim YK. (2015) Glucocorticoid receptor interacts with PNRC2 in a ligand-dependent manner to recruit UPF1 for rapid mRNA degradation. Proc Natl Acad Sci USA 112:E1540–E1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou CH, Shrestha S, Yang CD, Chang NW, Lin YL, Liao KW, Huang WC, Sun TH, Tu SJ, Lee WH, et al. (2018) miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res 46 (D1):D296–D302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton SA, Jones SW, Kurowska-Stolarska M, Clark AR. (2018) The role of microRNAs in glucocorticoid action. J Biol Chem 293:1865–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comings DE, Muhleman D, Dietz G, Sherman M, Forest GL. (1995) Sequence of human tryptophan 2,3-dioxygenase (TDO2): presence of a glucocorticoid response-like element composed of a GTT repeat and an intronic CCCCT repeat. Genomics 29:390–396. [DOI] [PubMed] [Google Scholar]
- Crick F. (1970) Central dogma of molecular biology. Nature 227:561–563. [DOI] [PubMed] [Google Scholar]
- D’Argenio D, Schumitzky A, Wang X. (2009) ADAPT 5 User’s Guide: Pharmacokinetic/Pharmacodynamic Systems Analysis Software. BioMedical Simulations Resource, Los Angeles, CA. [Google Scholar]
- de Morais SM, Schweikl H, Blaisdell J, Goldstein JA. (1993) Gene structure and upstream regulatory regions of human CYP2C9 and CYP2C18. Biochem Biophys Res Commun 194:194–201. [DOI] [PubMed] [Google Scholar]
- Duanmu Z, Kocarek TA, Runge-Morris M. (2001) Transcriptional regulation of rat hepatic aryl sulfotransferase (SULT1A1) gene expression by glucocorticoids. Drug Metab Dispos 29:1130–1135. [PubMed] [Google Scholar]
- Fang HL, Shenoy S, Duanmu Z, Kocarek TA, Runge-Morris M. (2003) Transactivation of glucocorticoid-inducible rat aryl sulfotransferase (SULT1A1) gene transcription. Drug Metab Dispos 31:1378–1381. [DOI] [PubMed] [Google Scholar]
- Garlatti M, Aggerbeck M, Bouguet J, Barouki R. (1996) Contribution of a nuclear factor 1 binding site to the glucocorticoid regulation of the cytosolic aspartate aminotransferase gene promoter. J Biol Chem 271:32629–32634. [DOI] [PubMed] [Google Scholar]
- Gotoh T, Chowdhury S, Takiguchi M, Mori M. (1997) The glucocorticoid-responsive gene cascade. Activation of the rat arginase gene through induction of C/EBPbeta. J Biol Chem 272:3694–3698. [DOI] [PubMed] [Google Scholar]
- Hazra A, DuBois DC, Almon RR, Jusko WJ. (2007a) Assessing the dynamics of nuclear glucocorticoid-receptor complex: adding flexibility to gene expression modeling. J Pharmacokinet Pharmacodyn 34:333–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazra A, DuBois DC, Almon RR, Snyder GH, Jusko WJ. (2008) Pharmacodynamic modeling of acute and chronic effects of methylprednisolone on hepatic urea cycle genes in rats. Gene Regul Syst Bio 2:1–19. [PMC free article] [PubMed] [Google Scholar]
- Hazra A, Pyszczynski N, DuBois DC, Almon RR, Jusko WJ. (2007b) Modeling receptor/gene-mediated effects of corticosteroids on hepatic tyrosine aminotransferase dynamics in rats: dual regulation by endogenous and exogenous corticosteroids. J Pharmacokinet Pharmacodyn 34:643–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazra A, Pyszczynski N, DuBois DC, Almon RR, Jusko WJ. (2007c) Pharmacokinetics of methylprednisolone after intravenous and intramuscular administration in rats. Biopharm Drug Dispos 28:263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter SR, Vonk A, Mullen Grey AK, Riddick DS. (2017) Role of glucocorticoid receptor and pregnane X receptor in dexamethasone induction of rat hepatic aryl hydrocarbon receptor nuclear translocator and NADPH-cytochrome P450 oxidoreductase. Drug Metab Dispos 45:118–129. [DOI] [PubMed] [Google Scholar]
- Jantzen HM, Strähle U, Gloss B, Stewart F, Schmid W, Boshart M, Miksicek R, Schütz G. (1987) Cooperativity of glucocorticoid response elements located far upstream of the tyrosine aminotransferase gene. Cell 49:29–38. [DOI] [PubMed] [Google Scholar]
- Jin JY, Almon RR, DuBois DC, Jusko WJ. (2003) Modeling of corticosteroid pharmacogenomics in rat liver using gene microarrays. J Pharmacol Exp Ther 307:93–109. [DOI] [PubMed] [Google Scholar]
- Jin JY, DuBois DC, Almon RR, Jusko WJ. (2004) Receptor/gene-mediated pharmacodynamic effects of methylprednisolone on phosphoenolpyruvate carboxykinase regulation in rat liver. J Pharmacol Exp Ther 309:328–339. [DOI] [PubMed] [Google Scholar]
- Jusko WJ. (1995) Pharmacokinetics and receptor-mediated pharmacodynamics of corticosteroids. Toxicology 102:189–196. [DOI] [PubMed] [Google Scholar]
- Jusko WJ. (2013) Moving from basic toward systems pharmacodynamic models. J Pharm Sci 102:2930–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamisoglu K, Acevedo A, Almon RR, Coyle S, Corbett S, Dubois DC, Nguyen TT, Jusko WJ, Androulakis IP. (2017) Understanding physiology in the continuum: integration of information from multiple -omics levels. Front Pharmacol 8:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamisoglu K, Sukumaran S, Nouri-Nigjeh E, Tu C, Li J, Shen X, Duan X, Qu J, Almon RR, DuBois DC, et al. (2015) Tandem analysis of transcriptome and proteome changes after a single dose of corticosteroid: a systems approach to liver function in pharmacogenomics. OMICS 19:80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirwan JR, Gunasekera W. (2017) Is there a renaissance of glucocorticoids in rheumatoid arthritis? Clin Pharmacol Ther 102:574–577. [DOI] [PubMed] [Google Scholar]
- Kong F, Liu Z, Jain VG, Shima K, Suzuki T, Muglia LJ, Starczynowski DT, Pasare C, Bhattacharyya S. (2017) Inhibition of IRAK1 ubiquitination determines glucocorticoid sensitivity for TLR9-induced inflammation in macrophages. J Immunol 199:3654–3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratschmar DV, Calabrese D, Walsh J, Lister A, Birk J, Appenzeller-Herzog C, Moulin P, Goldring CE, Odermatt A. (2012) Suppression of the Nrf2-dependent antioxidant response by glucocorticoids and 11β-HSD1-mediated glucocorticoid activation in hepatic cells. PLoS One 7:e36774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, Monsen E, Dabney AR, Storey JD. (2006) EDGE: extraction and analysis of differential gene expression. Bioinformatics 22:507–508. [DOI] [PubMed] [Google Scholar]
- Liu X, Zhao J, Liu Q, Xiong X, Zhang Z, Jiao Y, Li X, Liu B, Li Y, Lu Y. (2016a) MicroRNA-124 promotes hepatic triglyceride accumulation through targeting tribbles homolog 3. Sci Rep 6:37170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Beyer A, Aebersold R. (2016b) On the dependency of cellular protein levels on mRNA abundance. Cell 165:535–550. [DOI] [PubMed] [Google Scholar]
- Maier T, Güell M, Serrano L. (2009) Correlation of mRNA and protein in complex biological samples. FEBS Lett 583:3966–3973. [DOI] [PubMed] [Google Scholar]
- Makeyev EV, Zhang J, Carrasco MA, Maniatis T. (2007) The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol Cell 27:435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez SR, Ma Q, Dasgupta C, Meng X, Zhang L. (2017) MicroRNA-210 suppresses glucocorticoid receptor expression in response to hypoxia in fetal rat cardiomyocytes. Oncotarget 8:80249–80264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TT, Almon RR, Dubois DC, Jusko WJ, Androulakis IP. (2010) Comparative analysis of acute and chronic corticosteroid pharmacogenomic effects in rat liver: transcriptional dynamics and regulatory structures. BMC Bioinformatics 11:515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northemann W, Shiels BR, Braciak TA, Fey GH. (1989) Structure and negative transcriptional regulation by glucocorticoids of the acute-phase rat alpha 1-inhibitor III gene. Biochemistry 28:84–95. [DOI] [PubMed] [Google Scholar]
- Nouri-Nigjeh E, Sukumaran S, Tu C, Li J, Shen X, Duan X, DuBois DC, Almon RR, Jusko WJ, Qu J. (2014) Highly multiplexed and reproducible ion-current-based strategy for large-scale quantitative proteomics and the application to protein expression dynamics induced by methylprednisolone in 60 rats. Anal Chem 86:8149–8157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostlund Farrants AK, Blomquist P, Kwon H, Wrange O. (1997) Glucocorticoid receptor-glucocorticoid response element binding stimulates nucleosome disruption by the SWI/SNF complex. Mol Cell Biol 17:895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne SH. (2015) The utility of protein and mRNA correlation. Trends Biochem Sci 40:1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peshdary V, Atlas E. (2018) Dexamethasone induced miR-155 up-regulation in differentiating 3T3-L1 preadipocytes does not affect adipogenesis. Sci Rep 8:1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peshkin L, Wühr M, Pearl E, Haas W, Freeman RM, Jr, Gerhart JC, Klein AM, Horb M, Gygi SP, Kirschner MW. (2015) On the relationship of protein and mRNA dynamics in vertebrate embryonic development. Dev Cell 35:383–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phuc Le P, Friedman JR, Schug J, Brestelli JE, Parker JB, Bochkis IM, Kaestner KH. (2005) Glucocorticoid receptor-dependent gene regulatory networks. PLoS Genet 1:e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierreux CE, Stafford J, Demonte D, Scott DK, Vandenhaute J, O’Brien RM, Granner DK, Rousseau GG, Lemaigre FP. (1999) Antiglucocorticoid activity of hepatocyte nuclear factor-6. Proc Natl Acad Sci USA 96:8961–8966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan R, DuBois DC, Almon RR, Pyszczynski NA, Jusko WJ. (2002a) Fifth-generation model for corticosteroid pharmacodynamics: application to steady-state receptor down-regulation and enzyme induction patterns during seven-day continuous infusion of methylprednisolone in rats. J Pharmacokinet Pharmacodyn 29:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan R, DuBois DC, Almon RR, Pyszczynski NA, Jusko WJ. (2002b) Pharmacodynamics and pharmacogenomics of methylprednisolone during 7-day infusions in rats. J Pharmacol Exp Ther 300:245–256. [DOI] [PubMed] [Google Scholar]
- Schäcke H, Döcke WD, Asadullah K. (2002) Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther 96:23–43. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LK, Tandon A, Shah RR, Mav D, Scoltock AB, Cidlowski JA. (2013) Deep sequencing identification of novel glucocorticoid-responsive miRNAs in apoptotic primary lymphocytes. PLoS One 8:e78316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Xiao W, Leek JT, Tompkins RG, Davis RW. (2005) Significance analysis of time course microarray experiments. Proc Natl Acad Sci USA 102:12837–12842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh DS, Rechler MM. (1997) Hepatocyte nuclear factor 1 and the glucocorticoid receptor synergistically activate transcription of the rat insulin-like growth factor binding protein-1 gene. Mol Endocrinol 11:1822–1831. [DOI] [PubMed] [Google Scholar]
- Sun YN, Jusko WJ. (1998) Transit compartments versus gamma distribution function to model signal transduction processes in pharmacodynamics. J Pharm Sci 87:732–737. [DOI] [PubMed] [Google Scholar]
- Taylor AL, Watson CJE, Bradley JA. (2005) Immunosuppressive agents in solid organ transplantation: mechanisms of action and therapeutic efficacy. Crit Rev Oncol Hematol 56:23–46. [DOI] [PubMed] [Google Scholar]
- Tu C, Li J, Sheng Q, Zhang M, Qu J. (2014) Systematic assessment of survey scan and MS2-based abundance strategies for label-free quantitative proteomics using high-resolution MS data. J Proteome Res 13:2069–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia-Sanchez MA, Liu J, Hannon GJ, Parker R. (2006) Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev 20:515–524. [DOI] [PubMed] [Google Scholar]
- Valinezhad Orang A, Safaralizadeh R, Kazemzadeh-Bavili M. (2014) Mechanisms of miRNA-mediated gene regulation from common downregulation to mRNA-specific upregulation. Int J Genomics 2014:970607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C. (2013) Evolution. Protein expression under pressure. Science 342:1052–1053. [DOI] [PubMed] [Google Scholar]
- Vogel C, Marcotte EM. (2012) Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet 13:227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Shimano H, Nakagawa Y, Ide T, Yahagi N, Matsuzaka T, Nakakuki M, Takahashi A, Suzuki H, Sone H, et al. (2004) SREBP-1 interacts with hepatocyte nuclear factor-4 alpha and interferes with PGC-1 recruitment to suppress hepatic gluconeogenic genes. J Biol Chem 279:12027–12035. [DOI] [PubMed] [Google Scholar]